

ABSTRACT
A single liter of water contains hundreds, if not thousands, of bacterial and archaeal species, each of which typically makes up a very small fraction of the total microbial community (<0.1%), the so-called “rare biosphere.” How often, and via what mechanisms, e.g., clonal amplification versus horizontal gene transfer, the rare taxa and genes contribute to microbial community response to environmental perturbations represent important unanswered questions toward better understanding the value and modeling of microbial diversity. We tested whether rare species frequently responded to changing environmental conditions by establishing 20-liter planktonic mesocosms with water from Lake Lanier (Georgia, USA) and perturbing them with organic compounds that are rarely detected in the lake, including 2,4-dichlorophenoxyacetic acid (2,4-D), 4-nitrophenol (4-NP), and caffeine. The populations of the degraders of these compounds were initially below the detection limit of quantitative PCR (qPCR) or metagenomic sequencing methods, but they increased substantially in abundance after perturbation. Sequencing of several degraders (isolates) and time-series metagenomic data sets revealed distinct cooccurring alleles of degradation genes, frequently carried on transmissible plasmids, especially for the 2,4-D mesocosms, and distinct species dominating the post-enrichment microbial communities from each replicated mesocosm. This diversity of species and genes also underlies distinct degradation profiles among replicated mesocosms. Collectively, these results supported the hypothesis that the rare biosphere can serve as a genetic reservoir, which can be frequently missed by metagenomics but enables community response to changing environmental conditions caused by organic pollutants, and they provided insights into the size of the pool of rare genes and species.
IMPORTANCE A single liter of water or gram of soil contains hundreds of low-abundance bacterial and archaeal species, the so called rare biosphere. The value of this astonishing biodiversity for ecosystem functioning remains poorly understood, primarily due to the fact that microbial community analysis frequently focuses on abundant organisms. Using a combination of culture-dependent and culture-independent (metagenomics) techniques, we showed that rare taxa and genes commonly contribute to the microbial community response to organic pollutants. Our findings should have implications for future studies that aim to study the role of rare species in environmental processes, including environmental bioremediation efforts of oil spills or other contaminants.
KEYWORDS: biodegradation, community response, metagenomics, organic contaminants, rare biosphere
INTRODUCTION
Extant biodiversity is recognized as telling the evolutionary history of life while also providing an evolutionary scaffold for the future. Consequently, one of the great challenges for environmental microbiology is to better understand how the inventory of biodiversity determines the evolutionary path(s) that will shape the future. Microbial communities in terrestrial or aquatic habitats are typically composed of hundreds of distinct species (1–3), each of which typically makes up a rather small fraction of the total community, and these organisms contain hundreds of species-specific genes of unknown function (4, 5). The pool of low-abundance species has been termed the “rare biosphere” (6), although the definition of “rare” is typically based on arbitrary cutoffs in abundance, e.g., <0.1% of the total community (7, 8). A quantitative understanding of the contributions of this rare biosphere to the process of community response and adaptation within periods of time that are relevant for human activities (e.g., days to months) remains elusive.
It has been suggested that most bacterial species found in a given habitat represent important and ancient players of the indigenous community and contribute substantially to community function and resilience, for instance, by serving as a source of genomic innovation through the species-specific metabolic diversity they harbor (6, 9). It is also thought that the majority of this species diversity represents a sort of “biological detritus” accumulating from a combination of very efficient microbial dispersal and slow decay kinetics of individual cells (10). Furthermore, the high biodiversity observed in several habitats results, at least in part, from sequencing artifacts (11) or free DNA released from dead cells (12). These two contrasting views are not necessarily mutually exclusive; for instance, while the majority of rare species may never contribute to community function and adaptation, it can be hypothesized that a fraction of them do infrequently contribute to adaptation, depending on the specific selection pressures from the changing environment. Although few examples have been reported in which low-abundance species were shown to carry out major ecological roles (see, e.g., references 13–15), our understanding of the frequency and the mechanisms by which low-abundance species and genes, as opposed to abundant species, participate in the community response to perturbations is far from complete. Obtaining a quantitative view of the number of species, or sequence-based operational taxonomic units (OTUs), representing each of these two perspectives within representative ecosystems will lead to a better understanding and modeling of the extant biodiversity. Obtaining a quantitative understanding of the process will require following the temporal changes in the composition and activity of natural microbial communities after perturbations in well-replicated experiments/samples using high-resolution approaches.
To provide novel insights into the issues discussed above and test prevailing hypotheses about the ecological role of the rare biosphere, we set up parallel 20-liter laboratory mesocosms, in triplicate, containing a well-characterized planktonic community from Lake Lanier (Georgia, USA) [(16, 17) and challenged them with frequently used organic compounds that were not detectable at the limit of detection of high-performance liquid chromatography (HPLC) analysis in the lake water column at the time of sampling or other times. (Using compounds that are abundant in the lake would not have been as informative about the rare biosphere, since they typically select for abundant community members.) The compounds included 2,4-dichlorophenoxyacetic acid (2,4-D), a widely used herbicide; 1,3,7-trimethyluric acid (caffeine); and 4-nitrophenol (4-NP), a precursor compound of several fungicides and a decomposition product of pesticides. The tested compounds were added to the mesocosms at concentrations 10 to 20 times higher than the limit of detection (∼5 μM) to allow for robust determination of their biodegradation profiles. These compounds were also chosen because their biodegradation pathways and the underlying genes are known, which facilitated analysis. The mesocosms were sampled repeatedly for metagenomic analysis to follow the evolution of the bacterial communities and identify which populations responded to the treatment (e.g., became enriched or depleted over time). Parallel unamended mesocosms served as controls and references. The results allowed us to rigorously test the hypothesis that low-abundance members, as opposed to genes of the major components of the community, provided the metabolic diversity that enabled the community to respond to these changing conditions. Further, the data provided insights into the variability and redundancy of the responding low-abundance populations in different samples from this freshwater ecosystem, as well as the genetic diversity of the catabolic genes underlying the biodegradation of the added compounds.
RESULTS
Mesocosm biodegradation profiles.
Year-round metagenomic data sets, similar in sequencing effort to the data sets reported here from the mesocosms, from the exact same site and sampling depth have been determined previously (see, e.g., references 16 and 17). Analyses of those data sets showed that the (known) biodegradation genes for the above-mentioned organic compounds were not detectable, further confirming our HPLC results that the compounds are, in general, rare in the water column of the Lake Lanier, from where the inoculum water for the mesocosms originated. These previously determined data sets have also revealed the taxa that are abundant in the water column and their seasonal abundance patterns (see, e.g., reference 16), which were used as reference points in the assessment of the contributions of the rare versus abundant populations during the mesocosm experiments. These data sets also revealed that 30 to 40% of the total populations sampled in Lake Lanier at any time point represent rare species (17); thus, our sequencing effort adequately sampled the rare biosphere. The results from the 2,4-D mesocosms are preferentially reported below for simplicity purposes, since similar results were largely obtained with the caffeine and 4-NP mesocosms, unless noted otherwise.
The three replicate mesocosms revealed three distinct 2,4-D biodegradation profiles. In mesocosm I, 2,4-D was completely degraded within ∼10 days. After three 2,4-D respikes, degradation was robust and faster (∼2 days), even after doubling the 2,4-D concentration during the last respike (Fig. 1). In contrast, mesocosms II and III showed much slower 2,4-D biodegradation. Mesocosm II never completely degraded 2,4-D, even after a 40-day period, while mesocosm III demonstrated complete degradation within ∼21 days and weak degradation after one respike. Variability was also observed in the 4-NP and caffeine mesocosms, albeit degradation profiles were more similar among the three replicate mesocosms in these cases than in the 2,4-D mesocosms (Fig. S1). Degradation of 4-NP and caffeine took much longer, at least 20 days, and was often incomplete, while respike events typically showed even slower degradation.
FIG 1.
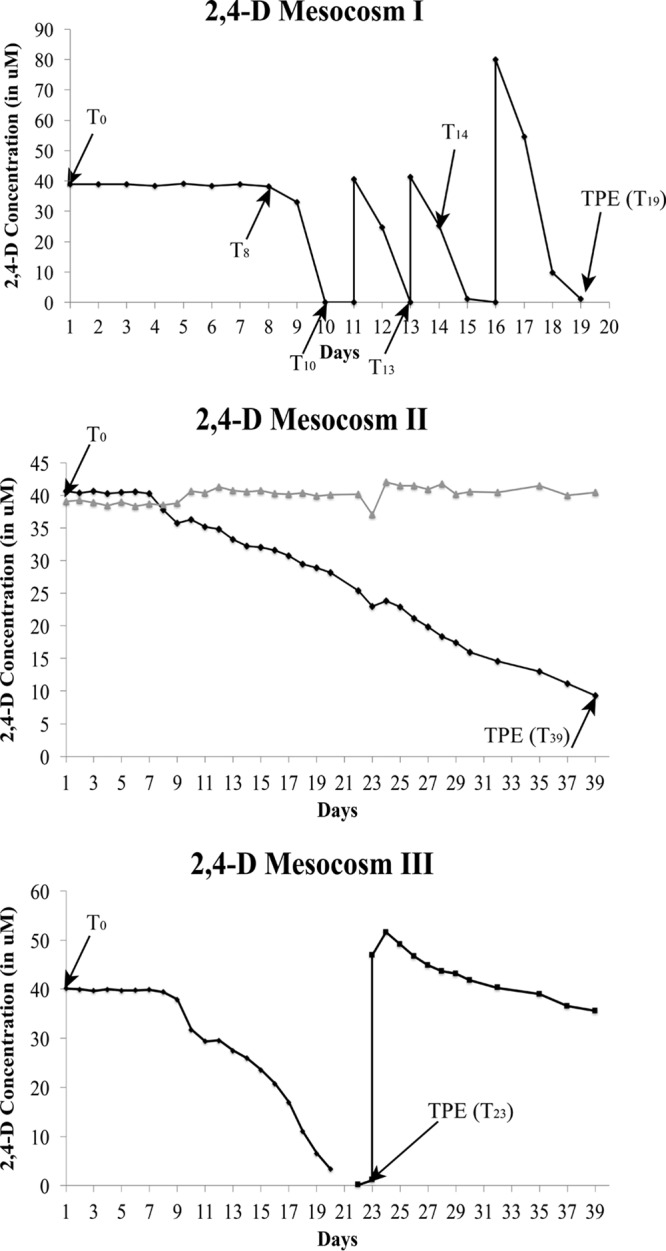
Degradation profiles of the triplicate 2,4-D mesocosms. The black line in each figure indicates the 2,4-D concentration over time, and the gray line represents the abiotic (negative) control (2,4-D in sterilized lake water). The black arrows indicate the time points at which DNA was extracted. The mesocosms were run in parallel, using the same mixed (homogenized) water as inoculum; hence, only one abiotic control was employed for all three mesocosms.
To test whether the observed differences in the 2,4-D degradation profiles were attributable to nutrient limitation (e.g., nitrogen) or change in pH (e.g., the final pH in mesocosms II and III was below 6), small volumes (∼2 ml) from mesocosms II and III were removed and supplemented with ¼-strength minimal salts basal (MSB) medium containing 7.6 mM (NH4)2SO4. The resulting microcosms showed complete 2,4-D removal in less than a day (data not shown). Mesocosm I apparently did not require the addition of nutrients, even though all mesocosms were inoculated with the same homogenized lake water. Therefore, the reason underlying the variation observed in degradation profiles, at least for the 2,4-D mesocosms, was presumably due to lack of nutrients (e.g., nitrogen) and/or lower pH, which apparently affected the response of the rare biosphere, e.g., the results implied that different rare taxa or genes, with different nutrient requirements and physiological adaptations, were activated in each mesocosm.
Degraders are members of the rare biosphere.
To determine whether degradation of the added organic compounds was due to abundant or low-abundance member(s) of the initial (inoculum) microbial community, 2,4-D degraders were isolated from the three mesocosms at the last sampling time point (i.e., time post-enrichment [TPE]) (mesocosm 1 [M1] at TPE, time [T] = 19 days; mesocosm 2 [M2] at TPE, T = 39 days; and mesocosm 3 [M3] at TPE, T = 23 days). One representative isolate per mesocosm based on colony morphology was chosen for further analysis and genome sequencing (morphologies were typically similar between colonies from the same mesocosm). Their genomes were sequenced and assigned to the Burkholderia, Sphingopyxis, and Variovorax genera for mesocosms I, II, and III, respectively, based on 16S rRNA gene sequence and whole-genome-based average nucleotide identity (ANI) (18) analysis. The relative abundances of the degraders during the mesocosm incubation were assessed based on the number of metagenomic reads from each sampling point mapping onto their genome sequence at high stringency (>99% nucleotide identity, excluding the rRNA gene operon, which is highly conserved). All three 2,4-D degraders recruited essentially no reads (Fig. 2) and were under the detection limit (∼0.0001% of total community, estimated based on spiking in silico a target genome into the mesocosm metagenome and requiring a minimum 0.1× coverage at the whole-genome level for robust detection) in the initial microbial community (T = 0 metagenome). However, all three 2,4-D degraders, especially the Burkholderia sp. strain KK1 population in 2,4-D mesocosm I, increased in abundance in their corresponding mesocosm by at least four orders of magnitude by the time the added 2,4-D was completely biodegraded (making up ∼0.24% of total community, or more, at T = 10 days). The abundance of Burkholderia sp. KK1 was even higher at the last sampling time point (TPE, T = 19 days), i.e., after three 2,4-D respikes, making up ∼0.82% of the total community. This level of enrichment was also consistent with theoretical calculations based on the energy available for growth on 2,4-D as a sole source of carbon and energy (see supplemental material).
FIG 2.
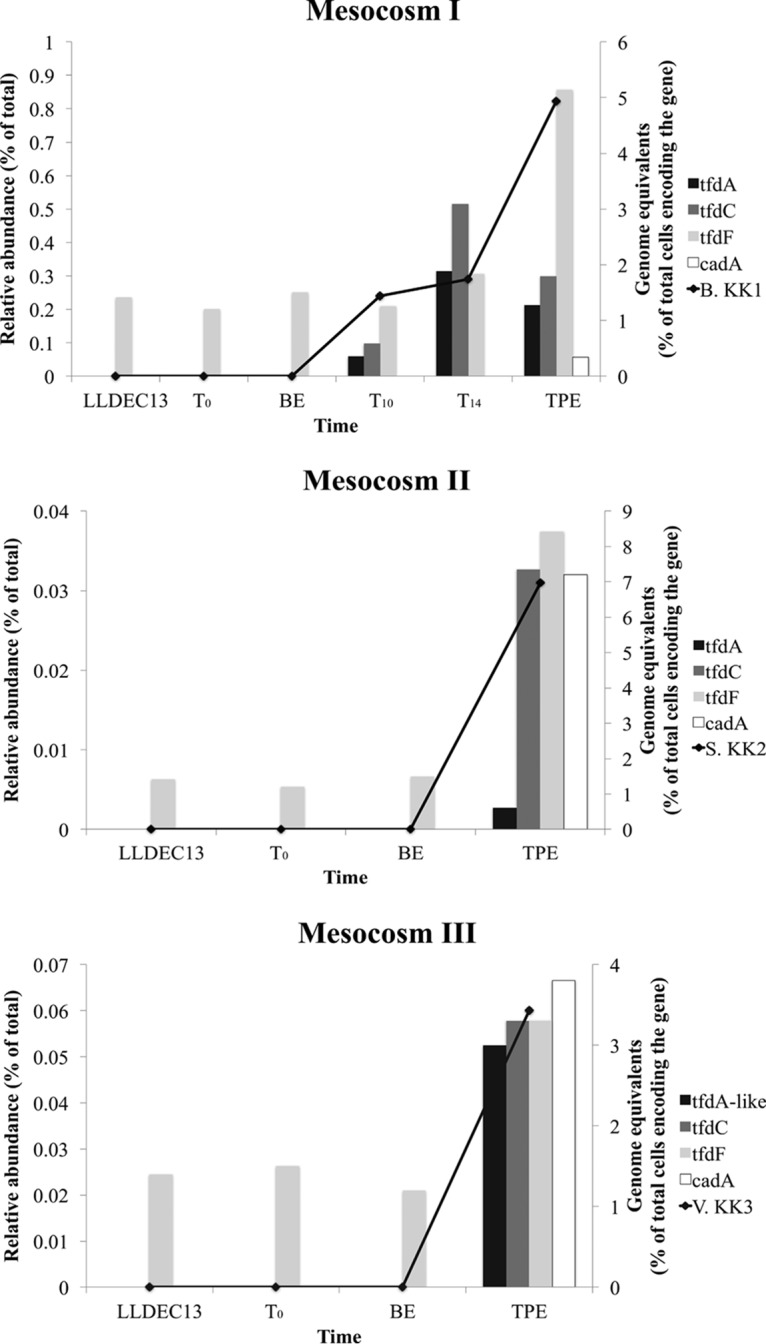
Relative abundance of 2,4-D degraders and genes from each mesocosm. The abundance of 2,4-D degraders was calculated as the fraction of total metagenomic reads mapping on the corresponding genome sequences (identity ≥99% and length ≥80 bp) in each metagenomic data set (black line, primary y axis). The abundance of 2,4-D genes was measured by genome equivalents, i.e., the percentage of total cells encoding the gene (bars, secondary y axis). BE, bottle effect; TPE, time post-enrichment, i.e., time for the last sampling time point from which the degraders were also isolated.
Similar results were observed for the 4-NP and caffeine mesocosms. The 4-NP degraders were isolated from 4-NP mesocosm I (4-NP-M1) at T = 17 days (T17) and were assigned to Pseudomonas. The predominant caffeine degraders were isolated from caffeine mesocosm I (Caff-M1) at T25 and assigned to Pelomonas. During enrichment, these degraders became abundant, similar to the results reported above for 2,4-D degraders, albeit the level of enrichment of the caffeine degrader was an order of magnitude less pronounced (Fig. S2). These results confirmed our hypothesis that the added compounds selected for rare members of the original lake microbial community.
Abundance and phylogeny of 2,4-D, 4-NP, and caffeine biodegradation genes.
The known genes responsible for 2,4-D degradation are the tfdABCEDF gene cluster (19) and the cadABCD gene cluster (20), and they were all present in at least one of our isolates. In brief, cadABCD encodes a protein complex that catalyzes the first step of the 2,4-D biodegradation pathway, which converts 2,4-D to 2,4-dichlorophenol (2,4-DCP), similar to tfdA; the tfdBCEDF cluster encodes enzymes for the subsequent steps. Hence, the tfdA and cadABCD genes represent robust biomarkers of 2,4-D degradation. The tfd gene cluster was present in the genome of the Burkholderia sp. strain KK1 isolate from mesocosm I and in assembled contigs from all mesocosms after perturbation. In fact, three tfd operons were identified in the genome of Burkholderia sp. KK1. Two of them, one complete and one that contained only the tfdBCE genes, showed 100% nucleotide identity to those in the previously reported pM7012 plasmid of Burkholderia sp. strain M701 (21). The third complete tfd gene cluster in the KK1 genome showed 100% nucleotide identity to that of the previously reported pJp4 plasmid of Ralstonia eutropha JMP134 (22). The cad gene cluster was present in the Sphingopyxis sp. KK2 isolate and was detected in lower abundance in metagenomes from mesocosm I at TPE (T = 19 days) than in those from in mesocosms II and III.
The abundances of 2,4-D biodegradation genes during enrichment were determined based on genome equivalents (see Materials and Methods for details). The results for tfdA genes are preferentially reported below for simplicity. Consistent with the metagenome-based abundances of the isolates reported above, no tfd gene was detectable in the original inoculum (T = 0 metagenome), while the percentage of total cells (i.e., genome equivalents) containing 2,4-D biodegradation genes increased over time (Fig. 2 for 2,4-D and Fig. S2 for 4-NP and caffeine catabolic genes). Phylogenetic analysis of the tfdA genes recovered in assembled contigs from all three 2,4-D mesocosms and isolates revealed that although the two tfdA alleles in the KK1 genome were present in the metagenomes, the most abundant tfdA alleles in the metagenome formed a new cluster that was divergent from previously identified tfdA genes or those present in Burkholderia sp. strain KK1. They showed 83% and 92% amino acid identity at maximum, respectively (and complete conservation of protein length, functional domains, etc.; Fig. 3). Therefore, additional 2,4-D degraders were present in the mesocosm compared to the isolates obtained (see also community composition analysis below). No further isolation efforts were performed in order to obtain these abundant populations in culture, since the goal was not to isolate all degraders in the mesocosms.
FIG 3.

Phylogeny of all tfdA and tfdA-like genes recovered from the three 2,4-D mesocosms and isolates. The tree was built using the neighbor-joining method as implemented in Geneious (version 8.1.8) based on an amino acid sequence alignment. The abundance (genome equivalents) of tfdA-like|contig02531-3 (originally identified from mesocosm III in TPE [T = 23 days] sample), tfdA|contig12396-2 (originally identified from mesocosm II in TPE [T = 39 days] sample), and tfdA|contig38919-2 (originally identified from mesocosm I at T = 10 days sample), as well as tfdA genes recovered in the isolate genomes are represented by a pie chart next to the gene name. Blue denotes tfdA genes contained in our isolates, and orange denotes tfdA or tfdA-like genes identified from metagenomes. Note that only one tfdA gene of strain KK1 can be detected in M1.T10 and M2.TPE metagenomes.
The phylogenetic analysis also revealed several additional “tfdA-like” genes present in the 2,4-D mesocosms. The genes are distant homologs (showing <40% amino acid identity; Fig. 3) of experimentally verified tfdA genes. These tfdA-like genes, also known as tfdAα (23), are ubiquitously found in both 2,4-D degraders and non-2,4-D degraders, and the proteins that these genes encode, TfdAα, likely do not contribute to 2,4-D biodegradation. For instance, it has been reported that TfdAα proteins show weak α-ketoglutarate-dependent 2,4-D dioxygenase activity (1/1,000 of specific activity) compared to that of the TfdA protein of pJp4 (24). Thus, these gene alleles were not discussed further, although several of them increased in abundance during our incubation, indicating that they might be involved in 2,4-D biodegradation or the by-products.
In addition to tfd genes, the cad gene cluster was also enriched during selective growth in all 2,4-D mesocosms, especially in mesocosms II and III (Fig. 2). The enzymes encoded by this complex are thought to catalyze the initial step of 2,4-D biodegradation in Bradyrhizobium sp. strain HW13 (analogous to the function of tfdA) (25). cad gene clusters were identified only in the Sphingopyxis sp. KK2 isolate obtained from mesocosm II among our isolates. Collectively, our results suggested that various alleles of 2,4-D biodegradation genes (tfd or cad) and distinct populations were enriched in the three mesocosms and that substantial gene functional redundancy may be present among the low-abundance organisms, even in relatively small volumes of lake water.
4-NP can be degraded by either the hydroquinone (HQ) (26) or the hydroxyquinol (BT) pathways (27). The HQ pathway is the predominant pathway in Gram-negative bacteria, such as Pseudomonas sp. strain WBC-3 (28). The HQ gene cluster pdcABCDEFG was found in two pieces, on contig26 (pdcABDE) and contig116 (pdcCDEFG) in the 4-NP-degrading Pseudomonas sp. strain KK4 isolate, showing 90.4% nucleotide identity to the previously described gene cluster. Several pdc genes were also identified in assembled metagenomic contigs from the 4-NP mesocosm I metagenome, and the abundances of all pdc genes increased from undetectable levels to about 1% of the total cells containing the genes during the incubation time, with the exception of pdcF (fig. S2). Notably, the coverage of all 4-NP genes (5.4×) was substantially higher than that of the isolate genome (3.1×), indicating that additional 4-NP degraders that were not isolated were present in the mesocosms (or multiple gene copies were encoded by the mesocosm population).
Caffeine degradation occurs via the N-demethylation pathway, which involves five enzymes encoded by ndmABCDE (29). The ndm genes were previously described in the caffeine degrader Pseudomonas putida CBB5, where they were contained on a 13.2-kb genomic DNA fragment (29, 30). Homologs were identified on contig10 in our Pelomonas sp. strain KK5 genome with ≥50% amino acid identity to the previously reported ndmABCDE genes. Caffeine degradation can also occur via the C-8 oxidation pathway (31). One of the key enzymes for the C-8 oxidation pathway is caffeine dehydrogenase (cdh), which contains three subunits (cdhABC) (32). The abundances of the ndmABCDE and cdhABC genes also increased during mesocosm incubation, although the level of increase was not consistent among all genes of the ndmABCDE operon, unlike the 4-NP and 2,4-D degradation genes (Fig. S2).
Quantitative PCR analysis of 2,4-D degradation genes.
To confirm that no major DNA sequencing or library creation biases affected our metagenome-based findings and likely obtain higher-sensitivity measurements of gene abundance, tfdA gene sequences in the genome of Burkholderia sp. strain KK1 were quantified in the 2,4-D mesocosm community DNA samples by quantitative PCR (qPCR). In general, the results were consistent with the metagenomic findings. In particular, at two early time points of the 2,4-D mesocosm I (T = 0 and 8 days), the abundances of tfdA genes were not quantifiable (i.e., were below the detection limit of 44 gene copies). At later time points (T = 14 days and TPE [T = 19 days]), the two tfdA genes increased in abundance by at least one order of magnitude (relative to the detection limit of 44 copies) and were robustly detected by qPCR (Fig. S3). These results confirmed that the tfdA genes were present in a rare fraction of the original lake microbial community and became abundant after perturbation.
Genetic basis of Burkholderia sp. strain KK1 2,4-D biodegradation genes.
Comparative analysis of the Burkholderia sp. strain KK1 contigs against available sequences (21, 22) suggested that all tfd genes were present on a putative megaplasmid (568,203 kb). Characteristic genes of plasmids, such as a Walker-type ATPase (33) and a centromere-binding protein (parB), were also found in contigs that were linked, based on paired-end read mapping or PacBio data, to the contigs that contain the tfd gene cluster. Considering that (i) the structure of the KK1 plasmid showed high similarity with pM7012 and pJp4 transmissible plasmids from Burkholderia sp. M701 (21) (two-way average amino acid identity [AAI], 99.25%; standard deviation [SD], 6.25%) and Ralstonia eutropha JMP134 (22) (one-way AAI, 47.97%; SD, 15.60%) (Fig. S4); and (ii) shared all tra genes required for transmissibility except for traI (relaxase), which is not always necessary for plasmid conjugation, the KK1 plasmid is also probably mobile (or was mobile in the recent past). Further, the ∼100-kb-long region that included the three tfd gene clusters (positions 376592 through 424188) also encoded many direct and inverted repeated sequences, insertion sequences, transposases, phage integrases, and many hypothetical proteins. Thus, it is appears as if this region represents a highly dynamic and mosaic part of the genome.
Furthermore, we isolated a Novosphingobium sp. (Alphaproteobacteria) able to degrade 2,4-D from our mesocosms. Its genome sequence revealed cad genes showing 100% nucleotide identity with a homolog from a previously described conjugative plasmid, pCADAB1, encoded by Sphingobium sp. ERG5 (Alphaproteobacteria) (20). The cad gene cluster identified in the Novosphingobium sp. was also flanked by mobile elements. Collectively, these results indicated that the 2,4-D degradation genes are transferred horizontally, either within Lake Lanier or, possibly, during the enrichment process in our mesocosms (see also Discussion below).
Microbial community taxonomy shifts during enrichment.
To assess community-wide responses to the added organic substrates, the taxonomic and functional gene content shifts at the whole-community level were also examined. First, we examined the average coverage of the corresponding microbial community by each metagenomic data set using Nonpareil, with default parameters (34), and found it to be between ∼0.55 and ∼0.9 (Fig. S5), meaning that at least 55% (for 2,4-D mesocosms, coverage was >73%) of the total extracted unique (nonredundant) DNA was sequenced. This level represents an appropriate coverage for statistically robust comparisons of gene and species relative abundance between metagenomic data sets (35). Based on both MyTaxa taxonomic assignments of metagenomic contigs and QIIME analysis of 16S rRNA gene fragments contained in the metagenomic reads, the community diversity of the 2,4-D mesocosms was much higher at the beginning than at later time points, presumably due to the strong selection pressure of the substrate and death due to restricted growth conditions (e.g., lack of primary production, 2,4-D toxicity to some community members). Overall, at least 2,640 OTU were estimated to be present in the T = 0 sample, but after enrichment (TPE [T = 19 days]), there were only 896 OTU remaining, an ∼66.1% reduction in total OTUs (Table S3). The Burkholderiaceae OTU showed among the strongest enrichment in mesocosm I, and the longest 16S rRNA gene sequence fragment (264 bp) representing this OTU showed 100% nucleotide identity to the Burkholderia sp. KK1 isolate (Fig. 4). Notably, the Comamonadaceae OTU also increased in abundance relative to the T = 0 metagenome. The representative 16S rRNA gene sequence of this OTU showed 99.6% nucleotide identity to the previously identified Delftia acidovorans strain P4a (36), which can completely degrade 2,4-D using its tfd gene clusters contained on a catabolic transposon (this sequence was not among the ones recovered in our metagenomic data set).
FIG 4.

Shifts in microbial community composition over time in the 2,4-D mesocosms. Results shown are based on total 16S rRNA gene-containing reads recovered from each metagenomic data sets and classified at the family level. Only taxa that recruited more than >2% of total reads are shown; white numbers represent relative abundance. The data sets are as follows: original lake (LLDEC13), 2,4-D mesocosm I at T = 0, 10, 14, and 19 days samples (M1.T0, M1.T10, M1.T14, and M1.TPE, respectively); 2,4-D mesocosm II at TPE (T = 39 days) sample (M2.TPE); 2,4-D mesocosm III at TPE (T = 23 days) sample (M3.TPE); bottle-effect metagenome (BE). The purple star denotes the family that the Burkholderia sp. KK1 isolate was assignable to (100% 16S rRNA gene identity). The blue and green stars denote the family that the Sphingopyxis sp. strain KK2 and the Variovorax sp. strain KK3 isolates were assignable to (99% and 100% 16S rRNA gene identity, respectively).
A total of 1,157 and 925 OTU were estimated to be present in TPE samples from mesocosms II and III (Table S3), which represented an ∼56.2% and ∼64.9% reduction in OTU richness compared to that at T = 0, respectively. The relatively smaller reduction in OTUs correlated with incomplete 2,4-D biodegradation (and hence, weaker selection pressure) in these mesocosms relative to mesocosm I, which robustly degraded 2,4-D with associated growth of several populations. In mesocosms II and III, the taxa that were enriched over time included Legionellaceae (Gammaproteobacteria), Comamonadaceae (represented by our isolates from Variovorax sp. strain KK3) (Betaproteobacteria), and Sphingomonadaceae (represented by our isolates from Sphingopyxis sp. [Alphaproteobacteria]), while Planctomycetaceae (Planctomycetes) and Acidimicrobiaceae (Actinobacteria) decreased in abundance (Fig. 4). Members of Sphingomonadaceae and Variovorax are known to encode 2,4-D biodegradation capabilities and were previously detected as the predominant 2,4-D degraders isolated from agricultural soils heavily treated with 2,4-D (37–39). These two groups were represented by our isolates, and their 16S rRNA gene sequences showed 98.9% nucleotide identity to previously identified degraders. On the other hand, Legionellaceae encompasses one genus, with about 50 named species, mostly of clinical relevance (40); hence, it is likely that this family also contained yet-to-be-described 2,4-D degraders or organisms that benefit from by-products of 2,4-D degradation. Therefore, it appears that the original lake inoculum contained more than one known and also some poorly characterized 2,4-D degraders, and all of them benefited from 2,4-D directly or indirectly, using by-products. (It should be noted, however, that no high-molecular-weight intermediates or by-products of the 2,4-D degradation pathway were detected by HPLC; however, if the intermediates are consumed rapidly, they typically cannot be detected).
For the 4-NP and caffeine mesocosms, the trends in total OTU shifts were similar to those of 2,4-D. Several OTUs encompassing known 4-NP degraders increased in abundance, such as Pseudomonadaceae (Gammaproteobacteria), represented by our Pseudomonas sp. isolates (at least 5-fold increase), Streptomycetaceae, and Acidimicrobiaceae (Actinobacteria), as well as taxa that do not include any known 4-NP isolates, such as other unclassified Actinobacteria (Fig. S6). This finding agreed with those of previous studies (28, 41) that identified Pseudomonas sp. strain WBC-3 as a 4-NP degrader, sharing 98.3% 16S rRNA gene sequence identity to our isolates. Notably, we have observed unclassified Actinobacteria that increased in abundance during 4-NP perturbation, which echoed several previous studies (42–44). However, the representative 16S rRNA gene sequence of the Actinobacteria OTU showed only 83% nucleotide identity to the previously described 4-NP isolates (Arthrobacter protophormiae), indicating that our 4-NP degraders belong to a novel Actinobacteria clade.
To date, only about 35 strains have been isolated and experimentally characterized as caffeine degraders that belong to phylogenetically diverse taxa of bacteria (45), such as Rhodococcus spp. and Pseudomonas sp. strain CBB1. These strains degrade caffeine via C-8 oxidation pathways, whereas Pseudomonas sp. strain CES, Pseudomonas putida CBB5 (46, 47), and Serratia marcescens degrade caffeine via N-demethylation pathways (48). For the caffeine mesocosms (Fig. S6), the abundance of Comamonadaceae (Betaproteobacteria) substantially increased (4-fold more abundant), and its longest 16S rRNA gene fragment identity to our isolates was 100%. Additional OTUs that increased in abundance in the post-enrichment metagenome included Burkholderiaceae (Betaproteobacteria), uncultivated members of Acidimicrobiaceae (Actinobacteria), and Proteobacteria (showing <85% 16S rRNA gene identity to any cultured taxon in the Greengenes database). Therefore, it appears that several caffeine degraders which were not represented by our isolates may also have contributed to the entire microbial community. More notably, Burkholderiaceae have not been reported as caffeine degraders yet (reviewed in reference 45), indicating that the corresponding OTUs might represent new caffeine degraders (or be associated with by-products).
Microbial community functional shifts during enrichment.
In order to compare community gene content shifts in the 2,4-D, 4-NP, and caffeine metagenomes before and after perturbation, we determined the relative coverage (×) of each protein-coding gene and compared the predicted protein functions based on Gene Ontology (GO) terms (49). For 2,4-D (Fig. 5), the most enriched genes (i.e., those showing >6-fold difference in abundance relative to T = 0) were associated with (i) cell motility, (ii) energy generation and maintenance, (iii) transporters, (iv) viral functions, and (v) several genes associated with 2,4-D biodegradation, e.g., catechol-1, 2 dioxygenase (19-fold difference), hydroxyquinol 1,2-dioxygenase (14-fold), 2,4-dichlorophenol 6-monooxygenase (11-fold), and chlorocatechol 1,2-dioxygenase (27-fold). The 2,4-D mesocosms II and III exhibited similar profiles, except that enriched genes also included genes encoding proteins involved in phosphate metabolism, which might represent a sign of phosphorus limitation in these mesocosms. Overall, however, the majority of gene functions did not change in abundance, e.g., 70.7% of total functions detected changed by less than 2-fold in abundance between T = 0 and TPE.
FIG 5.

Microbial community gene content shifts in 2,4-D mesocosm I. The relative coverage (x axis) of genes (y axis) was calculated by summing the length of all reads mapping on the gene (a minimum cutoff for a match of ≥80 bp alignment length and ≥97% nucleotide identity was used) and dividing it by the length of the gene sequence. The most differentially abundant genes between T0 (T = 0) and TPE (T = 19 days) metagenomes were grouped by their GO terms and are shown on the graph. The GO accession number is also provided, followed by the GO description. BE, bottle-effect metagenome (control incubation with no 2,4-D added); CoA, coenzyme A.
For 4-NP and caffeine (Fig. S7), several of the enriched genes were similar to those for 2,4-D, such as those for bacterium-type flagella, transmembrane transport activity, and viral activities. Enriched genes also included (i) phenol catabolic process (6-fold more abundant), presumably reflecting 4-NP biodegradation by-products; (ii) spore germination (54-fold more abundant), probably due challenging growth conditions; and (iii) caffeine-related catabolic pathways, including urate/purine-containing compound metabolism (39-fold more abundant), xanthine oxidase/dehydrogenase (6-fold more abundant), and demethylase activity (1.7-fold more abundant). Further, genes associated with maintenance of clustered regularly interspaced short palindromic repeat (CRISPR) elements and membrane signal transduction activity showed 5-fold and 8-fold higher abundance in caffeine and 4-NP mesocosms, respectively, likely reflecting active viral predation.
The results reported above were presumably attributable, at least in part, to the strength of the selection pressure applied by the addition of the organic compound and were not related to bottle effects from the incubation. Consistent with this interpretation, the data sets from the bottle-effect (BE) control, i.e., lake water incubated without the addition of the organic substrate, resembled the T = 0 (initial) community much more than those from the TPE (final sampling point) for the mesocosms amended with the three compounds used in this study, both in terms of taxon composition and gene functions (Fig. S8). For instance, the BE data sets had a small decrease, if any, in the number of OTUs detected relative to the T = 0 data set, compared to about 66% of the OTUs becoming undetectable in the 2,4-D mesocosms (Table S3), and no bacteriophage genes were strongly enriched in BE versus T = 0 data sets (Fig. 5 and S7). Further, the mesocosms showing robust biodegradation (e.g., 2,4-D mesocosm I) showed, in general, a greater reduction in OTU number than in mesocosms with low or incomplete biodegradation, consistent with lower competition among cooccurring microbial populations and selection pressures in the mesocosms with incomplete biodegradation. In agreement with these OTU diversity patterns, PCR-based 16S rRNA gene copy counts, and presumably cell counts (assuming no major change in the average rRNA copy number of the microbial community), were lower in TPE samples from the three 2,4-D mesocosms than in the T = 0 or BE samples, corroborating in part the strong selection pressure presented by 2,4-D addition and, probably, its toxicity to some community members (Fig. S9).
DISCUSSION
Bacteria able to grow on the compounds used here have been recovered from various habitats using standard enrichment and isolation procedures and have been shown to be widespread in nature (see, for instance, for 2,4-D references 24 and 37). What is lacking is a quantitative understanding of how often such organisms are members of the rare biosphere and how exactly they contribute to the microbial community response upon the addition of the organic compound, i.e., what molecular (e.g., clonal amplification and horizontal gene transfer) and ecological (e.g., competition and cooperation) mechanisms take place during the enrichment period until the disappearance of the added substrate. The results presented here provided some new insights into these issues.
The three 2,4-D mesocosms showed substantially different 2,4-D biodegradation profiles. Sequencing of time-series samples revealed that these profiles were due to variation in the pool of rare taxa in the inoculum for each mesocosm, even though the inocula originated from the same mixed (homogenized) lake water collected in a single sampling trip to Lake Lanier (i.e., a founder effect). All organisms enriched during the incubation period represented initially rare populations in situ, e.g., those below the detection limit of metagenomic analysis in the T = 0 sample or in the year-round data sets available (16, 17). These populations were usually assignable to different species in each replicate mesocosm and encoded distinct versions of the biodegradation enzymes, which were closely related to experimentally verified enzymes. For instance, we found that the microbial communities of the 2,4-D mesocosms II and III, which showed slow degradation, contained different degradation genes than those in mesocosm I (exhibiting robust degradation), including several homologs of tfdAα, which typically show weak 2,4-D degradation (24). 2,4-D mesocosms II and III were also enriched in phosphorus metabolism genes, a possible sign of phosphorus limitation in these mesocosms. The addition of nutrients, such as ¼-strength MSB liquid medium, a source of phosphorus among other nutrients, to microcosms originating from mesocosms II and III greatly increased the degradation rate. These findings show that not only were different taxa enriched in each mesocosm, but also, each of these taxa has different nutrient requirements and contains genes that have different biodegradation kinetics and/or regulation. No abundant organisms in T = 0 or promiscuous degradation enzymes, which could act on the added substrates cometabolically or after being mutated, were found to become strongly enriched during the incubation period. Collectively, these findings showed that the rare biosphere enabled a microbial community response to the changing conditions (i.e., promoted the degradation of the added organic compounds); however, the specific taxa and genes involved (those enriched during this process) were highly varied. Thus, the random presence/absence of degrading populations in relative small-volume inocula due to their low in situ abundance and/or random activation of the genes contained by these taxa apparently played a key role during community response, while the pool of rare species and taxa contributing to the response is large, at least based on the 2,4-D mesocosms. These findings were also consistent with recent studies that have highlighted the importance of stochastic processes, e.g., variation in abundance, extinction, speciation, and evolution, in mediating microbial community response to perturbations (see, e.g., references 50 and 51).
Another interesting observation was that at least three distinct tfd operons (Fig. 3) and several abundant OTUs/population coexisted after the addition of 2,4-D, without any one population outcompeting the remaining ones. These results could be attributable to different kinetics of the Tfd enzymes, e.g., enzymes with high versus low affinity for the substrate, or differential bacteriophage or protozoan predation of the abundant populations. The differential predation scenario has been well documented several times (33, 52–54), and preliminary findings reported by this study (discussed below) indicated that differential phage predation may be occurring in our mesocosms. However, whether or not this scenario fully applies to our mesocosm incubations remains to be experimentally tested.
Because the mesocosms were perturbed with 2,4-D, the microbial community was under strong selection pressure. The organisms that were capable of using the 2,4-D as a sole carbon and energy source for growth became more abundant and gained an advantage compared to other populations. For 2,4-D degraders, their degradation genes were often found on a large plasmid (e.g., the Burkholderia sp. strain KK1 plasmid, at ∼570 kb). Annotation of the KK1 plasmid showed that it carries all necessary tra genes for transmission. Comparison of the KK1 plasmid against those available in the public databases showed very high nucleotide identities (>99%) to plasmids that have been shown to be mobile (Fig. S4) and carried several repeat sequences and sequences encoding phage integrases and transposases, which suggests that the plasmid and its tfd gene clusters have undergone intensive genetic recombination events recently. Finally, the plasmid-to-chromosome ratio in strain Burkholderia sp. KK1, grown in isolation and harvested and sequenced at the mid- to-late-exponential phase, was ∼1.4, while the same ratio in the mesocosm increased over time: T = 0, not applicable [NA]; T = 10 days, 1.30; T = 14 days, 2.0; and TPE (T = 19 days), 2.9. These findings indicate that other members of the community might have acquired the plasmids during the incubation. The alternative hypothesis that the plasmid copy number increased due to selection pressure does not appear to be as parsimonious, because the ratio after the first 2,4-D spike-in event was close to the pure isolate ratio (1.3 versus 1.4). Therefore, it is intriguing to hypothesize that the KK1 plasmid is also mobile and was transferred horizontally during the incubation period, between cooccurring members of the community, due to strong selection pressure, thus mediating the microbial community response to the added 2,4-D. However, whether or not such a molecular mechanism played an important role during the incubation awaits experimental verification; related to this, it should be mentioned that sequencing of the early time point samples (e.g., T = 8 days) provided only a couple metagenomic reads mapping on the plasmid or the tfd gene clusters, and hence, it was not possible to assemble the biodegradation genes from these time points and determine the genetic background in which the genes resided. Our estimates based on Nonpareil (Fig. S5) indicated that about two orders of magnitude more sequencing would have been required (about 1 Tbp in order to provide almost complete community coverage), which is prohibitively expensive currently.
In addition to degradation genes, the relative abundances of several genes associated with phage proteins, flagellar motility, chemotaxis, and conjugative transfer increased in TPE samples relative to initial (t = 0) or the bottle-effect (BE) data sets (Fig. 5 and S7). These results were likely attributable to the stressful environmental conditions, e.g., toxicity of added organic compounds, lack of nutrients, and bacteriophage predation. Consistent with these interpretations, ∼66% of all OTUs decreased in abundance at the end of each mesocosm incubation but not in the data set from the bottle-effect incubation (Table S3). The degrading populations did not make more than 2 to 5% of the total at maximum; hence, the strong reduction in OTU numbers in the 2,4-D mesocosms relative to the control cannot be merely attributed to higher relative abundance (and hence, more frequent sampling by sequencing) of the responding populations but reflect, at least partly, true difference in OTU diversity. Under such stressful conditions, lysogenic bacteriophages can transition to a lytic phase (55–57). Moreover, the patterns observed might also be attributed, at least partly, to a “kill the winner” scenario (52). Under this scenario, the most successful population, i.e., the one with efficient biodegradation genes under the growth conditions of our mesocosms, is preferentially targeted by bacteriophages. The decrease in the most abundant (allelic) variety of the tfd operon at the last sampling time point after exponential growth in the three preceding sampling points that followed the addition of 2,4-D (Fig. 2, top) is consistent with the patterns expected under the kill the winner scenario. Cell motility is also expected to increase due to the nutrient-limiting conditions and the lack of continuous stirring of our mesocosms.
The concentrations of the organic compounds used in the mesocosms may appear relatively higher than their typical concentrations in the no-source environments, and these concentrations have certainly driven, in large part, the strong selection and OTU patterns described above. However, both the abundant and the rare members of the community experienced the same concentrations, and the concentrations were found not to be highly toxic based on our preliminary microcosm experiments. Therefore, our results should be still meaningful for the role of the rare biosphere in the community response to perturbations. Further, the high similarity between the communities at the initial state (T = 0) and the endpoint of the bottle-effect (BE) controls, especially compared to the communities that experienced the organic compounds (e.g., see Fig. 5 and S8), suggested that the OTU/gene patterns described are largely due to the selection pressure of the added compounds as opposed to the incubation conditions, time, or the bottle effect. The high similarity between the T = 0 and BE metagenomes also indicated that further replication of the control mesocosms was probably not necessary.
Overall, our results strongly support the hypothesis that the rare organisms and genes serve as a genetic reservoir that can respond quickly to environmental perturbations, such as the addition of organic pollutants or toxins. Our results also revealed that metagenomics, based on current best practices and costs, cannot always reveal the full metabolic potential of rare organisms and thus should be used in conjunction with other techniques, e.g., enrichment/isolation studies or qPCR, if the goal is to assess functions encoded by rare community members. The results and methodology reported here should have applications in future studies that aim to study the role of rare microbial community members in environmental processes, including environmental bioremediation efforts that involve biostimulation or natural attenuation.
MATERIALS AND METHODS
Site description and sampling.
Water samples were collected on 15 December 2013 from Lake Lanier, GA (34°15′43″N, 83°57′7″W). This seasonally stratified lake is a manmade reservoir located in the northern part of the state of Georgia, is approximately 150 km2 in surface area, and serves as the primary drinking water resource for the Atlanta metropolitan area. A horizontal sampler (Wildco Instruments, Yulee, FL, USA) was used to collect samples of planktonic microbial communities at a depth of 5 m below the surface, near the Browns Bridge. This depth was chosen because it represents the well-oxygenated and highly productive layer of the water column and has been extensively characterized previously by metagenomic surveys (see, e.g., references 16 and 17).
Mesocosm experiment setup, sampling strategy, and DNA extraction.
Glass bottles (20 liters) used for the mesocosms were rinsed with 10% freshly prepared hydrochloric acid, followed by three washes with distilled water and sterilization by autoclaving prior to addition of 18 liters of lake water. Aerobic mesocosms were established in triplicate and incubated at room temperature with gentle mixing in the dark. The initial added concentrations of 2,4-D, caffeine, and 4-NP were 40 μM, 100 μM, and 150 μM, respectively. The concentrations were chosen based on prior microcosm trials to not be highly toxic for the lake microbial communities and to allow effective monitoring by HPLC. Two controls were established for each compound: one abiotic control containing sterilized lake water (autoclaved for 180 min) supplemented with each of the three compounds, and a bottle-effect (BE) control with nonsterilized lake water, incubated under the same conditions without the addition of the compounds. A filtration system was used to collect samples for DNA sequencing, essentially as described previously (16). Briefly, a total of 1 liter or 5 liters of water was prefiltered through AP filters (∼5-μm pore size; Millipore, Billerica, MA, USA) and GF/A filters (∼1.6-μm pore size; Whatman, Little Chalfont, UK), and cells were collected on Sterivex filters (∼0.22-μm pore size; Millipore). Large-volume samples (5 liters) were taken right after the compounds disappeared for DNA and isolation work; small-volume samples (1 liter) were taken at regular intervals (about every 7 days) to provide additional intermediate time points for DNA sequencing, as needed, and for monitoring (see Fig. 1 and S1). Filters were stored at −80°C until used for DNA extraction. A phenol-chloroform DNA extraction protocol was used as described previously (16). DNA from metagenomes or isolates was prepared using the Illumina Nextera XT DNA library prep kit and sequenced on an in-house Illumina sequencer (either MiSeq or HiSeq 2500) using a 2 × 150-bp or 2 × 250 paired-end read strategy. DNA was extracted from pure cultures using the QIAamp DNA minikit (Qiagen, Valencia, CA).
HPLC analysis.
The concentrations of 2,4-D, 4-NP, and caffeine in the mesocosms were measured by high-performance liquid chromatography (HPLC). All mesocosms were mixed on stir plates for approximately 30 min before ∼1-ml samples were removed. HPLC was performed on an Agilent 1100 system (Santa Clara, CA, USA) equipped with a diode array detector, an autosampler, and an Ascentis Express C18 (reversed-phase) HPLC column (Bellevue, WA, USA). The HPLC analytical protocols were as follows. For 2,4-D, the mobile phase consisted of a 50:50 ratio of 0.5% trifluoroacetic acid in water to 0.05% trifluoroacetic acid in acetonitrile. For caffeine, the mobile phase consisted of a 65:35 ratio of 0.1% trifluoroacetic acid in water to 0.05% trifluoroacetic acid in acetonitrile. For 4-NP, the mobile phase consisted of a 90:10 ratio of 0.1% trifluoroacetic acid in water to 0.05% trifluoroacetic acid in acetonitrile. For all methods, the autosampler and column heaters were maintained at 4°C and 45°C, respectively.
Isolation and growth of bacteria.
For isolation procedures, 1-liter samples were removed from the last sampling time point (denoted TPE, for time post enrichment) from all 2,4-D mesocosms. The sample identifications (IDs) were: M1.TPE (19 days), M2.TPE (39 days), and M3.TPE (23 days); the 4-NP sample ID was 4-NP-M1 (17 days [T17]), and the caffeine sample ID was Caff-M1 (25 days [T25]). The 2,4-D degraders were isolated using 10-fold serial dilutions in 12-well plates filled with selective enrichment medium, a ¼-strength liquid minimal salts basal (MSB) medium (58), containing 7.6 mM (NH4)2SO4 as a nitrogen source and 40 μM 2,4-D. Individual colonies that appeared after 5 days of incubation were tested for the ability to degrade 2,4-D in liquid MSB with and without a carbon or nitrogen source. The confirmed colonies were restreaked consecutively at least four times to ensure purity. Similar procedures were used for caffeine and 4-NP degraders. Isolates were identified by sequencing of the 16S rRNA gene (16S) using universal primers (8F forward primer 5′-AGAGTTTGATCCTGGCTCAG-3′ and 1492R reverse primer 5′-GGTTACCTTGTTACGACTT-3′) and/or the whole genome.
Metagenome assembly and sequence analysis.
Metagenome and genome sequences were trimmed using SolexaQA (59), with a Phred score cutoff (-h) of 20 and a minimum fragment length of 50 bp, as described previously (60). Only paired reads with both sisters longer than 50 bp after trimming were used further (Tables S1 and S2). Genomic and metagenomic reads were assembled according to a previously described hybrid-assembly protocol (61). Burkholderia sp. strain KK1 was also sequenced using the PacBio technology at the Genomic Facility of Duke University and assembled with SPAdes (version 3.6.2), using the default settings (62), based on both Illumina and PacBio reads in order to obtain a complete genome sequence. Subsequently, the contigs obtained were reorganized and further joined using RAGOUT (63) and Burkholderia sp. strain RPE67 (accession no. PRJDB1660) as a reference for closing chromosome I.
Prodigal (version 2.6.2) (64) with the single model (-p single) was used to predict proteins in isolate genomes. Gene functional annotation was performed by a BLASTP search of the predicted protein sequences against the Swiss-Prot database (65) and the SEED database using subsystems categories (66). Only best matches of at least bit score 60 were considered for functional annotation. MyTaxa (67) was used for taxonomic classification of contigs, with default parameters. A phylogenomic approach was used to further corroborate the Swiss-Prot findings and identify 2,4-D, 4-NP, and caffeine degradation genes in the isolate genomes as follows. Well-characterized reference sequences of genes that encode 2,4-D, 4-NP, and caffeine catabolic enzymes were downloaded from GenBank (68). A BLASTP search was then performed with these reference protein sequences against the predicted metagenome (or genome) proteins, using default settings with an E value cutoff of 0.001 and an identity threshold of ≥35%. All matching sequences were further evaluated by visually inspecting neighbor-joining phylogenetic trees built using reference protein sequences from GenBank (e.g., whether the unknown/query sequences clustered with previously characterized/reference ones or formed distinct clades instead).
Metagenomic reads encoding 16S rRNA gene fragments were extracted using Parallel-Meta (version 2.4) (69), with default settings, except that the RDP database was used as a reference (option -d R). The 16S rRNA gene coding sequences were identified taxonomically using the QIIME pipeline (version 1.8.0) (70), with full-length closed-reference operational taxonomic unit (OTU) picking at 97% nucleotide sequence identity based on the August 2013 Greengenes release 13-5 database. Sequences that failed to align to reference taxa using the RDP Classifier at 50% confidence (71) were also removed from further analysis. Alpha diversity was measured by several methods, as previously implemented (72, 73), i.e., observed OTUs, Chao1 lower-boundary OTUs with 95% confidence intervals (CI), corrected Shannon index using the Chao-Shen estimator (74), and Bayesian-corrected Shannon index (Dirichlet Prior Bayesian Estimators of Entropy, parameter a = “ML” [maximum likelihood]) (Table S3). Beta diversity was measured by the Bray-Curtis dissimilarity metric.
Assessment of abundances of isolates and biodegradation genes.
The abundances of isolates and genes were assessed by BLASTN searches against their corresponding metagenomes using >150 bp for alignment length and ≥99% identity cutoff for a match. The relative abundances of isolates and genes were calculated as the ratio of the total length of all reads mapping on a reference isolate genome or gene sequence divided by the total length of the reference sequence (× coverage or sequencing depth). To calculate the genome equivalent for each gene/function, i.e., the fraction of total cells containing the gene, an in-house ruby script was used, essentially as described previously (60). Briefly, the script is used to detect and extract reads containing any of 91 essential protein-coding genes (universally conserved single-copy) (75) from the Genome Property database (entry ID, GenProp0799, named “bacterial core gene set, exactly 1 per genome”) (76). The median sequence depth for each single-copy gene was determined based on the number of reads mapped to a gene (cutoffs used, length ≥80 bp and identity ≥97%) divided by the length of the gene (reads per base pair). The genome equivalent of a target gene was estimated as the ratio of its sequence depth divided by the median sequence depth of the 91 marker genes. Differentially abundant genes or taxa among the samples were identified using the DESeq2 package (77).
tfdA primer design, plasmid cloning, and quantification of tfdA and 16S rRNA genes by qPCR.
Two copies of the tfdA gene, which encodes the enzyme for the first step in 2,4-D biodegradation pathway, were identified in the Burkholderia sp. strain KK1 assembled plasmid, and a primer set was designed based on the alignment of the two tfdA genes (Table S4). One tfdA gene, which showed 100% nucleotide identity to a homolog in the reference pM7012 plasmid (21), had a 100% identity match to both forward and reverse primers. The other tfdA gene, which had 100% identity to a homolog in the reference pJp4 plasmid (22), had 100% identity to the forward primer but had a 2-bp mismatch at the 5′ end of the reverse primer. Therefore, the primer set was expected to amplify both genes but not necessarily with the same efficiencies. Another version of the tfdA gene was identified from mesocosm I metagenomic contig01889 at TPE (t = 19 days) (denoted tfdA|contig01889-4); however, the alignment between the primer set and this metagenomic tfdA gene was low, especially for the reverse primer (95% nucleotide identity with forward primer and 42% identity with reverse primer).
The TOPO-TA cloning kit (pCR2.1-TOPO vector; Invitrogen, Carlsbad, CA, USA) was used to make the qPCR standard plasmid, and a single-copy tfdA (142-bp) insertion was confirmed by Sanger sequencing. The tfdA cloning was performed according to the methods described below. Briefly, the 142-bp fragment of the tfdA gene amplicon was cloned into the pCR2.1 vector, as directed by the manufacturer, and subsequently introduced into Escherichia coli DH5α cells by heat shock at 42°C for 30 s. Plasmid extraction was performed after 12 h of growth at room temperature on a shaker (∼200 rpm) with 2 ml of the overnight culture using the QIAprep miniprep kit. For qPCR, 10-fold serial dilutions of the standard plasmid were used to generate a standard curve over the dynamic range from 44 gene copies to 4.4 × 107 gene copies, respectively. The Power SYBR green PCR master mix (Thermo Fisher Scientific) was used in a total volume of 20 μl, with each reaction mixture containing 1× PCR master mix, 250 nM each primer, and 2 μl of template DNA. The amplification was carried out on an Applied Biosystems 7500 Fast real-time PCR system using the following parameters: 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Dissociation curves were generated using the following parameters: 15 s at 95°C, 1 min at 60°C, and 15 s at 95°C to ensure specificity of the PCR. A single peak was observed during the melting curve analysis. The PCR amplification efficiency was 96% and the slope and R2 of the standard curve were −3.47 and 0.999, respectively. The qPCR quantification of tfdA was first tested and validated with a serial dilution experiment of Burkholderia sp. strain KK1 genomic DNA before being applied to mesocosm DNA samples. To validate the qPCR assay, the Burkholderia sp. strain KK1 abundance estimate obtained by qPCR (1.96 × 108 cells/ml) was compared with the abundance determined by microscope-based direct cell counting using 4′,6-diamidino-2-phenylindole (DAPI) (1.19 × 108 cells/ml). The tfdA gene abundance in mesocosm DNA estimated by qPCR was calculated by taking the direct gene count from the instrument and factoring in the dilution factor of the DNA assayed, the volume of diluted DNA used per reaction (2 μl), the total volume of DNA extracted, and the volume of sample from which the DNA was extracted, as described previously (78). All unknown samples, standards, and a blank were assayed in triplicate. The average of the results from the triplicate experiments was used to quantify the abundance of tfdA genes obtained by qPCR (converted to number of tfdA gene copies per milliliter). The mesocosm DNA samples were diluted in water with a 32-fold dilution factor (DF), since the undiluted samples inhibited the qPCR amplification. Total bacterial 16S rRNA genes were also quantified by the same qPCR procedure described above in order to examine shifts in the total number of bacteria during perturbations (assuming a stable average 16S rRNA gene copy number) in the 2,4-D mesocosms. For the 16S rRNA gene qPCR assay, the efficiency was 94%, and the slope and R2 of the standard curve were −3.458 and 1, respectively. The 16S rRNA gene qPCR primers (Table S4) and standard plasmid (pBAV1-16S) used were described previously (78).
Accession number(s).
The metagenomic data used in this study were deposited in GenBank under the Lake Lanier BioProject no. PRJNA214105, with Sequence Read Archive (SRA) accession numbers as follows: SRR3568916 (original lake water; sample ID LLDEC13); SRR3570947 (bottle-effect control; sample ID BE); SRR3568955, SRR3569300, SRR3569349, SRR3569558, SRR3569624, and SRR3569810 (2,4-D mesocosms; sample IDs M1.T0, M1.T10, M1.T14, M1.TPE, M2.TPE, and M3.TPE, respectively); SRR3571025 (4-NP mesocosm; sample ID 4-NP-M1); and SRR3571354, SRR3571355, and SRR3571293 (caffeine mesocosms; sample IDs Caff-M1, Caff-M3, and Caff-BE, respectively). The whole-genome sequences of isolates can be found under the following accession numbers CP015999 to CP016006 (Burkholderia sp. strain KK1), LYVN00000000 (Sphingopyxis sp. strain KK2), LYVO00000000 (Variovorax sp. strain KK3), LYVP00000000 (Pseudomonas sp. strain KK4), and LYVQ00000000 (Pelomonas sp. strain KK5).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by U.S. National Science Foundation (award 1241046). D.T. acknowledges the support of the Onassis Scholarship Foundation.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.03321-16.
REFERENCES
- 1.Torsvik V, Goksoyr J, Daae FL. 1990. High diversity in DNA of soil bacteria. Appl Environ Microbiol 56:782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curtis TP, Sloan WT, Scannell JW. 2002. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci U S A 99:10494–10499. doi: 10.1073/pnas.142680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson KE, Paulsen IT, Heidelberg JF, Fraser CM. 2000. Status of genome projects for nonpathogenic bacteria and archaea. Nat Biotechnol 18:1049–1054. doi: 10.1038/80235. [DOI] [PubMed] [Google Scholar]
- 5.Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci U S A 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedrós-Alió C. 2012. The rare bacterial biosphere. Annu Rev Mar Sci 4:449–466. doi: 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- 8.Lynch MD, Neufeld JD. 2015. Ecology and exploration of the rare biosphere. Nat Rev Microbiol 13:217–229. doi: 10.1038/nrmicro3400. [DOI] [PubMed] [Google Scholar]
- 9.Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. 2011. Activity of abundant and rare bacteria in a coastal ocean. Proc Natl Acad Sci U S A 108:12776–12781. doi: 10.1073/pnas.1101405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falkowski PG, Fenchel T, DeLong EF. 2008. The microbial engines that drive Earth's biogeochemical cycles. Science 320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- 11.Huse SM, Welch DM, Morrison HG, Sogin ML. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dell'Anno A, Danovaro R. 2005. Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science 309:2179. doi: 10.1126/science.1117475. [DOI] [PubMed] [Google Scholar]
- 13.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5(4):e01371-14. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musat N, Halm H, Winterholler B, Hoppe P, Peduzzi S, Hillion F, Horreard F, Amann R, Jørgensen BB, Kuypers MMM. 2008. A single-cell view on the ecophysiology of anaerobic phototrophic bacteria. Proc Natl Acad Sci U S A 105:17861–17866. doi: 10.1073/pnas.0809329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hausmann B, Knorr K-H, Schreck K, Tringe SG, del Rio TG, Loy A, Pester M. 2016. Consortia of low-abundance bacteria drive sulfate reduction-dependent degradation of fermentation products in peat soil microcosms. ISME J 10:2365–2375. doi: 10.1038/ismej.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh S, Caro-Quintero A, Tsementzi D, DeLeon-Rodriguez N, Luo C, Poretsky R, Konstantinidis KT. 2011. Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl Environ Microbiol 77:6000–6011. doi: 10.1128/AEM.00107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsementzi D, Poretsky R, Rodriguez-R L-M, Luo C, Konstantinidis KT. 2014. Evaluation of metatranscriptomic protocols and application to the study of freshwater microbial communities. Environ Microbiol Rep 6:640–655. doi: 10.1111/1758-2229.12180. [DOI] [PubMed] [Google Scholar]
- 18.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 19.Top EM, Holben WE, Forney LJ. 1995. Characterization of diverse 2,4-dichlorophenoxyacetic acid-degradative plasmids isolated from soil by complementation. Appl Environ Microbiol 61:1691–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen TK, Xu Z, Gözdereliler E, Aamand J, Hansen LH, Sørensen SR. 2013. Novel insight into the genetic context of the cadAB genes from a 4-chloro-2-methylphenoxyacetic acid-degrading Sphingomonas. PLoS One 8:e83346. doi: 10.1371/journal.pone.0083346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakai Y, Ogawa N, Shimomura Y, Fujii T. 2014. A 2, 4-dichlorophenoxyacetic acid degradation plasmid pM7012 discloses distribution of an unclassified megaplasmid group across bacterial species. Microbiology 160:525–536. doi: 10.1099/mic.0.074369-0. [DOI] [PubMed] [Google Scholar]
- 22.Trefault N, De la Iglesia R, Molina AM, Manzano M, Ledger T, Pérez-Pantoja D, Sánchez MA, Stuardo M, González B. 2004. Genetic organization of the catabolic plasmid pJP4 from Ralstonia eutropha JMP134 (pJP4) reveals mechanisms of adaptation to chloroaromatic pollutants and evolution of specialized chloroaromatic degradation pathways. Environ Microbiol 6:655–668. doi: 10.1111/j.1462-2920.2004.00596.x. [DOI] [PubMed] [Google Scholar]
- 23.Hogan D, Buckley D, Nakatsu C, Schmidt T, Hausinger R. 1997. Distribution of the tfdA gene in soil bacteria that do not degrade 2,4-dichlorophenoxyacetic acid (2, 4-D). Microb Ecol 34:90–96. doi: 10.1007/s002489900038. [DOI] [PubMed] [Google Scholar]
- 24.Itoh K, Kanda R, Sumita Y, Kim H, Kamagata Y, Suyama K, Yamamoto H, Hausinger RP, Tiedje JM. 2002. tfdA-like genes in 2, 4-dichlorophenoxyacetic acid-degrading bacteria belonging to the Bradyrhizobium-Agromonas-Nitrobacter-Afipia cluster in α-Proteobacteria. Appl Environ Microbiol 68:3449–3454. doi: 10.1128/AEM.68.7.3449-3454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitagawa W, Takami S, Miyauchi K, Masai E, Kamagata Y, Tiedje JM, Fukuda M. 2002. Novel 2,4-dichlorophenoxyacetic acid degradation genes from oligotrophic Bradyrhizobium sp. strain HW13 isolated from a pristine environment. J Bacteriol 184:509–518. doi: 10.1128/JB.184.2.509-518.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spain JC, Gibson DT. 1991. Pathway for biodegradation of p-nitrophenol in a Moraxella sp. Appl Environ Microbiol 57:812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Sun W, Xu L, Zheng X, Chu X, Tian J, Wu N, Fan Y. 2012. Identification of the para-nitrophenol catabolic pathway, and characterization of three enzymes involved in the hydroquinone pathway, in Pseudomonas sp. 1-7. BMC Microbiol 12:27. doi: 10.1186/1471-2180-12-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Zhang J-J, Wang S-J, Zhang X-E, Zhou N-Y. 2005. Plasmid-borne catabolism of methyl parathion and p-nitrophenol in Pseudomonas sp. strain WBC-3. Biochem Biophys Res Commun 334:1107–1114. doi: 10.1016/j.bbrc.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Summers RM, Louie TM, Yu C-L, Gakhar L, Louie KC, Subramanian M. 2012. Novel, highly specific N-demethylases enable bacteria to live on caffeine and related purine alkaloids. J Bacteriol 194:2041–2049. doi: 10.1128/JB.06637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Summers RM, Seffernick JL, Quandt EM, Yu CL, Barrick JE, Subramanian MV. 2013. Caffeine junkie: an unprecedented glutathione S-transferase-dependent oxygenase required for caffeine degradation by Pseudomonas putida CBB5. J Bacteriol 195:3933–3939. doi: 10.1128/JB.00585-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohanty SK, Yu C-L, Das S, Louie TM, Gakhar L, Subramanian M. 2012. Delineation of the caffeine C-8 oxidation pathway in Pseudomonas sp. strain CBB1 via characterization of a new trimethyluric acid monooxygenase and genes involved in trimethyluric acid metabolism. J Bacteriol 194:3872–3882. doi: 10.1128/JB.00597-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu CL, Kale Y, Gopishetty S, Louie TM, Subramanian M. 2008. A novel caffeine dehydrogenase in Pseudomonas sp. strain CBB1 oxidizes caffeine to trimethyluric acid. J Bacteriol 190:772–776. doi: 10.1128/JB.01390-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Escobar-Páramo P, Faivre N, Buckling A, Gougat-Barbera C, Hochberg ME. 2009. Persistence of costly novel genes in the absence of positive selection. J Evol Biol 22:536–543. doi: 10.1111/j.1420-9101.2008.01673.x. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-R LM, Konstantinidis KT. 2014. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30:629–635. doi: 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-R LM, Konstantinidis KT. 2014. Estimating coverage in metagenomic datasets and why it matters. ISME J 8:2349–2351. doi: 10.1038/ismej.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann D, Kleinsteuber S, Müller RH, Babel W. 2003. A transposon encoding the complete 2,4-dichlorophenoxyacetic acid degradation pathway in the alkalitolerant strain Delftia acidovorans P4a. Microbiology 149:2545–2556. doi: 10.1099/mic.0.26260-0. [DOI] [PubMed] [Google Scholar]
- 37.Ka J, Holben WE, Tiedje JM. 1994. Genetic and phenotypic diversity of 2,4-dichlorophenoxyacetic acid (2,4-D)-degrading bacteria isolated from 2,4-D-treated field soils. Appl Environ Microbiol 60:1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tonso N, Matheson V, Holben W. 1995. Polyphasic characterization of a suite of bacterial isolates capable of degrading 2,4-D. Microb Ecol 30:3–24. [DOI] [PubMed] [Google Scholar]
- 39.Stibal M, Bælum J, Holben WE, Sørensen SR, Jensen A, Jacobsen CS. 2012. Microbial degradation of 2,4-dichlorophenoxyacetic acid on the Greenland ice sheet. Appl Environ Microbiol 78:5070–5076. doi: 10.1128/AEM.00400-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lory S. 2014. The family Legionellaceae, p 387–389. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes. Springer, New York, NY. [Google Scholar]
- 41.Zhang J-J, Liu H, Xiao Y, Zhang X-E, Zhou N-Y. 2009. Identification and characterization of catabolic para-nitrophenol 4-monooxygenase and para-benzoquinone reductase from Pseudomonas sp. strain WBC-3. J Bacteriol 191:2703–2710. doi: 10.1128/JB.01566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain RK, Dreisbach JH, Spain JC. 1994. Biodegradation of p-nitrophenol via 1,2,4-benzenetriol by an Arthrobacter sp. Appl Environ Microbiol 60:3030–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chauhan A, Chakraborti AK, Jain RK. 2000. Plasmid-encoded degradation of p-nitrophenol and 4-nitrocatechol by Arthrobacter protophormiae. Biochem Biophys Res Commun 270:733–740. doi: 10.1006/bbrc.2000.2500. [DOI] [PubMed] [Google Scholar]
- 44.Schäfer A, Harms H, Zehnder AJ. 1996. Biodegradation of 4-nitroanisole by two Rhodococcus spp. Biodegradation 7:249–255. doi: 10.1007/BF00058184. [DOI] [PubMed] [Google Scholar]
- 45.Summers RM, Mohanty SK, Gopishetty S, Subramanian M. 2015. Genetic characterization of caffeine degradation by bacteria and its potential applications. Microb Biotechnol 8:369–378. doi: 10.1111/1751-7915.12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu CL, Louie TM, Summers R, Kale Y, Gopishetty S, Subramanian M. 2009. Two distinct pathways for metabolism of theophylline and caffeine are coexpressed in Pseudomonas putida CBB5. J Bacteriol 191:4624–4632. doi: 10.1128/JB.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu CL, Summers RM, Li Y, Mohanty SK, Subramanian M, Pope RM. 2014. Rapid identification and quantitative validation of a caffeine-degrading pathway in Pseudomonas sp. CES. J Proteome Res 14:95–106. [DOI] [PubMed] [Google Scholar]
- 48.Mazzafera P, Olsson O, Sandberg G. 1996. Degradation of caffeine and related methylxanthines by Serratia marcescens isolated from soil under coffee cultivation. Microb Ecol 31:199–207. [DOI] [PubMed] [Google Scholar]
- 49.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prach K, Walker LR. 2011. Four opportunities for studies of ecological succession. Trends Ecol Evol 26:119–123. doi: 10.1016/j.tree.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 51.Zhou J, Deng Y, Zhang P, Xue K, Liang Y, Van Nostrand JD, Yang Y, He Z, Wu L, Stahl DA, Hazen TC, Tiedje JM, Arkin AP. 2014. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci U S A 111:E836–E845. doi: 10.1073/pnas.1324044111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Valera F, Martin-Cuadrado A-B, Rodriguez-Brito B, Pašić L, Thingstad TF, Rohwer F, Mira A. 2009. Explaining microbial population genomics through phage predation. Nat Rev Microbiol 7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 53.Weitz JS, Wilhelm SW. 2012. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol Rep 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fuhrman J, Schwalbach M. 2003. Viral influence on aquatic bacterial communities. Biol Bull 204:192–195. doi: 10.2307/1543557. [DOI] [PubMed] [Google Scholar]
- 55.Middelboe M, Jorgensen N, Kroer N. 1996. Effects of viruses on nutrient turnover and growth efficiency of noninfected marine bacterioplankton. Appl Environ Microbiol 62:1991–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pradeep Ram AS, Sime-Ngando T. 2008. Functional responses of prokaryotes and viruses to grazer effects and nutrient additions in freshwater microcosms. ISME J 2:498–509. doi: 10.1038/ismej.2008.15. [DOI] [PubMed] [Google Scholar]
- 57.Payet JP, Suttle CA. 2013. To kill or not to kill: the balance between lytic and lysogenic viral infection is driven by trophic status. Limnol Oceanogr 58:465–474. doi: 10.4319/lo.2013.58.2.0465. [DOI] [Google Scholar]
- 58.Stanier R. 1942. The Cytophaga group: a contribution to the biology of myxobacteria. Bacteriol Rev 6:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez-R L-M, Overholt WA, Hagan C, Huettel M, Kostka JE, Konstantinidis KT. 2015. Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J 9:1928–1940. doi: 10.1038/ismej.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo C, Tsementzi D, Kyrpides NC, Konstantinidis KT. 2012. Individual genome assembly from complex community short-read metagenomic datasets. ISME J 6:898–901. doi: 10.1038/ismej.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kolmogorov M, Raney B, Paten B, Pham S. 2014. Ragout—a reference-assisted assembly tool for bacterial genomes. Bioinformatics 30:i302–309. doi: 10.1093/bioinformatics/btu280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O'Donovan C, Redaschi N, Suzek B. 2006. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo C, Rodriguez-R LM, Konstantinidis KT. 2014. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42:e73. doi: 10.1093/nar/gku169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. 2004. GenBank: update. Nucleic Acids Res 32:D23–D26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su X, Pan W, Song B, Xu J, Ning K. 2014. Parallel-META 2.0: enhanced metagenomic data analysis with functional annotation, high performance computing and advanced visualization. PLoS One 9:e89323. doi: 10.1371/journal.pone.0089323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez-R LM, Konstantinidis KT. 2016. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1. [DOI] [Google Scholar]
- 73.Hausser J, Strimmer K. 2009. Entropy inference and the James-Stein estimator, with application to nonlinear gene association networks. J Mach Learn Res 10:1469–1484. [Google Scholar]
- 74.Chao A, Shen T-J. 2003. Nonparametric estimation of Shannon's index of diversity when there are unseen species in sample. Environ Ecol Stat 10:429–443. doi: 10.1023/A:1026096204727. [DOI] [Google Scholar]
- 75.Dupont CL, Rusch DB, Yooseph S, Lombardo M-J, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL, Lasken RS, Nealson K, Friedman R, Venter JC. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. doi: 10.1038/ismej.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haft DH, Selengut JD, Brinkac LM, Zafar N, White O. 2005. Genome Properties: a system for the investigation of prokaryotic genetic content for microbiology, genome annotation and comparative genomics. Bioinformatics 21:293–306. doi: 10.1093/bioinformatics/bti015. [DOI] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ritalahti KM, Amos BK, Sung Y, Wu Q, Koenigsberg SS, Löffler FE. 2006. Quantitative PCR targeting 16S rRNA and reductive dehalogenase genes simultaneously monitors multiple Dehalococcoides strains. Appl Environ Microbiol 72:2765–2774. doi: 10.1128/AEM.72.4.2765-2774.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.