

Abstract
This work aimed to investigate the co-grinding effects of β-cyclodextrin (β-CD) and cucurbit[7]uril (CB[7]) on crystalline zaltoprofen (ZPF) in tablet formulation. Crystalline ZPF was prepared through anti-solvent recrystallization and fully analyzed through single-crystal X-ray diffraction. Co-ground dispersions and mono-ground ZPF were prepared using a ball grinding process. Results revealed that mono-ground ZPF slightly affected the solid state, solubility, and dissolution of crystalline ZPF. Co-ground dispersions exhibited completely amorphous states and elicited a significant reinforcing effect on drug solubility. UV-vis spectroscopy, XRPD, FT-IR, DSC, ssNMR, and molecular docking demonstrated the interactions in the amorphous product. Hardness tests on blank tablets with different β-CD and CB[7] contents suggested the addition of β-CD or CB[7] could enhance the compressibility of the powder mixture. Disintegration tests showed that CB[7] could efficiently shorten the disintegrating time. Dissolution tests indicated that β-CD and CB[7] could accelerate the drug dissolution rate via different mechanisms. Specifically, CB[7] could accelerate the dissolution rate by improving disintegration and β-CD showed a distinct advantage in solubility enhancement. Based on the comparative study on β-CD and CB[7] for tablet formulation combined with co-grinding, we found that CB[7] could be considered a promising drug delivery, which acted as a disintegrant.
Cyclodextrins (CDs) and cucurbit[n]urils (CB[n]s) are two types of macrocycles commonly used in supramolecular chemistry to improve drug properties, such as solubility, stability, and bioavailability1,2,3,4. Natural CDs are cyclic oligosaccharides containing six (α-CD), seven (β-CD), and eight (γ-CD) glucose units by α-(1,4) linkages. CB[n]s are generally synthetic pumpkin-like rigid macrocycles comprising n (n = 5, 6, 7, 8, 10) glycoluril units doubly-bridged by methylene linkers5. More recently, other twisted CB[n]s (n = 13, 14, 15) were synthesized and separated6,7. β-CD (Fig. 1a) was firstly found in 18918 and has been studied for over 100 years as pharmaceutical excipient9. However, CB[7] (Fig. 1b) was discovered in 200010 and still has wide research prospects. Currently, most reported studies on CB[7] have focused on the inclusion mechanism with drugs at a molecular level11,12,13. Further developments of CB[7]-containing drug dosage forms, such as tablets for oral delivery, have been seldom described. To our best knowledge, reports discussing the dosage forms of CB[n]s are limited, except for Wheate et al. who have investigated five different dosage forms: CB[6] in oral solid tablet14, CB[6] in topical cream15, CB[6,7,8]s in eye drop16, CB[7] in implantable hydrogel17 and CB[6,7]s in nasal insert18. Although CB[7] and β-CD exhibit similar cavity size, water solubility, and low toxicity19,20,21, they manifest different inclusion behaviors because of their structural distinctions22. Thus, we comparatively investigated their features in solid-state dispersion and tablet formulation.
Figure 1.

The chemical structure of (a) β-CD, (b) CB[7] and (c) ZPF.
Poorly soluble zaltoprofen (ZPF, logP = 4.25, pKa = 4.21; Fig. 1c) was chosen as a model drug because its commercial formulation is oral tablet and its crystal form is easily obtained. It is a nonsteroidal anti-inflammatory drug (NSAID) that is more potent for the treatment of inflammatory conditions accompanied by pain than other NSAIDs23. Numerous methods, including co-evaporation, co-crystallization, freeze-drying, spray-drying, and co-grinding24,25,26, have been widely utilized to prepare drug–excipient dispersions. Among these methods, co-grinding drugs with macrocycles is regarded as the most suitable for scale-up and environmentally friendly industrial production27. Nevertheless, when utilizing co-grinding method, there is one thing worthy of being considered. Solely grinding drug without excipient (mono-grinding) may also destroy the drug crystal structure by shear force, even into amorphous forms28,29, then make more or less effect. Thus, to verify the effects of co-grinding on the drug properties were caused by the adding of macrocycles, the effect of mono-grinding on drug must be excluded.
This study aimed to understand the co-grinding effect of β-CD and CB[7] on the physicochemical properties of crystalline ZPF and to explore their corresponding influence on tablet formulation. Crystalline ZPF was prepared through anti-solvent recrystallization, and its structure was determined by single-crystal X-ray diffraction. Co-ground dispersions (ZPF−CB[7], ZPF−β-CD) were prepared by co-grinding process, and the mono-ground ZPF was also obtained under the same grinding condition as control. Interactions in co-ground dispersions were investigated by using UV-vis spectroscopy, X-ray powder diffraction (XRPD), Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), solid-state NMR spectroscopy (ssNMR), scanning electron microscopy (SEM) and molecular simulation. The effects of mono-ground ZPF, physical mixtures, and co-ground dispersions on the solubility of crystalline ZPF in water, simulated gastric fluid (SGF), and simulated intestinal fluid (SIF) were also evaluated. Blank tablets with various contents (0%–50%) of β-CD and CB[7] were designed to reveal their specific roles in tablet hardness and disintegration performance. The influence of co-ground dispersions on ZPF tablets was further analyzed through disintegration and dissolution tests. This work provided a basis for the development of new dosage forms by co-grinding method and explored the features of using CB[7] as a tablet excipient in pharmaceutical production.
Results
Single-crystal X-ray diffraction
Anti-solvent recrystallization method was utilized to obtain crystalline ZPF. Specifically, crystalline ZPF for X-ray diffraction (Fig. S1A) was obtained through dissolving commercial ZPF in dichloromethane, and n-hexane (V/V = 3:7) was gradually added without stirring at room temperature. X-ray diffraction data were collected on an Xcalibur E X-ray single crystal diffractometer and the structure was solved with Olex230, and refined with SHELXS-9731 using least square minimization. The determined crystal structure is illustrated in Fig. S1B, and the corresponding crystallographic data are summarized in Table S1 (CCDC No. 1520356). Each ZPF ligand is hydrogen-bonded to an uncoordinated carboxy oxygen atom of an adjacent dimer [O–H···O 2.653(2) Å], resulting in a wavy network that stacks via π-π interactions into a 3D architecture (Fig. S2). Microcrystalline ZPF (Fig. S1C) used for tablet formulation was prepared by the same solvent system but with vigorous stirring with n-hexane addition. The microcrystalline ZPF produced in this manner consists of particles with sizes ranging from 10 μm to 130 μm with a mean of 50 μm.
UV-vis spectroscopy and molecular docking study
The interactions between ZPF and CB[7] in solution have been determined by UV-vis spectroscopy previously32, and the corresponding binding constant that calculated by Benesi–Hildebrand equation was 276 M−1. The interactions between ZPF and β-CD in solution were also evaluated by Benesi–Hildebrand equation here. The absorbance intensity of ZPF (Fig. 2A) shows a hyperchromic effect with increasing β-CD concentrations, suggesting the formation of inclusion complex between ZPF and β-CD. From the Benesi–Hildebrand plot, the binding constant of ZPF-β-CD complex was calculated to be 341 M−1.
Figure 2.

(A) Absorption spectra of ZPF in the presence of various concentrations of β-CD from 0, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0 × 10−3 M; Insert: the Benesi–Hildebrand plot of 1/∆A against 1/[β-CD]. (B) Docking conformation of CB[7] and ZPF. (C,D) Two main docking conformations of β-CD and ZPF.
Molecular docking was used to predict the most favorable structures and interaction details of ZPF-CB[7] and ZPF-β-CD complexes. Due to the symmetrical portals of CB[7], there is only one kind of docking conformation of ZPF-CB[7] complex (Fig. 2B). Two main conformations that ZPF molecule entered into the cavity of β-CD from the wide portal (Fig. 2C) and narrow (Fig. 2D) portal respectively were obtained. Regardless of the different structures of macrocycles, the polycyclic moiety of ZPF molecule, including C = O group and phenyl group, was encapsulated both in the hydrophobic cavity of CB[7] and β-CD, and the carboxyl group formed hydrogen bonds with macrocycles. The diverse inclusion conformations and stronger hydrogen bonds between ZPF and β-CD may result in the larger binding constant.
Characterization of the microcrystalline and mono-ground ZPF, physical mixtures, and binary co-ground dispersions
XRPD measurements demonstrated that the characteristic XRPD peaks of microcrystalline ZPF were evidently broadened by grinding solely into mono-ground ZPF for 5 h (Fig. 3A). This finding suggested that the molecularly ordered arrangement of microcrystalline ZPF was partly disrupted and showed a tendency of becoming amorphous. However, ZPF–CB[7] and ZPF–β-CD dispersions exhibited completely amorphous states, indicating that the amorphization of ZPF can be promoted by co-grinding with CB[7] and β-CD. By contrast, the XRPD patterns of physical mixtures presented a simple aggregate of patterns from the corresponding ZPF and macrocycles.
Figure 3.

(A) XRPD patterns and (B) FT-IR spectra of microcrystalline ZPF, mono-ground ZPF and their corresponding physical mixtures and co-ground dispersions (ZPF−CB[7], ZPF−β-CD).
In the FT-IR spectra (Fig. 3B), the characteristic absorption peaks of the mono-ground ZPF were almost the same as that of the microcrystalline ZPF, which presented in the spectra of physical mixtures as well. However, the peaks at 1703 and 1667 cm−1, belonging to the stretching vibration of C = O groups, and the peaks at 1588 and 1458 cm−1, assigned to the skeleton vibrations of phenyl groups, disappeared after the formation of amorphous dispersions, which demonstrates that the phenyl ring and carbonyl of ZPF interacted with CB[7] and β-CD. This finding is consistent with the result of molecular docking study.
The DSC curves of microcrystalline ZPF and mono-ground ZPF exhibited a sharp endothermic peak at 138 °C and 131 °C, respectively, which correspond to their melting points (Fig. 4A). When microcrystalline ZPF was ground into mono-ground ZPF by shear force, the melting points varied based on the different solid forms of ZPF. The thermogram of β-CD displayed broad endothermic peaks at approximately 100 °C and 300 °C, which correspond to the release of water molecules and the melting point, respectively. For the DSC curve of CB[7], only a water-released peak was observed because the melting point of CB[7] is beyond 400 °C33. The thermograms of physical mixtures ZPF + CB[7] and ZPF + β-CD show the presence of either microcrystalline ZPF or mono-ground ZPF melting peak, indicating that no interaction occurred when the drug and macrocycles were simply mixed. Conversely, the melting peak of ZPF disappeared in both ZPF–CB[7] and ZPF–β-CD amorphous dispersions curves, which indicated that ZPF was embedded by CB[7] and β-CD.
Figure 4.

(A) DSC curves of microcrystalline ZPF, mono-ground ZPF and their corresponding physical mixtures and co-ground dispersions. (B) 13C CP/MAS NMR spectra of microcrystalline ZPF, mono-ground ZPF, CB[7], β-CD, ZPF−β-CD and ZPF−CB[7]. Numbering for assignments corresponds to Figure 1. (C) SEM images of (a) microcrystalline ZPF, (b) mono-ground ZPF, (c) ZPF−CB[7], and (d) ZPF−β-CD at 500 × magnification.
ssNMR provides a direct way to probe the interactions in solid complexes and identify polymorphic forms34. Fig. 4B illustrates the 13C CP-MAS spectra of microcrystalline ZPF, mono-ground ZPF, CB[7], β-CD, and their corresponding amorphous solid dispersions. The 13C CP-MAS spectrum of microcrystalline ZPF exhibited typical crystalline systems with multiple and sharp resonances for each type of carbon. Comparatively broadening signals were observed in the spectrum of mono-ground ZPF, which suggested that the molecular arrangement and intermolecular interactions were partly disrupted after grinding. However, the spectra of ZPF–CB[7] and ZPF–β-CD amorphous dispersions showed the broad featureless resonances of the macrocycle carbon atoms, and the sharp signals of ZPF were distinctly broadened and even merging into the base line after they co-ground with CB[7] and β-CD. This observation demonstrates the loss of overall crystallinity or drug inclusion with macrocycles35. Consistent with the XRPD results, this finding confirms their amorphous nature and disordered arrangement and further indicates that solid phase transformation occurs and the dispersions are new solid forms.
The SEM images present the morphology of microcrystalline ZPF, mono-ground ZPF, ZPF–CB[7], and ZPF–β-CD (Fig. 4C). The ZPF crystals showed a prismatic form, whereas the mono-ground ZPF was a granulated aggregation with a much smaller size. ZPF–CB[7] and ZPF–β-CD dispersions also contained smaller particle sizes and exhibited rough, fluffy, irregular surfaces. Furthermore, ZPF co-ground with CB[7] produced more dispersive complexes than β-CD did.
Solubility studies
The effects of mono-grinding and co-grinding with CB[7] and β-CD on the solubility of microcrystalline ZPF are summarized in Table 1. Grinding ZPF alone for 5 h did not significantly improve the solubility of microcrystalline ZPF. β-CD-containing physical mixtures and co-ground dispersions showed increased ZPF solubility in pure water and SGF. However, no drug solubility improvement was observed for CB[7]-containing physical mixtures in water and SGF. Hence, the solubilization effect of CB[7] is weaker than that of β-CD. CBs are known to preferentially bind guests with positive charges, whereas CDs prefer neutral guest molecules19. With neutral ZPF molecules existing in water and SGF, β-CD played a major role in complexation. Interestingly, the co-ground dispersion with CB[7] shows increased ZPF solubility. The solubilization effect of ZPF-CB[7] may be caused by the interactions between CB[7] and ZPF, combining with the disordered molecular arrangement. In SIF, an adverse effect was even observed in the solubility of ZPF by adding β-CD or CB[7]. The different behaviors can be explained by the corresponding ionization equilibria of drugs in the media of different pH values. The ionized and negatively charged ZPF molecules composed of a great proportion in SIF were hard to embed into the hydrophobic cavities of CB[7] or β-CD. A main reason for the decreased solubility in SIF is probably that once a neutral ZPF molecule is complexed with CB[7] and β-CD, the ionization equilibrium shifts and the amount of ionized ZPF molecule is reduced, then the solubility is accordingly reduced. Overall, co-ground dispersions with CB[7] and β-CD were determined to have higher ZPF solubility than its corresponding physical mixtures because of the decreased crystallinity and formed inclusion caused by co-grinding.
Table 1. Solubility of microcrystalline ZPF, mono-ground ZPF and the corresponding physical mixtures (ZPF + CB[7] and ZPF + β-CD) and co-ground dispersions (ZPF−CB[7], ZPF−β-CD) in pure water, SGF and SIF at 37 ± 0.1 °C after 72 h.
| Samples | Solubility (mg/mL) |
||
|---|---|---|---|
| Water | SGF | SIF | |
| Microcrystalline ZPF | 0.018 ± 0.003 | 0.003 ± 0.001 | 2.23 ± 0.132 |
| Mono-ground ZPF | 0.021 ± 0.005 | 0.005 ± 0.001 | 2.37 ± 0.051 |
| Microcrystalline ZPF + CB[7] | 0.023 ± 0.004 | 0.008 ± 0.002 | 1.19 ± 0.035& |
| Microcrystalline ZPF + β-CD | 0.090 ± 0.008* | 0.022 ± 0.004# | 1.45 ± 0.041& |
| Mono-ground ZPF + CB[7] | 0.025 ± 0.005 | 0.009 ± 0.003 | 1.28 ± 0.079& |
| Mono-ground ZPF + β-CD | 0.093 ± 0.007* | 0.022 ± 0.006# | 1.49 ± 0.086& |
| ZPF−CB[7] | 0.076 ± 0.006* | 0.052 ± 0.005# | 1.23 ± 0.082& |
| ZPF−β-CD | 0.101 ± 0.013* | 0.063 ± 0.006# | 1.58 ± 0.051& |
Values are presented as mean ± SD (n = 3), *p < 0.05 compared with microcrystalline ZPF group in water; #p < 0.05 compared with microcrystalline ZPF group in SGF; &p < 0.05 compared with microcrystalline ZPF group in SIF.
Tablet hardness and disintegration testing
The thickness of blank tablet without CB[7] or β-CD (F0; detailed blank tablet formulation are given in Supplementary Table S2) was set to 2.6 mm to achieve hardness within 4–6 kg. To investigate the effects of β-CD and CB[7] on the disintegrating time of tablet, the hardness must kept consistent (4–6 kg) by adjusting the tablet thickness. The adjusted thickness and corresponding disintegrating time are summarized in Table 2. The tablet thickness was increased gradually as the CB[7] and β-CD contents increased. Therefore, the addition of β-CD or CB[7] instead of lactose enhanced the compressibility of the powder mixture, and the compressibility of CB[7] was better than that of β-CD. Testing the disintegrating time of various β-CD-containing tablets showed that the addition of β-CD almost had no effect on the disintegrating time. Nevertheless, increasing the CB[7] content beyond 15% significantly shortened the disintegrating time. The excellent behavior of CB[7] in disintegration makes it especially suitable as a disintegrant in the preparation of immediate-release tablets.
Table 2. The tablet thickness and disintegrating time of blank tablets containing 0%, 5%, 15%, 25% and 50% CB[7] or β-CD in pure water.
| F0 | β-CD |
CB[7] |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 0% | 5% | 15% | 25% | 50% | 5% | 15% | 25% | 50% | |
| Tablet thickness (mm) | 2.6 | 2.6 | 2.8 | 3.0 | 3.1 | 2.8 | 3.0 | 3.2 | 3.7 |
| Disintegrating time (min) | 36 | 34 | 33 | 30 | 32 | 32 | 5 | 2 | <1 |
The disintegrating time of various ZPF tablets (each tablet containing 20% CB[7] or β-CD, and 80 mg ZPF; detailed ZPF tablet formulation are given in Supplementary Table S3) in water, SGF and SIF were evaluated (Table 3). In SGF, each ZPF tablet exhibits the same short disintegrating time. In water and SIF, it is obvious that the addition of CB[7], either in a free state or a co-ground state in tablets showed significant improvement on disintegrating time. Addition of β-CD and mono-grinding ZPF would not make any influence on the disintegration of microcrystalline ZPF tablet.
Table 3. The disintegrating time of various ZPF tablets (each tablet containing 20% CB[7] or β-CD, and 80 mg ZPF) in water, SGF and SIF.
| Samples | Disintegrating time (min) |
||
|---|---|---|---|
| Water | SGF | SIF | |
| Microcrystalline ZPF tablet | 31 | 1 | 27 |
| Mono-ground ZPF tablet | 26 | 1 | 21 |
| Microcrystalline ZPF + CB[7] tablet | <1 | <1 | <1 |
| Microcrystalline ZPF + β-CD tablet | 33 | <1 | 36 |
| Mono-ground ZPF + CB[7] tablet | <1 | <1 | <1 |
| Mono-ground ZPF + β-CD tablet | 29 | <1 | 31 |
| ZPF−CB[7] tablet | <1 | <1 | <1 |
| ZPF−β-CD tablet | 30 | <1 | 33 |
ZPF tablet dissolution test
The cumulative release curves of microcrystalline ZPF, mono-ground ZPF, and their corresponding physical mixtures (ZPF + CB[7], ZPF + β-CD) and co-ground dispersions (ZPF−CB[7], ZPF−β-CD) tablets in pure water, SGF, and SIF are shown in Fig. 5. Overall, the dissolution rate of microcrystalline ZPF tablet (left in Fig. 5) was not influenced observably by grinding ZPF without macrocycles into mono-ground ZPF (right in Fig. 5). Generally, improving tablet disintegration and solubility of drug would correspondingly increase the drug dissolution rate from tablet. In pure water, CB[7]-containing tablets performed much shorter disintegrating time than that of β-CD-containing tablets (Table 3). But contrary, the dissolution rate of CB[7]-containing tablets were slower than that of β-CD-containing tablets. β-CD-containing tablets exhibited a more significant enhancement in dissolution rate as well in SGF. This finding suggests that although CB[7]-containing tablets disintegrated quickly, the solubilizing ability of β-CD played a major role in increasing the dissolution rate when the dissolution medium is pure water or SGF. Nevertheless, CB[7]-containing tablets showed the fastest dissolution rate in SIF. This drug release performance demonstrated that when the drug solubility was not improved in SIF, the excellent behavior in disintegration of CB[7] accelerated the drug dissolution rate. Thus, β-CD and CB[7] could both accelerate the drug dissolution rate via different mechanisms. Specifically, CB[7] could accelerate the dissolution rate by improving disintegration and β-CD showed a distinct advantage in solubility enhancement. Additionally, the drug release percentage of ZPF–CB[7] tablets was evidently greater in SIF (nearly 100%) than in SGF (<10% after 180 min). This finding suggested that the intestinal environment could trigger the release of prepared ZPF–CB[7] tablets. Furthermore, the effects of CB[7] on the dissolution of positively charged drugs should be evaluated systematically in the future because of the expected synergistic improvement in both solubility and disintegration.
Figure 5.

Mean (n = 6) release curves of microcrystalline ZPF, mono-ground ZPF, and their corresponding physical mixtures (ZPF + CB[7], ZPF + β-CD) and co-ground dispersions (ZPF−CB[7], ZPF−β-CD) tablets in (A) pure water, (B) SGF and (C) SIF at 37 ± 0.1 °C under 50 rpm stirring.
Discussion
In summary, the amorphous dispersions of ZPF co-ground with β-CD and CB[7] were successfully prepared through a ball grinding process to investigate their effects on the physicochemical properties of crystalline ZPF. Mono-ground ZPF was also prepared under the same grinding condition to eliminate the influence of polymorphism. Multiple characterization techniques, specifically UV-vis spectroscopy, molecular docking, XRPD, FI-IR, DSC, and ssNMR demonstrated the interactions between ZPF and macrocycles. The molecular arrangement of mono-ground ZPF was incompletely disordered and this arrangement slightly affected the solubility, disintegration, and dissolution rate of crystalline ZPF. Solubility and dissolution of crystalline ZPF can be significantly enhanced from co-ground dispersions (ZPF–CB[7] and ZPF–β-CD). Various blank tablets containing 0%–50% β-CD and CB[7] were prepared through direct powder compression to examine the effects of β-CD and CB[7] on oral tablets. Hardness tests revealed that the addition of β-CD and CB[7] increased the compressibility of the powder mixture, and the same weight of CB[7] elicited a stronger effect on compressibility than β-CD did. Disintegration tests demonstrated that more than 15% CB[7] effectively shortened the disintegrating time and acted as a disintegrant. In vitro dissolution tests showed that CB[7] accelerated the dissolution rate mainly by shortening the disintegrating time. β-CD displayed a distinct advantage in solubility enhancement. This study indicated that amorphous co-ground dispersions can effectively improve intrinsic drug solubility and dissolution. This study also revealed the enormous potential of CB[7] in the preparation of immediate-release tablets for future applications.
Methods
Materials
Glycoluril (98%) and paraformaldehyde powder (96%) were purchased from J&K Chemical (Beijing, China). β-CD (98%) and concentrated hydrogen chloride (HCl, 37%) were obtained from Chengdu Kelong Chemical Co., Ltd. (Chengdu, China). ZPF (98%) was purchased from Guangzhou Ciming Biological Technology Co., Ltd. (Guangzhou, China). All pharmaceutical excipients were kindly supplied by Sichuan Huirui Pharmaceutical Co., Ltd. (Chengdu, China). Tri-distilled water was used throughout the experiments.
CB[7] synthesis
CB[7] was synthesized by a modified procedure of Bardelang et al.36 1H NMR spectrum and MALDI-TOF-MS of synthetic CB[7] are shown in Figs S3 and S4, respectively. Elemental analysis for C42H42N28O14(H2O)10 is stated as follows: C 37.56, H 4.65, N 29.20%; found: C 37.61, H 4.64, N 29.09%.
Recrystallization and grinding
Crystalline ZPF was obtained by recrystallization from anti-solvent system (dichloromethane/n-hexane). Mono-ground ZPF, co-ground dispersions ZPF–β-CD, and ZPF–CB[7] were prepared by grinding 1500 mg ZPF solely, 298 mg ZPF and 1135 mg β-CD, 298 mg ZPF and 1343 mg CB[7] respectively in a 50 mL agate jar with grinding balls (mixture of 10 × 10 mm, 20 × 5 mm; agate) for 5 h using a planetary mill (LNMN-QM 0.4L, Heishan Xinlitun Agate Handicrafts Co., Ltd., China) at room temperature. The rotation speed of the solar disk was set to 400 rpm, with alternate grinding periods (5 min) and pause periods (1 min). For comparison, corresponding binary physical mixtures with same ratio at 1:1 were prepared by simply mixing ZPF and β-CD or CB[7] for 5 min by using a vortex blender.
UV-vis spectroscopy and molecular docking study
UV-vis spectroscopy was performed by using a TU-1901 UV-vis spectrophotometer (Persee, China) from 200–400 nm. Absorption spectra were recorded with various concentrations of β-CD (0, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0 × 10−3 M) adding to a constant ZPF concentration (1 × 10−4 M). The interactions between β-CD and ZPF were determined by Benesi–Hildebrand equation:37
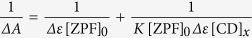 |
where ΔA is the change in absorbance of ZPF at 337 nm in the presence and absence of β-CD; Δε is the change in molar extinction coefficients between ZPF and its inclusion complex; [ZPF]0 and [CD]x are the concentrations of ZPF and β-CD, respectively; and K is the binding constant that could be determined from the plot of 1/ΔA versus 1/[CD]x.
Theoretical studies are available to predict the favorable conformations of inclusion complexes and give insight into the interaction details. The structure of β-CD (CCDC No. 1107192) and CB[7] (CCDC No. 1428201) were abstracted from Cambridge Structural Database, and all attached water molecules were removed. The structure of ZPF was abstracted from the crystal structure that we solved. After which, molecular docking was conducted using Lamarckian genetic algorithm in AutoDock 4.0 program package (Accelrys). Docking simulations were performed with 200 runs, 2,500,000 energy evaluations, 27,000 numbers of generations, and 150 GA populations.
Solid-state characterizations
XRPD analysis was performed at room temperature using an X’Pert PRO diffractometer (PANalytical Co., Ltd., Netherlands) with a PIXcel 1D detector and Cu Kα radiation (λ = 1.54056 Å, generator setting: 40 kV and 40 mA). Diffraction data were collected at a step size of 0.01313° and a counting time of 30 ms/step.
FT-IR spectra were recorded on a FT-IR spectrometer (Nicolet 6700, Thermo Fisher Scientific) according to the KBr disc technique. Measurements were performed within the 4000 cm−1 to 400 cm−1 scanning range.
Differential scanning calorimetry (DSC) was conducted using a TA Q200 thermal analyzer (TA Instruments Co., New Castle, DE, USA). Samples in 5–8 mg were sealed in Al crucibles for DSC analysis at a 10 °C/min heating rate from 30 °C to 400 °C under nitrogen.
High resolution solid state 13C spectra of microcrystalline and mono-ground ZPF, CB[7], β-CD, and co-ground systems ZPF–CB[7] and ZPF–β-CD were recorded with a ramp cross polarization/magic angle spinning (CP-MAS) sequence. All ssNMR experiments were performed on a Bruker AV II-500 MHz NMR spectrometer operating at Larmor frequencies of 125.76 MHz for 13C and 500.13 MHz for 1H and equipped with a 4 mm (od) MAS probe at 303 K. 13C CP/MAS NMR chemical shifts were referenced with respect to tetramethylsilane at 0 ppm, using adamantine as secondary external standard. A spectral width of 37.9 kHz, recycle delay of 3 s, and acquisition time of 49.9 ms were used.
SEM was used to evaluate the morphological characteristics of the samples. Samples were coated with an electrically conducting thin layer of gold, then examined using a JSM-7500F SEM (JEOL, Japan) at 5.0 kV.
Solubility studies
The effects of mono-ground ZPF, physical mixtures, and co-ground dispersions of β-CD and CB[7] on the solubility of crystalline ZPF were studied at 37.0 ± 0.1 °C in pure water, simulated gastric fluid (SGF; consisting of 2.0 g NaCl and 7.0 mL concentrated HCl per 1.0 L of water; pH = 1.1), and simulated intestinal fluid (SIF; consisting of 6.8 g of KH2PO4 and 0.89 g of NaOH in 1.0 L of water; pH = 6.8). An excess of crystalline and mono-ground ZPF, physical mixtures, and co-ground dispersions were added into water and buffered aqueous solutions. Suspensions were sonicated for 15 min then placed for 72 h in a water bath. After reaching equilibrium, the remaining solid was removed by filtration through a 0.45 μm membrane filter. The clear filtrates were suitably diluted and analyzed by UV–vis spectrophotometry at 337 nm. Each analysis was repeated three times.
Blank tablets and ZPF tablets formulation
The formulation code and composition of blank tablets and ZPF tablets are shown in Table S2 and S3, respectively. The blank tablet formulation was designed referring to the report of Wheate14 as initial formulation to evaluate the effect of increasing content of β-CD and CB[7] on the tablets. Briefly, blank tablets containing 0%–50% lactose, corresponding 50%–0% β-CD or CB[7], 42.5% Avicel, 1% magnesium stearate, 2.5% talc, and 4% carboxyl methyl cellulose (CMC) were prepared by direct powder compression. ZPF tablets, containing 80 mg crystalline ZPF, mono-ground ZPF in the absence and presence of 20% (60 mg) β-CD or CB[7], and co-ground ZPF–β-CD/ZPF–CB[7] dispersions were prepared. When ZPF and macrocycles were added, lactose content was proportionally reduced, given that lactose is used as a diluent/bulking agent38. Before compression, powders were mixed thoroughly and all tablets at a weight of approximately 300 mg were prepared using a rotary tablet press (ZPS008, Shanghai Tianxiang & Chentai Pharmaceutical Machinery Co., Ltd., Shanghai, China) with a 9 mm diameter curved mold.
Tablet hardness, disintegration and dissolution testing
Tablet hardness was measured using a conventional tablet hardness tester (0–20 kg, Shanghai Huanghai Drug Testing Instrument Co., Ltd.). Disintegrating time for each tablet was determined using a BJ–4 A intelligent disintegration apparatus (Tianjin Chuang Xing Electronic Equipment Manufacturing Co., Ltd.) by measuring the time for six tablets to disintegrate fully in 900 mL water, SGF, or SIF, at 37.0 ± 0.1 °C. Dissolution test was performed at 50 rpm and 37.0 ± 0.1 °C according to the paddle method, using 900 mL of water, SGF, and SIF as dissolution media. Aliquots (5 mL) from the sample solutions were obtained using a syringe at a prescribed time, filtered through a 0.45 μm hydrophilic membrane, and analyzed in a UV-vis spectrophotometer at 337 nm.
Statistical analysis
Data were expressed as mean ± standard deviation. Statistical comparisons between the means of individual groups were performed through one-way ANOVA with Duncan’s test. Differences were considered statistically significant when *p < 0.05. SPSS 11.0 software (SPSS, Chicago, IL, USA) was used for statistical analysis.
Additional Information
How to cite this article: Li, S. et al. Co-grinding Effect on Crystalline Zaltoprofen with ß-cyclodextrin/Cucurbit[7]uril in Tablet Formulation. Sci. Rep. 7, 45984; doi: 10.1038/srep45984 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by the Applied Basic Research Project of Sichuan Province (Grant No. 2014JY0042), the National Development and Reform Commission and Education of China (Grant No. 2014BW011) and the Large-scale Science Instrument Shareable Platform Construction of Sichuan Province (Grant No. 2015JCPT0005-15010102).
Footnotes
The authors declare no competing financial interests.
Author Contributions S.L. and H.L. conceived the experiment; S.L. synthesized CB[7] and performed the solubility, disintegration and dissolution experiment with K.X.; X.L., J.H. and H.Y. designed the tablet formulation and made tablets; H.L. and J.H. performed the solid-state characterizations; S.L., X.L. and K.X. wrote the manuscript. All authors discussed the results and commented on the manuscript.
References
- Mendes C. et al. Inclusion complexes of hydrochlorothiazide and beta-cyclodextrin: Physicochemical characteristics, in vitro and in vivo studies. Eur. J. Pharm. Sci. 83, 71–78 (2016). [DOI] [PubMed] [Google Scholar]
- Davis M. E. & Brewster M. E. Cyclodextrin-based pharmaceutics: past, present and future. Nat. Rev. Drug Discov. 3, 1023–1035 (2004). [DOI] [PubMed] [Google Scholar]
- Ma D. et al. Acyclic cucurbit[n]uril molecular containers enhance the solubility and bioactivity of poorly soluble pharmaceuticals. Nat. Chem. 4, 503–510 (2012). [DOI] [PubMed] [Google Scholar]
- Ahmed S. A., Chatterjee A., Maity B. & Seth D. Supramolecular interaction of a cancer cell photosensitizer in the nanocavity of cucurbit[7]uril: A spectroscopic and calorimetric study. Int. J. Pharm. 492, 103–108 (2015). [DOI] [PubMed] [Google Scholar]
- Kim K. et al. Functionalized cucurbiturils and their applications. Chem. Soc. Rev. 36, 267–279 (2007). [DOI] [PubMed] [Google Scholar]
- Cheng X. J. et al. Twisted cucurbit[14]uril. Angew. Chem. Int. Edit. 52, 7252–7255 (2013). [DOI] [PubMed] [Google Scholar]
- Li Q. et al. Twisted Cucurbit[n]urils. Org. Lett. 18, 4020–4023 (2016). [DOI] [PubMed] [Google Scholar]
- Villiers A. Sur la fermentation de la fécule parl’action du ferment butyrique. C. R. Acad. Sci. 112, 536–538 (1891). [Google Scholar]
- Kurkov S. V. & Loftsson T. Cyclodextrins. Int. J. Pharm. 453, 167–180 (2013). [DOI] [PubMed] [Google Scholar]
- Kim J. et al. New cucurbituril homologues: syntheses, isolation, characterization, and X-ray crystal structures of cucurbit[n]uril (n = 5, 7, and 8). J. Am. Chem. Soc. 122, 540–541 (2000). [Google Scholar]
- Liu Q. et al. Host–guest interactions of thiabendazole with normal and modified cucurbituril: 1H NMR, phase solubility and antifungal activity studies. Supramol. Chem. 27, 386–392 (2015). [Google Scholar]
- Nuzzo A., Scherman O. A., Mazzei P. & Piccolo A. pH–controlled release of auxin plant hormones from cucurbit[7]uril macrocycle. Chem. Biol. Technol. Agric. 1, 1–8 (2014). [Google Scholar]
- Saleh N., Khaleel A., Al-Dmour H., al-Hindawi B. & Yakushenko E. Host–guest complexes of cucurbit[7]uril with albendazole in solid state. J. Therm. Anal. Calorim. 111, 385–392 (2013). [Google Scholar]
- Walker S., Kaur R., McInnes F. J. & Wheate N. J. Synthesis, processing and solid state excipient interactions of cucurbit[6]uril and its formulation into tablets for oral drug delivery. Mol. Pharm. 7, 2166–2172 (2010). [DOI] [PubMed] [Google Scholar]
- Seif M., Impelido M. L., Apps M. G. & Wheate N. J. Topical cream-based dosage forms of the macrocyclic drug delivery vehicle cucurbit[6]uril. Plos One 9, e85361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. J., Apps M. G. & Wheate N. J. Chemical factors affecting cucurbit[n]uril formulation into ocular dosage forms: excipient binding, solubility, corneal permeability and antibiotic encapsulation. Supramol. Chem. 26, 648–656 (2014). [Google Scholar]
- Oun R., Plumb J. A. & Wheate N. J. A cisplatin slow-release hydrogel drug delivery system based on a formulation of the macrocycle cucurbit[7]uril, gelatin and polyvinyl alcohol. J. Inorg. Biochem. 134, 100–105 (2014). [DOI] [PubMed] [Google Scholar]
- Walker S. Novel methods to deliver platinum (II) chemotherapeutics to the nose. Ph.D. Thesis, university of Strathclyde: Glasgow, Scotland (2013).
- Lagona J., Mukhopadhyay P., Chakrabarti S. & Isaacs L. The cucurbit[n]uril family. Angew. Chem. Internat. Edit. 44, 4844–4870 (2005). [DOI] [PubMed] [Google Scholar]
- Lee J. W., Samal S., Selvapalam N., Kim H. J. & Kim K. Cucurbituril homologues and derivatives: new opportunities in supramolecular chemistry. Accounts Chem. Res. 36, 621–630 (2003). [DOI] [PubMed] [Google Scholar]
- Uzunova V. D., Cullinane C., Brix K., Nau W. M. & Day A. I. Toxicity of cucurbit[7]uril and cucurbit[8]uril: an exploratory in vitro and in vivo study. Org. Biomol. Chem. 8, 2037–2042 (2010). [DOI] [PubMed] [Google Scholar]
- Pinjari R. V., Khedkar J. K. & Gejji S. P. Cavity diameter and height of cyclodextrins and cucurbit[n]urils from the molecular electrostatic potential topography. J. Incl. Phenom. Macro. 66, 371–380 (2010). [Google Scholar]
- Okamoto T. NSAID zaltoprofen improves the decrease in body weight in rodent sickness behavior models: proposed new applications of NSAIDs. Int. J. Mol. Med. 9, 369–372 (2002). [PubMed] [Google Scholar]
- Salustio P. J., Feio G., Figueirinhas J. L., Pinto J. F. & Marques H. M. C. The influence of the preparation methods on the inclusion of model drugs in a beta-cyclodextrin cavity. Eur. J. Pharm. Biopharm. 71, 377–386 (2009). [DOI] [PubMed] [Google Scholar]
- Passos J. J. et al. Double continuous injection preparation method of cyclodextrin inclusion compounds by spray drying. Chem. Eng. J. 228, 345–351 (2013). [Google Scholar]
- Bandarkar F. S. & Vavia P. R. An optimized commercially feasible milling technique for molecular encapsulation of meloxicam in β-cyclodextrin. Drug Dev. Ind. Pharm. 37, 1318–1328 (2011). [DOI] [PubMed] [Google Scholar]
- Ramos A. I. et al. Chloramphenicol·cyclodextrin inclusion compounds: co-dissolution and mechanochemical preparations and antibacterial action. CrystEngComm 15, 2822–2834 (2013). [Google Scholar]
- Xu K. et al. Two solid forms of tauroursodeoxycholic acid and the effects of milling and storage temperature on solid-state transformations. Int. J. Pharm. 486, 185–194 (2015). [DOI] [PubMed] [Google Scholar]
- Descamps M., Aumelas A., Desprez S. & Willart J. F. The amorphous state of pharmaceuticals obtained or transformed by milling: Sub-Tg features and rejuvenation. J. Non-Cryst. Solids 407, 72–80 (2015). [Google Scholar]
- Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K. & Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 42, 339–341 (2009). [Google Scholar]
- Sheldrick M. G. SHELXS-97: Program for Crystal Structure Solution. Göttingen, Germany: University of Göttingen (1997). [Google Scholar]
- Li S. et al. Interactions of cucurbit[6,7]urils with human serum albumin and their effects on zaltoprofen transportation. RSC Adv. 6, 85811–85819 (2016). [Google Scholar]
- Day A., Arnold A. P., Blanch R. J. & Snushall B. Controlling factors in the synthesis of cucurbituril and its homologues. J. Org. Chem. 66, 8094–8100 (2001). [DOI] [PubMed] [Google Scholar]
- Paradowska K. & Wawer I. Solid-state NMR in the analysis of drugs and naturally occurring materials. J. Pharmaceut. Biomed. 93, 27–42 (2014). [DOI] [PubMed] [Google Scholar]
- Petrovski Ž. et al. Synthesis of ferrocenyldiimine metal carbonyl complexes and an investigation of the Mo adduct encapsulated in cyclodextrin. New J. Chem. 29, 347–354 (2005). [Google Scholar]
- Bardelang D. et al. Cucurbit[n]urils (n = 5–8): a comprehensive solid state study. Cryst. Growth Des. 11, 5598–5614 (2011). [Google Scholar]
- Kuntz Jr I. D., Gasparro F. P., Johnston Jr M. D. & Taylor R. P. Molecular interactions and the Benesi-Hildebrand equation. J. Am. Chem. Soc. 90, 4778–4781 (1968). [Google Scholar]
- Rowe R. C., Sheskey P. J. & Quinn M. E. Lactose monohydrate. In Handbook of pharmaceutical excipients, 6th ed.; 364–370 pp Pharmaceutical Press: London (2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.