

ABSTRACT
The aim of this study is to obtain a systems-level understanding of the interactions between Dehalococcoides and corrinoid-supplying microorganisms by analyzing community structures and functional compositions, activities, and dynamics in trichloroethene (TCE)-dechlorinating enrichments. Metagenomes and metatranscriptomes of the dechlorinating enrichments with and without exogenous cobalamin were compared. Seven putative draft genomes were binned from the metagenomes. At an early stage (2 days), more transcripts of genes in the Veillonellaceae bin-genome were detected in the metatranscriptome of the enrichment without exogenous cobalamin than in the one with the addition of cobalamin. Among these genes, sporulation-related genes exhibited the highest differential expression when cobalamin was not added, suggesting a possible release route of corrinoids from corrinoid producers. Other differentially expressed genes include those involved in energy conservation and nutrient transport (including cobalt transport). The most highly expressed corrinoid de novo biosynthesis pathway was also assigned to the Veillonellaceae bin-genome. Targeted quantitative PCR (qPCR) analyses confirmed higher transcript abundances of those corrinoid biosynthesis genes in the enrichment without exogenous cobalamin than in the enrichment with cobalamin. Furthermore, the corrinoid salvaging and modification pathway of Dehalococcoides was upregulated in response to the cobalamin stress. This study provides important insights into the microbial interactions and roles played by members of dechlorinating communities under cobalamin-limited conditions.
IMPORTANCE The key chloroethene-dechlorinating bacterium Dehalococcoides mccartyi is a cobalamin auxotroph, thus acquiring corrinoids from other community members. Therefore, it is important to investigate the microbe-microbe interactions between Dehalococcoides and the corrinoid-providing microorganisms in a community. This study provides systems-level information, i.e., taxonomic and functional compositions and dynamics of the supportive microorganisms in dechlorinating communities under different cobalamin conditions. The findings shed light on the important roles of Veillonellaceae species in the communities compared to other coexisting community members in producing and providing corrinoids for Dehalococcoides species under cobalamin-limited conditions.
KEYWORDS: Dehalococcoides, reductive dechlorination, corrinoid, Veillonellaceae, metagenome, metatranscriptome
INTRODUCTION
Chlorinated solvents, such as tetra- and trichloroethene (PCE and TCE, respectively), are common groundwater contaminants in the United States (1, 2). Bioremediation is a cost-effective and environmentally friendly approach to clean up solvent-contaminated field sites (3, 4). Dehalococcoides mccartyi is the only known bacterium capable of carrying out complete reductive dechlorination of chloroethenes to ethene (5, 6). Numerous studies have been performed on the physiology (7–10), genomics (11–15), and metabolic pathways (16–19) of this bacterium to optimize bioremediation processes. Corrinoids are a group of cyclic compounds containing four pyrrole rings, many of which are vitamins and/or enzyme cofactors. D. mccartyi requires hydrogen, acetate, and corrinoid cofactors (e.g., cobalamin, also known as vitamin B12) during reductive dechlorination (5). However, all sequenced strains lack the genetic potential for forming hydrogen or acetate via fermentation, as well as the capabilities of de novo synthesis of corrinoid cofactors (6, 11, 20). Consequently, D. mccartyi must acquire each of these essential substrates and nutrients from the environment or through interactions with other coexisting organisms. In a typical groundwater bioremediation site amended with low-cost organic substrates, like whey and molasses, hydrogen and acetate can readily be provided by fermenters (21–23). Since a number of environmental microorganisms are capable of de novo corrinoid biosynthesis (24), corrinoid cofactors may also be available for Dehalococcoides in an environmental microbial community. D. mccartyi strains are able to utilize corrinoid forms other than cobalamin and modify them into cobalamin in the presence of the favorable lower ligand dimethylbenzimidazole (DMB) (9, 25, 26). Therefore, understanding the capabilities of microorganisms supportive of D. mccartyi and how they interact will provide important guidance for manipulations of site conditions toward better bioremediation strategies.
Previous community studies focused primarily on the physiological and transcriptional responses of D. mccartyi to different growth conditions (27–31), with limited knowledge of the corresponding responses of the coexisting supportive microorganisms, particularly at the transcriptional level. Total RNA sequencing (metatranscriptomics) can complement total DNA sequencing (metagenomics) by providing information about active community members and the taxonomic or functional dynamics over a time course or across different environmental conditions (32). Metagenomics or 16S rRNA gene amplicon sequencing, sometimes followed by metatranscriptomics, has been successfully applied to a wide range of environmental microbiomes, including ocean surface waters (33, 34), acid mine drainage (35), permafrost (32), and microbial fuel cells (36). Microbial compositions and functional genes of Dehalococcoides-containing dechlorinating communities have been reported in several metagenomic studies (19, 36, 37), and the active community members and the genes involved in carbon metabolism in the community have been investigated by BrdU-incorporated DNA sequencing (38). Metatranscriptomics sequencing can further provide insights into a comprehensive profile of functional activity and dynamics, which is important for identifying the roles played by the supportive microorganisms in dechlorinating communities under various environmental conditions.
The goal of this study was to get a systems-level understanding of taxonomic and functional compositions in dechlorinating communities without exogenous cobalamin and the roles of supportive microorganisms in corrinoid supplying for D. mccartyi. We constructed enrichment cultures from contaminated groundwater that were able to reductively dechlorinate TCE using lactate as the primary electron donor, with and without exogenous cobalamin cofactor. The microbial compositions and functional potentials were first analyzed by metagenomic sequencing. Draft genomes were binned from the metagenomes representing dominant community members of the enrichment community. Subsequently, the active members and their functional roles in the community were revealed by metatranscriptomics, reverse transcription-quantitative PCR (RT-qPCR), and microarrays.
RESULTS
Metagenomic analysis.
A total of 101,964,968 reads from both TCE-dechlorinating enrichments with cobalamin (HiTCEB12) and without cobalamin (HiTCE) metagenomes were filtered out after quality control and dynamic trimming, with an average length of 100 bp. Using de novo assembly, 16,404 contigs with an average length of 2,463 bp were obtained. The coverages of the assembled contigs for the HiTCEB12 and HiTCE metagenomes were 89.7% and 83.0%, respectively, which indicate a reliable accuracy of the de novo assembly. The assembled contigs consist of 49,488 predicted open reading frames (ORFs). Nine clusters of metagenome bins were retrieved using a modified differential coverage method (39) (Fig. 1). Seven metagenome bins (bin1 to -6 and bin9) with relatively high coverage and long contig length were subjected to further analysis, including those related to Dehalococcoides, Veillonellaceae, Desulfovibrio, Sedimentibacter, Spirochaetaceae, Bacteroides, and Clostridium (Fig. 1 and Table 1), which are referred to here as bin-genomes. The bin-genome sizes, in terms of the number of predicted ORFs, were comparable to those of fully sequenced genomes of related taxa (Table 1) (http://www.ncbi.nlm.nih.gov/). Dehalococcoides, Desulfovibrio, and Sedimentibacter bin-genomes exhibited an average of more than 90% amino acid sequence identity with the corresponding sequenced genomes, whereas the other bin-genomes exhibited less than 70% amino acid sequence identity, indicating that they represent novel species (Table 1). Detailed gene annotations of each bin-genome are listed in Table S2 of the supplemental material.
FIG 1.

Metagenome binning using differential coverages of HiTCE and HiTCEB12 (note that the taxonomic assignment of the bin-genomes was based on BLAST results of the unique 16S rRNA gene sequence included in each metagenome bin against SILVA structural RNA database, with resolution at the genus or family level.).
TABLE 1.
Summary of bin-genome reconstruction from metagenomes and corresponding transcripts detected in metatranscriptomes
| Bin-genome | Size (Mbp) | No. of predicted ORFs | Estimated genome completeness (%) | Avg amino acid sequence identity (%) | No. of transcripts detected in metatranscriptomes: |
|||
|---|---|---|---|---|---|---|---|---|
| HiTCEB12_T1 | HiTCE_T1 | HiTCE_T2 | HiTCE_T3 | |||||
| Bin1_Dehalococcoides | 1.3 | 1,431 | 99.3 | 95 | 1,300 | 1,308 | 1,333 | 1,334 |
| Bin2_Veillonellaceae | 3.6 | 3,856 | 99.9 | 66 | 808 | 910 | 991 | 466 |
| Bin3_Desulfovibrio | 3.4 | 3,390 | 91.1 | 91 | 222 | 221 | 1,483 | 641 |
| Bin4_Sedimentibacter | 3.5 | 3,659 | 93.3 | 90 | 274 | 278 | 286 | 89 |
| Bin5_Spirochaetaceae | 2.7 | 2,682 | 92.1 | 56 | 892 | 891 | 853 | 571 |
| Bin6_Bacterioides | 2.4 | 2,203 | 96.4 | 68 | 728 | 724 | 494 | 449 |
| Bin9_Clostridium | 2.6 | 2,871 | 99.0 | 67 | 57 | 60 | 216 | 63 |
| Unbinned | 29,396 | 1,530 | 1,551 | 196 | 239 | |||
| Total | 49,488 | 5,811 | 5,943 | 5,852 | 3,852 | |||
Differential gene expression from metatranscriptomic sequencing.
In addition to the metagenomes, which represent the taxonomic composition and metabolic potentials of each taxonomic group (i.e., bin-genome), the metatranscriptomes provide additional information on the dynamics of these taxonomic groups and their functional activities. Over 70% of the detected gene transcripts from the metatranscriptomes were assigned to the seven analyzed bin-genomes (Table 1), indicating that they represent the most active community members. In order to investigate the structural and functional activity dynamics in the community when no exogenous cobalamin was available, we first compared the metatranscriptomes of HiTCE (without B12) and HiTCEB12 (with B12) on day 2. Although similar numbers of transcripts were detected in the two metatranscriptomes (Table 1), approximately 550 functional genes exhibited more than 5-fold greater transcript abundance in HiTCE than in HiTCEB12 (Fig. 2A). Moreover, about 75% of those upregulated genes were assigned to the Veillonellaceae bin-genome (Fig. 2A). Similar taxonomic distribution was obtained using BLASTx to annotate the upregulated gene sequences (Fig. 2B).
FIG 2.

Taxonomic distribution by genomic reconstruction (A) and by BLASTx and MEGAN5 (B) of genes upregulated (>5-fold) in HiTCE compared to HiTCEB12. Numbers of upregulated genes are indicated next to the names; the family with the most upregulated genes is highlighted in bold in panel B..
Community dynamics were also analyzed by metatranscriptomic sequencing over a time series in the HiTCE enrichment. The transcript abundances of most annotated genes between the two replicates at day 13 (T3) were at similar levels, as was the case for the remaining 16S rRNA if it is assumed that the removal of structural RNA was the same among samples (Fig. S3). Therefore, the averaged transcript abundance at T3 was used for analysis. Veillonellaceae cells were most active on day 2 (T1), while Desulfovibrio dominated on day 9 (T2). On day 13 (T3), the activities of all the other community members decreased substantially, resulting in a relatively higher activity of Dehalococcoides (Fig. 3). This suggests that the Veillonellaceae taxon likely plays a key role in corrinoid exchange during the first 2 days in the culture without exogenous cobalamin.
FIG 3.
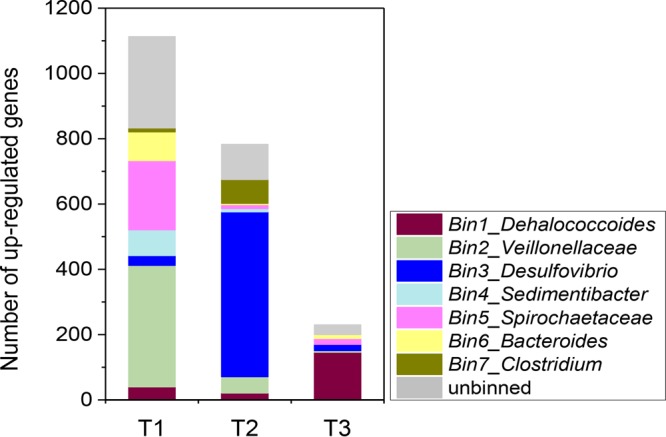
Taxonomic distribution of the upregulated genes in HiTCE (>5-fold difference) based on metatranscriptomic analysis over a time course.
Corrinoid biosynthesis.
The corrinoid biosynthesis pathway is one of the most complex pathways in nature, containing more than 30 enzymatic steps (24). In the Dehalococcoides bin-genome, only three upstream corrinoid biosynthesis genes were detected (Fig. 4 and Table S3), consistent with its reported inability to carry out de novo corrinoid synthesis (40, 41). Nevertheless, a full set of genes involved in the downstream corrinoid salvaging and remodeling pathway is present in the Dehalococcoides bin-genome (Fig. 4), suggesting its potential for corrinoid remodeling when corrinoids are made available by others (9). Three of the non-Dehalococcoides bin-genomes, Veillonellaceae, Desulfovibrio, and Sedimentibacter, possess near-complete de novo corrinoid biosynthesis pathways (Fig. 4 and Table S3), indicating their potential for providing corrinoids to Dehalococcoides. However, transcripts of those corrinoid biosynthesis genes were not positively detected in the metatranscriptomes. Only the cysG (gene_id_944) and cobS_2 (gene_id_5485) transcripts in the Veillonellaceae bin-genome were detected in both enrichments at T1, and higher transcript abundances (>5-fold) were observed in HiTCE than in HiTCEB12 (Fig. 4 and Table S4).
FIG 4.

Corrinoid biosynthesis pathways in 4 bin-genomes.
Corrinoid-dependent metabolic pathways.
Considering the complexity and energy cost of de novo corrinoid biosynthesis, corrinoids would presumably be synthesized only if they were required as cofactors for enzymes in essential metabolic pathways. Therefore, we analyzed the presence of known corrinoid-dependent metabolic pathways in the bin-genomes, as well as their transcriptional activities, in order to further identify the active corrinoid producers.
One known corrinoid-dependent metabolic pathway is the methylmalonyl-coenzyme A (CoA) pathway used for propionate formation during lactate fermentation. The methylmalonyl-CoA mutase in this pathway is a corrinoid-dependent isomerase. This pathway was annotated in the Veillonellaceae, Sedimentibacter, Spirochaetaceae, and Bacteroides bin-genomes. Those in the Veillonellaceae and Spirochaetaceae bin-genomes were actively transcribed (Fig. 5 and Table S5). In the Veillonellaceae bin-genome, the transcripts of the two genes encoding the corrinoid-dependent methylmalonyl-CoA mutase and methylmalonyl-CoA carboxyltransferase were less abundant in HiTCE than in HiTCEB12, whereas the other genes in this pathway exhibited higher expression in HiTCE, especially for lactate permease. In contrast, in the Spirochaetaceae bin-genome, similar transcript abundances were observed for HiTCE and HiTCEB12. In addition, the transcriptional dynamics of the methylmalonyl-CoA pathway over a time course was observed. The activity in Veillonellaceae was relatively higher at T2, while that in Spirochaetaceae was higher at T1 (Fig. 5 and Table S5).
FIG 5.

Heatmap of RPKM values of selected genes in the Veillonellaceae bin-genome.
Another corrinoid-dependent metabolic pathway that might activate corrinoid de novo synthesis in corrinoid producers is methionine synthesis. The corrinoid-dependent methionine synthase (MetH) is a methyltransferase. It was detected in all of the non-Dehalococcoides bin-genomes, except for the Clostridium bin-genome (Table S2). However, only the one in Veillonellaceae was actively transcribed and exhibited higher abundances in HiTCE (reads per kilobase per million [RPKM], 12) than in HiTCEB12 (RPKM, 6) (Fig. 5). One gene encoding the alternative corrinoid-independent methionine synthase (MetE) was also detected in the Desulfovibrio bin-genome, suggesting the ability to synthesize methionine when corrinoids are limited. However, the corresponding gene transcript was not detected in the culture, suggesting that corrinoids were available to the community in some form. Other corrinoid-dependent methyltransferases important for energy metabolism and carbon cycle in anaerobic acetogenic bacteria include vanillate O-demethylase, a corrinoid iron-sulfur protein, and its corresponding methyltransferase (AcsE), which is involved in the Wood-Ljungdahl pathway for CO2/CO fixation (42). However, we were not able to detect genes encoding those methyltransferases in any of the non-Dehalococcoides bin-genomes (data not shown).
Cobalt and corrinoid transport.
Cobalt is the key metal element in a corrinoid molecule, which is transported into cells via ATP-binding cassette (ABC) transporters for de novo corrinoid synthesis. The uptake of cobalt may also indicate corrinoid biosynthesis activities and help identify active corrinoid producers. We discovered four operons annotated as cobalt ABC transporters (cbiMQO) in the Veillonellaceae bin-genome and one in the Desulfovibrio, but not in the other non-Dehalococcoides, bin-genomes. Three of the four cobalt transporters in Veillonellaceae were transcribed with higher abundance in HiTCE than in HiTCEB12, while the cbiQ transcript in Desulfovibrio was less abundant in HiTCE (Fig. 5 and Table S4). In addition, the transcripts of cobalt ABC transporters were detected only at T1, indicating that corrinoid biosynthesis occurred at an early phase, and its activity decreased afterwards when corrinoids reached an adequate level.
We also analyzed the presence and transcriptional activities of the corrinoid ABC transporter operon (btuFCD) in all bin-genomes. As the btuFCD operon exhibits similarity with other ABC transporters leading to potential misannotations, we manually annotated the btuFCD homologous genes in each bin-genome by conducting a BLAST search against a customized protein database containing the known amino acid sequences of the BtuFCD operon from all bacterial species extracted from the NCBI nonredundant database. A 30% amino acid sequence identity was set as the cutoff value. Genes homologous to btuFCD were detected in the Veillonellaceae, Desulfovibrio, Sedimentibacter, and Dehalococcoides bin-genomes. However, only the Dehalococcoides btuD gene (gene_id_5163) transcript was detected in the metatranscriptomes, with about 2 times more abundance in the enrichment without exogenous cobalamin than that with exogenous cobalamin (Table S4).
Sporulation-related genes in the Veillonellaceae bin-genome.
Interestingly, a distinctive difference between the metatranscriptomes of HiTCE and HiTCEB12 was related to sporulation (Fig. 5). For all of the detected Veillonellaceae transcripts, abundances were substantially higher (up to 28-fold) in HiTCE than HiTCEB12 at T1, but were dramatically lower at T2 and T3 (Fig. 5 and Tables S4 and S5). Although the other two Firmicutes bin-genomes, those of Sedimentibacter and Clostridium, also possess a suite of sporulation-related genes, the sporulation activities were insignificant, with only 2 to 4 genes actively transcribed (Tables S4 and S5).
Complementary molecular approaches for differential gene expression analyses.
Although the detected activities of corrinoid-dependent metabolic pathways and cobalt transport strongly suggest the need for de novo corrinoid synthesis within the HiTCE community, transcripts of genes involved in the corrinoid biosynthesis pathway in the Veillonellaceae bin-genome were not detected in the metatranscriptome in large numbers. In order to query the expression of low-abundance mRNAs of the corrinoid biosynthesis pathway in Veillonellaceae, specific qPCR primers were designed and applied to the same RNA samples used for sequencing. With this complementary approach, we were able to detect and quantify the relative expression of the corrinoid biosynthesis genes in the Veillonellaceae bin-genome. Since the transcript abundances of the tceA gene among all samples were at similar levels, we used tceA as the reference gene for the relative RT-qPCR analysis. Corrinoid biosynthesis activities of Veillonellaceae in HiTCE at T1 were higher than in HiTCEB12 under the same dechlorination activity conditions (Fig. 6). Interestingly, consistent with the metatranscriptomic data (Fig. 4 and Table S4), one gene annotated as cobS_1 (gene_id_5485) exhibited higher relative expression in HiTCE, while another gene, annotated as cobS_2 (gene_id_6498), had a higher transcript abundance in HiTCEB12 (Fig. 6) in this qPCR analysis.
FIG 6.
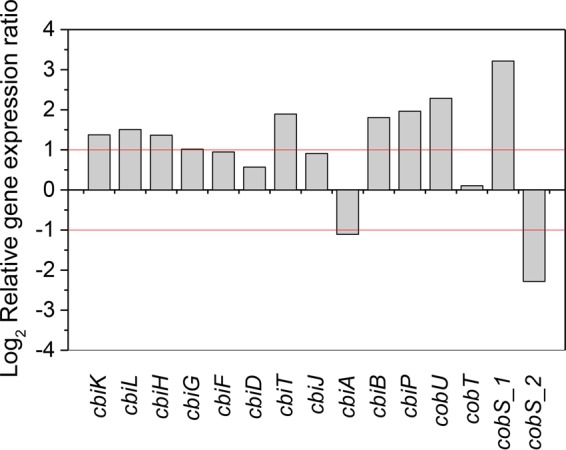
Relative gene expression ratios (HiTCE/HiTCEB12 at T1) by RT-qPCR targeting corrinoid biosynthesis genes in the Veillonellaceae bin-genome (red lines indicate >2-fold difference; reference gene, tceA, representing dechlorination activity).
Microarrays are another complementary approach to analyze differential gene expression. To further validate the metatranscriptomic sequencing results related to the Dehalococcoides bin-genome, we compared differential gene expression obtained from a Dehalococcoides genus-wide microarray to that obtained from the metatranscriptomic sequencing. The targeted D. mccartyi genomes of the microarray include strain 195, which is the closest genome to the identified Dehalococcoides bin-genome. According to microarray results, 96 genes were upregulated and 2 genes were downregulated in HiTCE compared to HiTCEB12. Eighty-six out of the 96 upregulated genes and one of the two downregulated genes were detected in the metatranscriptomes (Table S6), although the transcript abundance ratios of HiTCE to HiTCEB12 according to RNA sequencing were less significant than the signal intensity ratios by RNA microarray. The results of microarray and RNA sequencing were more consistent when there was a >4-fold difference (Table S6). We further examined the Dehalococcoides genes involved in corrinoid salvaging and remodeling pathways. In general, metatranscriptomes and microarrays give similar gene regulation trends. Consistently upregulated genes include cbiZ (DET0653), cobD (DET0655), cobT (DET0657), and btuCD (DET0651 and DET0652), although several genes (i.e., cbiP, butF, DET1175, and DET1176) did not show differential expression based on metatranscriptome but were significantly upregulated according to microarray analysis (Fig. S4). Since we included technical replicates only for the RNA samples of HiTCE at T3 (due to relatively high cost of sequencing at the time of this study and the budget limitation), the statistical significance of the comparison between metatranscriptomic and microarray results was not able to be obtained.
DISCUSSION
Metagenomic and metatranscriptomic analyses provide powerful cultivation independent tools to disentangle microbial interactions, given that most environmental microorganisms are excluded from lab cultivation and many grow in important syntrophy. This study is the first combined application, to our knowledge, of metagenomic and metatranscriptomic sequencing to dechlorinating communities. Here, we compared enrichments with and without exogenous cobalamin to uncover the phylogenetic composition, functional potentials, activities, and dynamics of the communities, with a focus on the function of nutrient exchanges, including corrinoids. We acknowledge that the lack of biological replicates for metatranscriptomic sequencing makes the comparative analyses without statistical significance. Nevertheless, we still carried out some qualitative and semiquantitative comparison of gene expression using the RNA sequencing data. We then conducted qPCR and microarray analyses with three biological replicates as a validation.
Among the bin-genomes with de novo corrinoid biosynthesis pathways, we observed the most differential gene expression of Veillonellaceae bin-genome at an early stage in the enrichment without exogenous cobalamin compared to that with cobalamin, including genes related to cobalt transport and de novo corrinoid biosynthesis. Veillonellaceae is one of the three bin-genomes capable of de novo corrinoid synthesis that also exhibited activities of corrinoid-dependent metabolic pathways, such as propionate-forming methylmalonyl CoA and MetH-mediated methionine synthesis (42). Moreover, the Veillonellaceae bin-genome in HiTCE exhibited the highest activity on day 2 (T1), when the corrinoid production reached maximum production (41). In addition, p-cresolyl cobamide was the dominant corrinoid form produced endogenously by supportive microorganisms in this enrichment (41), and the phenolic corrinoids are known to be produced by Veillonellaceae species (i.e., Pelosinus and Sporomusa), similar to the Veillonellaceae bin-genome detected in this study (43, 44). Collectively, the above-mentioned evidence suggests that the cells associated with the Veillonellaceae bin-genome are the corrinoid cofactor (i.e., p-cresolyl cobamide) suppliers for Dehalococcoides in this community. Pelosinus and Sporomusa species in the Veillonellaceae family are among the commonly detected species in dechlorinating communities (22, 40, 45, 46).
In the p-cresolyl cobamide molecule, the lower ligand cannot form the coordination to the cobalt ion (base-off conformation) (43). Only several corrinoid-dependent enzymes can use corrinoids with base-off conformation as cofactors, and the corrinoid-dependent methylmalonyl CoA mutase in the propionate-producing methylmalonyl CoA pathway is one such enzyme (42). Therefore, p-cresolyl cobamide is most likely produced and utilized by the Veillonellaceae bin-genome as the corrinoid cofactor for the methylmalonyl CoA pathway to generate propionate from lactate fermentation.
The de novo synthesis and utilization of corrinoid cofactors generally occur intracellularly. Therefore, it is still an enigma how corrinoids are made available extracellularly to other community members, such as Dehalococcoides. Possibilities include active secretion and passive release via lysis. So far, no active mechanisms of corrinoid export have been identified (47). The transcript abundances of corrinoid ABC transporter genes (btuFCD) in the Veillonellaceae bin-genome were rather low, and no difference was observed between the enrichments with and without added cobalamin, suggesting little active corrinoid exchange via corrinoid ABC transporters. Nevertheless, the sporulation activity of Veillonellaceae was substantially induced in the enrichment without exogenous cobalamin on day 2. This suggests that the cell lysis during sporulation likely served as a possible mechanism of the corrinoid release. Similarly, some nutrients may also be released during the nocturnal sporulation of an Epulopiscium species symbiont, which is beneficial to its host, the surgeonfish (48). Once released into the environment, corrinoids could be salvaged by D. mccartyi for remodeling to the preferred cobalamin form with the corresponding lower ligand dimethylbenzimidazole (DMB), as reported previously (9, 41, 44). Sporulation is an important strategy for spore-forming species within the phylum Firmicutes to survive under adverse conditions, presumably triggered by nutrient depletion or the presence of toxic compounds (49, 50). As the enrichments with and without exogenous cobalamin encountered the same level of TCE toxicity and the same lactate depletion after the first 2 days, it seems to be other conditions, possibly the limited cobalamin, that stimulated the sporulation of the Veillonellaceae cells in the enrichment without cobalamin. Therefore, regarding the interactions between the corrinoid suppliers and the dechlorinators in the investigated community without exogenous cobalamin, it is not unreasonable to speculate that during the first 2 days, corrinoids were produced by Veillonellaceae cells via corrinoid-dependent fermentation pathways and then passively released to the culture suspension during the induced sporulation, which made them available for Dehalococcoides to utilize for dechlorination afterwards.
If Dehalococcoides was obtaining cobalamin by modifying other corrinoid forms using the lower ligand DMB when there was no exogenous cobalamin, the availability of DMB in the community would become essential. DMB was detected in the enrichment cultures (41), indicating that some community members were able to produce it anaerobically. An anaerobic DMB synthesis operon (bzaABCDE) has been identified very recently (51), but we were not able to detect homologous genes in our metagenomes (data not shown), so the DMB producers here remain unknown.
High-throughput DNA/RNA sequencing allows us to obtain the compositions, activities, and dynamics of complex communities at systems levels. In this study, we complemented metagenomic and metatranscriptomic sequencing with qPCR and microarray analyses to gain a comprehensive understanding of the microbial communities involved in reductive dechlorination. The corrinoid biosynthesis genes are not highly expressed, probably because the pathway complexity makes it energy demanding, and corrinoid cofactors are needed in only trace amounts to function (24). Here, RT-qPCR using specific primers targeting corrinoid synthesis genes identified from the Veillonellaceae bin-genome, as well as Dehalococcoides microarrays, seemed to be more sensitive in detecting the differential gene expression than metatranscriptomic sequencing, by avoiding the interference of structural RNAs and mRNAs of housekeeping genes. All these complementary tools are important for the differential gene expression analyses, especially for the rare genes in complex communities. We also recognize the limitation of all RNA-based function and activity analyses, as mRNA abundance does not necessarily reflect the actual metabolic activity. Nevertheless, valuable hypotheses regarding the research questions can still be derived from the mRNA-level differential gene expression and then be further tested by gene modification, as well as protein- and metabolite-level analyses.
In summary, metagenomic and metatranscriptomic sequencing, together with downstream qPCR and microarray analyses, gave a comprehensive view of the genomic information and phylogenetic and functional dynamics of supportive microorganisms in TCE-dechlorinating enrichments without exogenous cobalamin. Veillonellaceae with corrinoid biosynthesis pathways are important for corrinoid supply to Dehalococcoides. The induced sporulation activity of Veillonellaceae species is likely a response to exogenous corrinoid limitation and might contribute to the release of de novo-synthesized corrinoids. Our findings provide insights into the ecological interactions between Dehalococcoides and other community members and lead to further hypothesis-driven validations for a better understanding of dechlorinating communities.
MATERIALS AND METHODS
Enrichment cultures.
Two TCE-dechlorinating enrichments with (HiTCEB12) and without (HiTCE) exogenous cobalamin were constructed using contaminated groundwater and were stably maintained in 160-ml serum bottles (with 60 ml headspace of 90%/10% [vol/vol] N2/CO2) at 34°C in the dark, and subcultured (5% [vol/vol] transfer to fresh defined medium) every 2 weeks for over 3 years prior to this study. Lactate was supplied as the primary electron donor (20 mM on day 0, 2.5 mM on days 6 and 8, 25 mM in total) (Fig. S1), along with high concentrations of TCE (∼770 μM) as a primary electron acceptor, which completely inhibited methanogens. The composition of the defined medium used and the details of growth conditions are described elsewhere (40). Both HiTCEB12 and HiTCE enrichments were performed in triplicate for TCE measurement and microarray analysis.
Cell harvesting.
For metagenomic sequencing, 40-ml cell samples from one bottle of HiTCEB12 or HiTCE culture were collected on the last day (day 13) of incubation. For metatranscriptomic sequencing, in order to compare the activities of specific microorganisms in the enrichments, particularly corrinoid-related activities between the enrichment with exogenous cobalamin and without, a 40-ml sample was obtained on day 2 (T1) from one bottle each of HiTCEB12 and HiTCE (Fig. S1), when the corrinoids were observed to be actively produced (41). Moreover, a 40-ml sample from the second bottle of HiTCE enrichment on day 9 (T2) and two 40-ml samples from the third bottle of HiTCE on day 13 (T3) (Fig. S1) were also sampled in order to obtain the temporal dynamics of gene expression. The two HiTCE samples taken on day 13 were sequenced as technical duplicates to test the reliability of the sequencing technique. All cell samples were collected by centrifugation at 15,000 × g and 4°C for 10 min and stored at −80°C prior to use.
HMW DNA isolation.
High-molecular-weight (HMW) DNA samples were isolated according to the steps described in the Bacterial DNA isolation cetyltrimethylammonium bromide (CTAB) protocol on the Joint Genome Institute (JGI) website (52). The isolated HMW DNA was visualized using gel electrophoresis (0.7% agarose gels) for integrity check and was then quantified by a NanoPhotometer P-300 (Implen, Inc., Westlake Village, CA). The DNA samples were adjusted to 1 μg/10 μl using nuclease-free water and stored at −20°C until used for metagenomic sequencing.
RNA isolation.
Total RNA was isolated using the acidic phenol-chloroform protocol described previously (44). Briefly, cell pellets were resuspended in 250 μl of lysis buffer (50 mM sodium acetate, 10 mM EDTA [pH 5.1]), 100 μl of 10% sodium dodecyl sulfate, and 1.0 ml of buffer-equilibrated phenol (pH 4.3) (Sigma-Aldrich, St. Louis, MO). After bead beating, the aqueous lysate was extracted twice with one volume of acid (pH 4.3) phenol-chloroform-isoamyl alcohol (25:24:1) and once with one volume of chloroform-isoamyl alcohol (24:1) (Sigma-Aldrich). RNA was then precipitated, collected, washed, vacuum-dried, and resuspended in 100 μl of nuclease-free water. DNA contamination was removed by DNase I treatment using a Turbo DNA-free kit (Ambion, Life Technologies, Grand Island, NY), according to the manufacturer's instructions. Purified RNA was stored at −80°C until library construction.
Library preparation for metatranscriptomic sequencing.
First, mRNA in the total RNA samples was enriched by removing the biotinylated structural RNA using streptavidin-coated magnetic beads. Second, first-strand cDNA was synthesized by using SuperScript II reverse transcriptase, and the second strand of cDNA was then synthesized by NEBNext mRNA second-strand synthesis module (New England BioLabs, Inc., Ipswich, MA), according to the manufacturer's instructions. Third, the double-stranded cDNA was then subjected to the T7 linear amplification for deep sequencing (LADS) procedure developed by Hoeijmakers et al. (53). The detailed procedure is provided in the supplemental material.
High-throughput sequencing.
The overall workflow of the high-throughput DNA and RNA sequencing is outlined in Fig. S2. The genomic DNA and double-stranded cDNA (∼100 ng per sample) were submitted to the QB3 sequencing facility of the University of California, Berkeley (http://qb3.berkeley.edu/gsl/), for Illumina HiSeq 2000 sequencing (paired-end, 150 bp).
Metagenomic and metatranscriptomic analyses.
Raw sequencing reads were trimmed and screened according to sequencing quality (see the supplemental methods). The DNA reads after quality control were subjected to de novo assembly using CLC Genomics Workbench version 6 (CLC bio, Boston, MA) and genome reconstruction (i.e., metagenome binning) by a bidimensional binning process, as reported previously (39) (see also the supplemental methods). A genome evaluation software, CheckM, was used to evaluate the genome completeness using maker genes (54). The RNA reads were mapped against the assembled DNA sequences, and transcript abundances were determined as reads per kilobase pair transcript per million total reads mapped (RPKM) values. Transcript abundances of HiTCE and HiTCEB12 2d-samples, as well as those of HiTCE time course samples, were then compared (see details in the supplemental material).
RT-qPCR.
For genes involved in corrinoid de novo biosynthesis that are usually present in low transcript abundance, specific qPCR primers were designed using Primer-BLAST (Table S1). The relative gene expression ratios were determined using two-step RT-qPCR. The first-strand cDNA was synthesized from the same RNA samples submitted for Illumina sequencing using the SuperScript III kit (Invitrogen), according to the manufacturer's instructions. The cDNA was then relatively quantified by qPCR using Fast SYBR green PCR reagent (Applied Biosystems, Life Technologies, Grand Island, NY), according to the manufacturer's instructions.
The relative gene expression ratio was calculated using equation 1:
| (1) |
where ΔCTHiTCE(ref − target) and ΔCTHiTCEB12(ref − target) are the differences in threshold cycle (CT) values of a reference gene and the targeted corrinoid-related gene in the HiTCE and HiTCEB12 samples, respectively. As the transcript abundances of the tceA gene in HiTCE and HiTCEB12 at T1 were at about the same level, tceA was used as the reference gene for the relative gene expression ratio calculation.
Microarray analysis.
The custom-designed microarray (Affymetrix, Santa Clara, CA, USA) targeting four sequenced D. mccartyi genomes, those of strains CBDB1, BAV1, 195, and VS, as well as 348 outside genes, is described elsewhere (31). Biological triplicates were performed. The genomic DNA (gDNA) (40) and RNA microarrays were processed according to the instructions provided in chapter 4 of the Affymetrix GeneChip Expression Analysis technical manual (55), and data analysis is provided in the supplemental information.
Microarray (DNA and RNA) data were analyzed using Affymetrix GeneChip software and the MAS5 algorithm. Each microarray was normalized by scaling the signal intensities of the positive-control spiked mix to a target signal intensity of 2,500 to allow comparison between microarrays. A gene (DNA microarray) or gene transcript (RNA microarray) was considered “present” in a culture if the probe set across all three replicated samples had signal intensities greater than 140 (DNA microarray) or 250 (RNA microarray) and P values less than 0.05.
Accession number(s).
The assembled DNA sequences and predicted ORF sequences were deposited in the MG-RAST database under the project identification (ID) 7649. Raw reads of metagenomics and metatranscriptomic sequencing were deposited in the SRA database under PRJNA344005 (SRP090641), and the accession numbers for each of the metagenomes/metatranscriptomes are SAMN05817823 (HiTCEB12_DNA), SAMN05817824 (HiTCE_DNA), SAMN05817822 (HiTCEB12_2d_RNA), SAMN05817821 (HiTCE_2d_RNA), SAMN05817818 (HiTCE_9d_RNA), SAMN05817819 (HiTCE_13d_RNA_rep1), and SAMN05817820 (HiTCE_13d_RNA_rep2). The microarray data are deposited into the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE94143.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (grant CBET-1336709, 1/14–12/16), NIEHS Superfund (grants R42 ES004705-19 and R01 ES024255-01), and the Danish Research Council FTP (grant 09-069890). The Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley carried out Illumina sequencing with the support of NIH S10 Instrumentation grants S10RR029668 and S10RR027303. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under contract DE-AC02-05CH11231.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.03508-16.
REFERENCES
- 1.Doherty RE. 2000. A history of the production and use of carbon tetrachloride, tetrachloroethylene, trichloroethylene and 1,1,1-trichloroethane in the United States: part 1—historical background; carbon tetrachloride and tetrachloroethylene. Environ Forensics 1:69–81. doi: 10.1006/enfo.2000.0010. [DOI] [Google Scholar]
- 2.Moran MJ, Zogorski JS, Squillace PJ. 2007. Chlorinated solvents in groundwater of the United States. Environ Sci Technol 41:74–81. doi: 10.1021/es061553y. [DOI] [PubMed] [Google Scholar]
- 3.Stroo HF, Leeson A, Marqusee JA, Johnson PC, Ward CH, Kavanaugh MC, Sale TC, Newell CJ, Pennell KD, Lebron CA, Unger M. 2012. Chlorinated ethene source remediation: lessons learned. Environ Sci Technol 46:6438–6447. doi: 10.1021/es204714w. [DOI] [PubMed] [Google Scholar]
- 4.Lovley DR. 2003. Cleaning up with genomics: applying molecular biology to bioremediation. Nat Rev Microbiol 1:35–44. doi: 10.1038/nrmicro731. [DOI] [PubMed] [Google Scholar]
- 5.Maymó-Gatell X, Chien YT, Gossett JM, Zinder SH. 1997. Isolation of a bacterium that reductively dechlorinates tetrachloroethene to ethene. Science 276:1568–1571. doi: 10.1126/science.276.5318.1568. [DOI] [PubMed] [Google Scholar]
- 6.Löffler FE, Yan J, Ritalahti KM, Adrian L, Edwards EA, Konstantinidis KT, Müller JA, Fullerton H, Zinder SH, Spormann AM. 2013. Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int J Syst Evol Microbiol 63:625–635. doi: 10.1099/ijs.0.034926-0. [DOI] [PubMed] [Google Scholar]
- 7.He JZ, Ritalahti KM, Aiello MR, Löffler FE. 2003. Complete detoxification of vinyl chloride by an anaerobic enrichment culture and identification of the reductively dechlorinating population as a Dehalococcoides species. Appl Environ Microbiol 69:996–1003. doi: 10.1128/AEM.69.2.996-1003.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee PKH, Conrad ME, Alvarez-Cohen L. 2007. Stable carbon isotope fractionation of chloroethenes by dehalorespiring isolates. Environ Sci Technol 41:4277–4285. doi: 10.1021/es062763d. [DOI] [PubMed] [Google Scholar]
- 9.Yi S, Seth EC, Men YJ, Allen RH, Alvarez-Cohen L, Taga ME. 2012. Versatility in corrinoid salvaging and remodeling pathways supports the corrinoid-dependent metabolism of Dehalococcoides mccartyi. Appl Environ Microbiol 78:7745–7752. doi: 10.1128/AEM.02150-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cupples AM, Spormann AM, McCarty PL. 2003. Growth of a Dehalococcoides-like microorganism on vinyl chloride and cis-dichloroethene as electron acceptors as determined by competitive PCR. Appl Environ Microbiol 69:953–959. doi: 10.1128/AEM.69.2.953-959.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seshadri R, Adrian L, Fouts DE, Eisen JA, Phillippy AM, Methe BA, Ward NL, Nelson WC, Deboy RT, Khouri HM, Kolonay JF, Dodson RJ, Daugherty SC, Brinkac LM, Sullivan SA, Madupu R, Nelson KT, Kang KH, Impraim M, Tran K, Robinson JM, Forberger HA, Fraser CM, Zinder SH, Heidelberg JF. 2005. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Science 307:105–108. doi: 10.1126/science.1102226. [DOI] [PubMed] [Google Scholar]
- 12.Hug LA, Salehi M, Nuin P, Tillier ER, Edwards EA. 2011. Design and verification of a pangenome microarray oligonucleotide probe set for Dehalococcoides spp. Appl Environ Microbiol 77:5361–5369. doi: 10.1128/AEM.00063-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee PKH, Cheng D, Hu P, West KA, Dick GJ, Brodie EL, Andersen GL, Zinder SH, He JZ, Alvarez-Cohen L. 2011. Comparative genomics of two newly isolated Dehalococcoides strains and an enrichment using a genus microarray. ISME J 5:1014–1024. doi: 10.1038/ismej.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adrian L, Rahnenfuhrer J, Gobom J, Holscher T. 2007. Identification of a chlorobenzene reductive dehalogenase in Dehalococcoides sp. strain CBDB1. Appl Environ Microbiol 73:7717–7724. doi: 10.1128/AEM.01649-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson DR, Nemir A, Andersen GL, Zinder SH, Alvarez-Cohen L. 2009. Transcriptomic microarray analysis of corrinoid responsive genes in Dehalococcoides ethenogenes strain 195. FEMS Microbiol Lett 294:198–206. doi: 10.1111/j.1574-6968.2009.01569.x. [DOI] [PubMed] [Google Scholar]
- 16.Morris RM, Sowell S, Barofsky D, Zinder S, Richardson R. 2006. Transcription and mass-spectroscopic proteomic studies of electron transport oxidoreductases in Dehalococcoides ethenogenes. Environ Microbiol 8:1499–1509. doi: 10.1111/j.1462-2920.2006.01090.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhuang WQ, Yi S, Feng XY, Zinder SH, Tang YJJ, Alvarez-Cohen L. 2011. Selective utilization of exogenous amino acids by Dehalococcoides ethenogenes strain 195 and its effects on growth and dechlorination activity. Appl Environ Microbiol 77:7797–7803. doi: 10.1128/AEM.05676-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang YJJ, Yi S, Zhuang WQ, Zinder SH, Keasling JD, Alvarez-Cohen L. 2009. Investigation of carbon metabolism in “Dehalococcoides ethenogenes” strain 195 by use of isotopomer and transcriptomic analyses. J Bacteriol 191:5224–5231. doi: 10.1128/JB.00085-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuang WQ, Yi S, Bill M, Brisson VL, Feng X, Men Y, Conrad ME, Tang YJ, Alvarez-Cohen L. 2014. Incomplete Wood-Ljungdahl pathway facilitates one-carbon metabolism in organohalide-respiring Dehalococcoides mccartyi. Proc Natl Acad Sci U S A 111:6419–6424. doi: 10.1073/pnas.1321542111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schipp CJ, Marco-Urrea E, Kublik A, Seifert J, Adrian L. 2013. Organic cofactors in the metabolism of Dehalococcoides mccartyi strains. Philos Trans R Soc B Biol Sci 368:20120321. doi: 10.1098/rstb.2012.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freeborn RA, West KA, Bhupathiraju VK, Chauhan S, Rahm BG, Richardson RE, Alvarez-Cohen L. 2005. Phylogenetic analysis of TCE-dechlorinating consortia enriched on a variety of electron donors. Environ Sci Technol 39:8358–8368. doi: 10.1021/es048003p. [DOI] [PubMed] [Google Scholar]
- 22.Duhamel M, Edwards EA. 2006. Microbial composition of chlorinated ethene-degrading cultures dominated by Dehalococcoides. FEMS Microbiol Ecol 58:538–549. doi: 10.1111/j.1574-6941.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- 23.Macbeth TW, Cummings DE, Spring S, Petzke LM, Sorenson KS Jr. 2004. Molecular characterization of a dechlorinating community resulting from in situ biostimulation in a trichloroethene-contaminated deep, fractured basalt aquifer and comparison to a derivative laboratory culture. Appl Environ Microbiol 70:7329–7341. doi: 10.1128/AEM.70.12.7329-7341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martens JH, Barg H, Warren MJ, Jahn D. 2002. Microbial production of vitamin B12. Appl Microbiol Biotechnol 58:275–285. doi: 10.1007/s00253-001-0902-7. [DOI] [PubMed] [Google Scholar]
- 25.Yan J, Ritalahti KM, Wagner DD, Loffler FE. 2012. Unexpected specificity of interspecies cobamide transfer from Geobacter spp. to organohalide-respiring Dehalococcoides mccartyi strains. Appl Environ Microbiol 78:6630–6636. doi: 10.1128/AEM.01535-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan J, Simsir B, Farmer AT, Bi M, Yang Y, Campagna SR, Loffler FE. 2016. The corrinoid cofactor of reductive dehalogenases affects dechlorination rates and extents in organohalide-respiring Dehalococcoides mccartyi. ISME J 10:1092–1101. doi: 10.1038/ismej.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He JZ, Holmes VF, Lee PKH, Alvarez-Cohen L. 2007. Influence of vitamin B12 and cocultures on the growth of Dehalococcoides isolates in defined medium. Appl Environ Microbiol 73:2847–2853. doi: 10.1128/AEM.02574-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahm BG, Morris RM, Richardson RE. 2006. Temporal expression of respiratory genes in an enrichment culture containing Dehalococcoides ethenogenes. Appl Environ Microbiol 72:5486–5491. doi: 10.1128/AEM.00855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waller AS, Hug LA, Mo K, Radford DR, Maxwell KL, Edwards EA. 2012. Transcriptional analysis of a Dehalococcoides-containing microbial consortium reveals prophage activation. Appl Environ Microbiol 78:1178–1186. doi: 10.1128/AEM.06416-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West KA, Johnson DR, Hu P, DeSantis TZ, Brodie EL, Lee PKH, Feil H, Andersen GL, Zinder SH, Alvarez-Cohen L. 2008. Comparative genomics of “Dehalococcoides ethenogenes” 195 and an enrichment culture containing unsequenced “Dehalococcoides” strains. Appl Environ Microbiol 74:3533–3540. doi: 10.1128/AEM.01835-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.West KA, Lee PK, Johnson DR, Zinder SH, Alvarez-Cohen L. 2013. Global gene expression of Dehalococcoides within a robust dynamic TCE-dechlorinating community under conditions of periodic substrate supply. Biotechnol Bioeng 110:1333–1341. doi: 10.1002/bit.24819. [DOI] [PubMed] [Google Scholar]
- 32.Hultman J, Waldrop MP, Mackelprang R, David MM, McFarland J, Blazewicz SJ, Harden J, Turetsky MR, McGuire AD, Shah MB, VerBerkmoes NC, Lee LH, Mavrommatis K, Jansson JK. 2015. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 521:208–212. doi: 10.1038/nature14238. [DOI] [PubMed] [Google Scholar]
- 33.Mason OU, Hazen TC, Borglin S, Chain PS, Dubinsky EA, Fortney JL, Han J, Holman HY, Hultman J, Lamendella R, Mackelprang R, Malfatti S, Tom LM, Tringe SG, Woyke T, Zhou J, Rubin EM, Jansson JK. 2012. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J 6:1715–1727. doi: 10.1038/ismej.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, DeLong EF. 2008. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci U S A 105:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen LX, Hu M, Huang LN, Hua ZS, Kuang JL, Li SJ, Shu WS. 2015. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J 9:1579–1592. doi: 10.1038/ismej.2014.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishii S, Suzuki S, Norden-Krichmar TM, Tenney A, Chain PS, Scholz MB, Nealson KH, Bretschger O. 2013. A novel metatranscriptomic approach to identify gene expression dynamics during extracellular electron transfer. Nat Commun 4:1601. doi: 10.1038/ncomms2615. [DOI] [PubMed] [Google Scholar]
- 37.Brisson VL, West KA, Lee PKH, Tringe SG, Brodie EL, Alvarez-Cohen L. 2012. Metagenomic analysis of a stable trichloroethene-degrading microbial community. ISME J 6:1702–1714. doi: 10.1038/ismej.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.David MM, Cecillon S, Warne BM, Prestat E, Jansson JK, Vogel TM. 2015. Microbial ecology of chlorinated solvent biodegradation. Environ Microbiol 17:4835–4850. doi: 10.1111/1462-2920.12413. [DOI] [PubMed] [Google Scholar]
- 39.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 40.Men YJ, Lee PKH, Harding KC, Alvarez-Cohen L. 2013. Characterization of four TCE-dechlorinating microbial enrichments grown with different cobalamin stress and methanogenic conditions. Appl Microbiol Biotechnol 97:6439–6450. doi: 10.1007/s00253-013-4896-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Men Y, Seth EC, Yi S, Crofts TS, Allen RH, Taga ME, Alvarez-Cohen L. 2015. Identification of specific corrinoids reveals corrinoid modification in dechlorinating microbial communities. Environ Microbiol 17:4873–4884. doi: 10.1111/1462-2920.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banerjee R, Ragsdale SW. 2003. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem 72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 43.Stupperich E, Eisinger HJ, Krautler B. 1989. Identification of phenolyl cobamide from the homoacetogenic bacterium Sporomusa ovata. Eur J Biochem 186:657–661. doi: 10.1111/j.1432-1033.1989.tb15256.x. [DOI] [PubMed] [Google Scholar]
- 44.Men Y, Seth EC, Yi S, Allen RH, Taga ME, Alvarez-Cohen L. 2014. Sustainable growth of Dehalococcoides mccartyi 195 by corrinoid salvaging and remodeling in defined lactate-fermenting consortia. Appl Environ Microbiol 80:2133–2141. doi: 10.1128/AEM.03477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penny C, Gruffaz C, Nadalig T, Cauchie HM, Vuilleumier S, Bringel F. 2015. Tetrachloromethane-degrading bacterial enrichment cultures and isolates from a contaminated aquifer. Microorganisms 3:327–343. doi: 10.3390/microorganisms3030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang YR, Pesaro M, Sigler W, Zeyer J. 2005. Identification of microorganisms involved in reductive dehalogenation of chlorinated ethenes in an anaerobic microbial community. Water Res 39:3954–3966. doi: 10.1016/j.watres.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Seth EC, Taga ME. 2014. Nutrient cross-feeding in the microbial world. Front Microbiol 5:350. doi: 10.3389/fmicb.2014.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flint JF, Drzymalski D, Montgomery WL, Southam G, Angert ER. 2005. Nocturnal production of endospores in natural populations of epulopiscium-like surgeonfish symbionts. J Bacteriol 187:7460–7470. doi: 10.1128/JB.187.21.7460-7470.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Talukdar PK, Olguin-Araneda V, Alnoman M, Paredes-Sabja D, Sarker MR. 2014. Updates on the sporulation process in Clostridium species. Res Microbiol 166:225–235. doi: 10.1016/j.resmic.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Higgins D, Dworkin J. 2012. Recent progress in Bacillus subtilis sporulation. FEMS Microbiol Rev 36:131–148. doi: 10.1111/j.1574-6976.2011.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hazra AB, Han AW, Mehta AP, Mok KC, Osadchiy V, Begley TP, Taga ME. 2015. Anaerobic biosynthesis of the lower ligand of vitamin B12. Proc Natl Acad Sci U S A 112:10792–10797. doi: 10.1073/pnas.1509132112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feil WS, Feil H, Copeland A. 2012. Bacterial genomic DNA isolation using CTAB. Joint Genome Institute, Department of Energy, Walnut Creek, CA: http://1ofdmq2n8tc36m6i46scovo2e.wpengine.netdna-cdn.com/wp-content/uploads/2014/02/JGI-Bacterial-DNA-isolation-CTAB-Protocol-2012.pdf. [Google Scholar]
- 53.Hoeijmakers WA, Bartfai R, Francoijs KJ, Stunnenberg HG. 2011. Linear amplification for deep sequencing. Nat Protoc 6:1026–1036. doi: 10.1038/nprot.2011.345. [DOI] [PubMed] [Google Scholar]
- 54.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thermo Fisher Scientific. 2017. Affymetrix GeneChip Expression Analysis technical manual, with specific protocols for using the GeneChip hybridization, wash, and stain kit. Affymetrix, Santa Clara, CA, USA: http://tools.thermofisher.com/content/sfs/manuals/expression_analysis_technical_manual.pdf. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.