

Abstract
The present study aimed to investigate the effects of lipoprotein-associated phospholipase A2 (Lp-PLA2) on endothelial dysfunction in an in vitro cell model of atherosclerosis, and to determine whether AMP-activated protein kinase (AMPK) mediates the effects of Lp-PLA2 on endothelial dysfunction. A total of 392 patients with coronary artery disease (CAD), including various sub-conditions, were recruited, and the plasma concentrations of Lp-PLA2 were evaluated. In addition, an in vitro model of atherosclerosis was established by exposing human umbilical vein endothelial cells (HUVECs) to oxidized low-density lipoprotein (oxLDL). SB-435495 was used to inhibit Lp-PLA2, and compound C was used to suppress AMPK expression. Lp-PLA2, AMPKα and phosphorylated-AMPKα (T172) expression in HUVECs were evaluated using western blot analysis. The concentrations of nitric oxide (NO), endothelin 1 (ET-1), intercellular adhesion molecule 1 (ICAM-1) and platelet/endothelial cell adhesion molecule 1 (PECAM-1) in cell culture supernatant were determined using commercially available ELISA kits. MTT assays were employed to indicate changes in cell viability. The current study found the plasma Lp-PLA2 levels were elevated in the CAD patients with stable angina pectoris, unstable angina pectoris, acute coronary syndromes and acute myocardial infarction, compared with a healthy control population. In addition, the in vitro results showed that Lp-PLA2 expression levels were elevated in oxLDL-exposed HUVECs. Lp-PLA2 suppression could increase cell viability, induce the production of NO and decrease the secretion of ET-1, in addition to suppressing the expression of cell adhesion molecules, including ICAM-1 and PECAM-1 in oxLDL-exposed HUVECs. The expression of AMPKα and phosphorylated-AMPKα (T172) was regulated by Lp-PLA2, and AMPK suppression was able to reverse the effects of Lp-PLA2 with regard to cell viability, endothelial vasorelaxation capacity and the secretion of adhesion molecules in oxLDL-exposed HUVECs. In conclusion, the present study provides initial evidence that Lp-PLA2 is able to cause endothelial dysfunction in an in vitro model of atherosclerosis, and the effects of Lp-PLA2 on endothelial dysfunction was at least partially a result of the downregulation of AMPKα, thus contributing to the progression of atherosclerosis.
Keywords: lipoprotein-associated phospholipase A2, endothelial dysfunction, atherosclerosis, AMP-activated protein kinase
Introduction
Atherosclerosis, which is characterized by the formation of atherosclerotic plaques in large- and medium-sized arteries, is the most common cause of mortality from cardiovascular diseases in Western countries (1). The pathological process of atherosclerosis is complex, and numerous components of the metabolic, immune and vascular systems are involved in the formation of atherosclerotic plaques (2,3). However, the pathomechanism of atherosclerotic vascular disease has yet to be fully elucidated.
Vascular endothelial cells (VECs) are found in the inner layer of the vessel wall. They serve as a barrier between the blood and the surrounding tissues, and serve important functions in the cardiovascular system (4,5). The association between endothelial dysfunction and the mechanism of atherosclerosis has gained marked interest (6) and the present study investigates endothelial dysfunction in relation to atherosclerosis.
Lipoprotein-associated phospholipase A2 (Lp-PLA2) is a Ca2+-independent secreted protein that belongs to the PLA2 superfamily. It is produced by a wide range of inflammatory and non-inflammatory cells, and circulates in plasma bound mainly to low-density lipoprotein (LDL) and promotes vascular inflammation (7–9). Despite the inflammatory potential, the association of Lp-PLA2 with endothelial dysfunction has been demonstrated in a number of epidemiological studies (10,11). Yang et al (10) demonstrated that circulating Lp-PLA2 is a strong predictor of coronary artery endothelial dysfunction in humans. Local coronary production of Lp-PLA2 is also associated with endothelial dysfunction in patients with early coronary atherosclerosis (11). However, relatively few studies have investigated the role of Lp-PLA2 in endothelial dysfunction and the associated molecular mechanism in vitro.
AMP-activated protein kinase (AMPK) is an important regulator of carbohydrate and lipid metabolism, and it appears to be a therapeutic target for improving endothelial function in patients with cardiovascular disease (12–14).
In the present study, the effects of Lp-PLA2 on endothelial dysfunction in an in vitro cell model of atherosclerosis were initially investigated. In addition, whether AMPK mediates the effects of Lp-PLA2 on endothelial dysfunction was also assessed.
Materials and methods
Study participants
The study enrolled 392 patients who had been diagnosed with coronary artery disease (CAD) in Tianjin Chest Hospital (Tianjin, China) and 66 healthy controls. All patients signed a consent form agreeing to participate in the present study, and the Ethics Committee of Tianjin Thoracic Hospital approved the protocol. Among the patients with CAD, 10 had acute coronary syndromes (ACS), 69 had acute myocardial infarction (AMI), 55 had stable angina pectoris (SAP) and the remaining 258 patients had unstable angina pectoris (UAP). The mean age of patients was 62±5 years, and the mean age of healthy controls was 59±6 years. Fasting blood samples were drawn by vein-puncture in the morning.
Cell culture and treatments
Human umbilical vascular endothelium, (HUVECs; catalogue no. CRL-1730; American Type Culture Collection (ATCC), Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.). The cells were maintained at 37°C in a humidified 5% CO2, 95% air atmosphere, and the cell culture media was changed every 2–3 days. Oxidized low density lipoprotein (oxLDL) was purchased from Yiyuan Biotech Co., Ltd. (Guangzhou, China) and diluted in DMEM to 200 µg/ml to treat cells. The Lp-PLA2 inhibitor, SB-435495 was obtained from GlaxoSmithKline (Collegeville, PA, USA) and used at a concentration of 5 µM. Compound C, an AMPK inhibitor, was purchased from Merck Millipore (Darmstadt, Germany) and used at a concentration of 20 µM.
Measurement of nitric oxide (NO)
NO production of HUVECs was determined using a NO Assay kit (Beyotime Institute of Biotechnology, Haimen, China) according to the manufacturer's protocol. Cell culture supernatant of HUVECs (50 µl), Griess Reagent (50 µl) and Griess Reagent (50 µl) were added to each well of the 96-well plates. The plates were incubated at room temperature for 2 min, and the optical density (OD) was measured at 540 nm using a microplate reader (MK3; Thermo Fisher Scientific, Inc.). NO production was then calculated by comparing the OD of the samples with the standard curve.
ELISA assay
Plasma levels of Lp-PLA2 were detected using a commercial ELISA kit: Lp-PLA2 Assay kit (Kangerke Biotech Co., Ltd., Tianjin, China). The concentrations of endothelin 1 (ET-1), intercellular adhesion molecule 1 (ICAM-1) and platelet/endothelial cell adhesion molecule 1 (PECAM-1) in the cell culture supernatant were determined using Human ET-1 ELISA kit (TW Reagent Company, Shanghai, China), Human ICAM-1 ELISA kit (Huijia Biotechnology, Xiamen, Fujian, China), and Human PECAM-1 ELISA kit (Meixuan Biological Technology Co., Ltd, Shanghai, China). The microtiter plate provided in each of the kits had been pre-coated with an antibody specific to ET-1, ICAM-1 or PECAM-1. Standards (50 µl) or samples (10 µl) were then added to the wells, followed by the addition of horseradish peroxidase (HRP)-conjugated antibody, as stated in manufacturer protocols of aforementioned ELISA kits. Subsequent to incubation, a tetramethylbenzidine substrate solution was added to each well and the enzyme-substrate reaction was terminated by the addition of stop buffer. The color change was measured at a wavelength of 450 nm (MK3). The concentration of the samples was determined by comparing the OD of the samples to the standard curve.
MTT assay
An MTT metabolic assay was considered to indicate the changes in cell viability. The cells (1.5×103 cells/well) were plated into the 96-well plates and cultured at 37°C overnight. After treatment for 24, 48 and 72 h, 10 µl MTT stock solution (0.5 mg/ml; Beyotime Institute of Biotechnology) was added to each well, and the cells were incubated at 37°C for 4 h. Next, 150 µl dimethyl sulfoxide was added to each well and mixed thoroughly with a pipette. After incubation at 37°C for 10 min, the absorbance at 570 nm was measured using a microplate reader (MK3).
Western blot analysis
The cells were washed with phosphate-buffered saline (PBS) and lysed in cell lysis buffer [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 50 mM NaF, 2 mM EDTA, 1 mM Na3VO4, 1% NP-40, 1 µg/ml Leupeptin, 1 µg/ml Pepstatin and 1 mM PMSF]. After centrifugation at 12,000 × g for 10 min at 4°C, the supernatant was subjected to 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Merck Millipore). The membranes were blocked in Tris-buffered saline containing 5% nonfat milk and 0.05% Tween-20 at 4°C overnight. Thereafter, the blots were probed with Lp-PLA2 rabbit polyclonal antibody (1:800; ab96619; Abcam, Cambridge, MA, USA), AMPKα rabbit monoclonal antibody (1:400; 2603), phospho-AMPKα (Thr172) rabbit monoclonal antibody (1:400; 2535; Cell Signaling Technology, Inc., Danvers, MA, USA) at 37°C for 2 h. After washing with PBS, the membranes were incubated with HRP-conjugated donkey anti-rabbit IgG (1:2,000; ab16284; Abcam) at 37°C for 1 h. Immunoreactive bands were detected by an enhanced chemiluminescence western blotting kit (Pierce Protein Biology; Thermo Fisher Scientific, Inc., Rockford, IL, USA). ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to calculate band density.
Statistical analysis
Statistical analysis was performed using SPSS 19.0 software (SPSS IBM, Armonk, NY, USA). All data are representative of a minimum of three independent experiments, and are expressed as the mean ± standard deviation. Differences between two groups were compared using the Student's t-test. One-way analysis of variance, followed by a least significant difference test was used to compare the differences among three groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Plasma Lp-PLA2 levels were upregulated in patients with CAD
The plasma Lp-PLA2 levels were detected in 392 patients with CAD and 66 healthy controls. It was found that, compared with the control subjects, the plasma Lp-PLA2 levels were significantly increased in patients with SAP, UAP, ACS and AMI, as well as the total CAD (P<0.05; Fig. 1).
Figure 1.
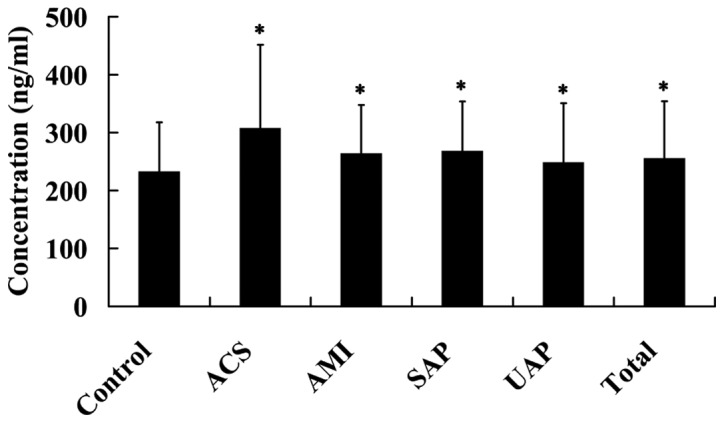
Plasma Lp-PLA2 levels in patients with CAD. *P<0.05 vs. normal group. Lp-PLA2, lipoprotein-associated phospholipase A2; CAD, coronary artery disease; SAP, stable angina pectoris; USAP, unstable angina pectoris; ACS, acute coronary syndromes; AMI, acute myocardial infarction.
oxLDL induced endothelial dysfunction in HUVECs
As shown in Fig. 2A, oxLDL exerted an inhibitory effect on cell proliferation. Compared with the cells in the normal group, cell viability was significantly decreased in the oxLDL group (P<0.05).
Figure 2.
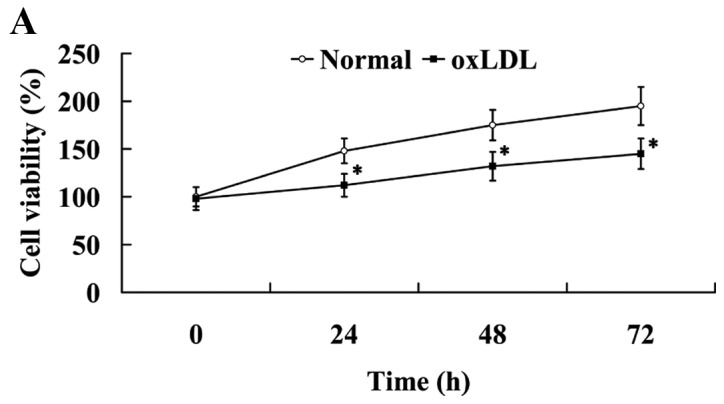
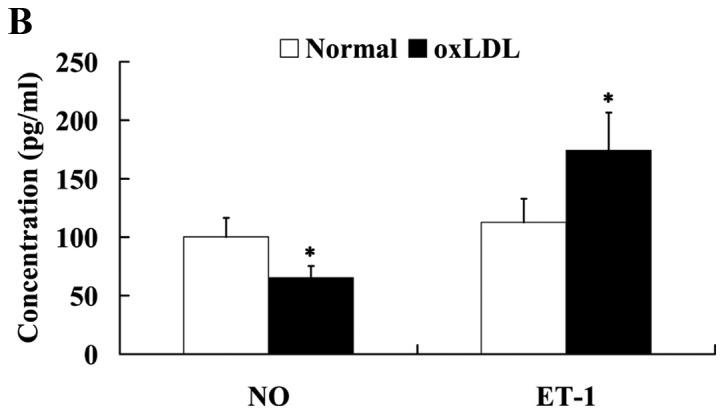
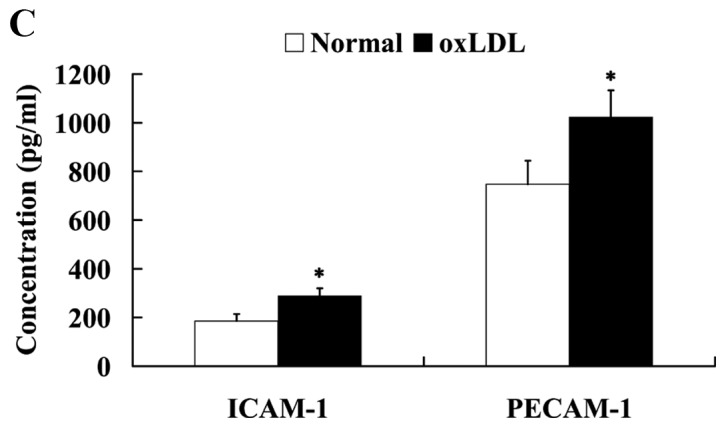
oxLDL induced the endothelial dysfunction in human umbilical vein endothelial cells. Effect of oxLDL on (A) cell viability, (B) NO and ET-1 expression and (C) on ICAM-1 and PECAM-1 expression levels. *P<0.05 vs. normal group. OxLDL, oxidized low density lipoprotein; NO, nitric oxide; ET-1, endothelin 1; ICAM-1, intercellular adhesion molecule 1; PECAM-1, platelet/endothelial cell adhesion molecule 1.
The HUVECs were treated with 200 µg/ml oxLDL for 24 h, and then the cells were harvested for western blot analysis. The expression levels of vasorelaxation factor NO and vasoconstrictor ET-1 were determined by western blot analysis to assess endothelial vasorelaxation capacity. It was observed that NO expression was significantly decreased (P<0.05), while ET-1 expression was significantly increased in HUVECs following treatment with oxLDL (P<0.05; Fig. 2B).
The expression levels of ICAM-1 and PECAM-1 were evaluated to determine the secretion of adhesion molecules by HUVECs. As shown in Fig. 2C, treatment with oxLDL significantly increased the expression levels of ICAM-1 and PECAM-1 proteins in HUVECs (P<0.05).
oxLDL induced Lp-PLA2 expression in HUVECs
The effect of oxLDL on Lp-PLA2 expression was determined using western blot analysis. As indicated in Fig. 3, compared with the normal cells, Lp-PLA2 protein expression levels were significantly upregulated in the cells treated with oxLDL (P<0.05).
Figure 3.
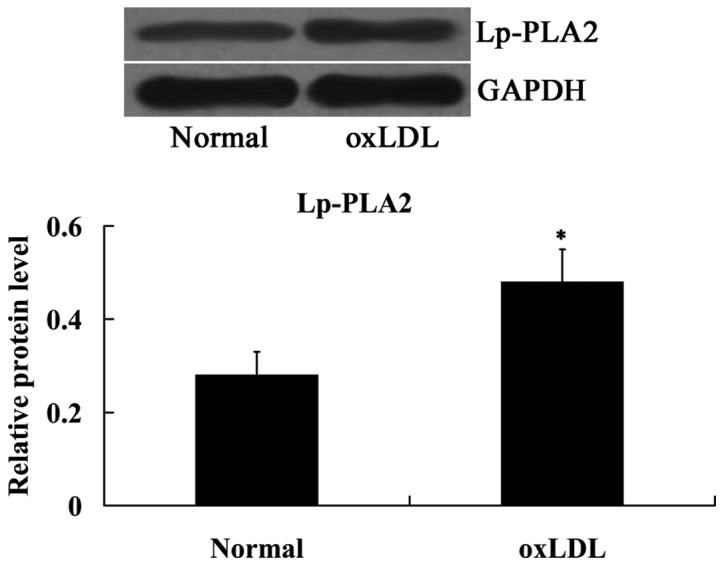
oxLDL induced Lp-PLA2 expression in human umbilical vein endothelial cells. *P<0.05 vs. normal group. OxLDL, oxidized low density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2.
oxLDL inhibited AMPKα in HUVECs
The expression levels of AMPKα and phosphorylated-AMPKα (T172) in oxLDL-treated HUVECs were also measured using western blot analysis. Fig. 4 indicated that the relative protein expression levels of AMPKα and phosphorylated-AMPKα (T172) were decreased significantly in oxLDL-exposed HUVECs, compared with normal cells (P<0.05).
Figure 4.
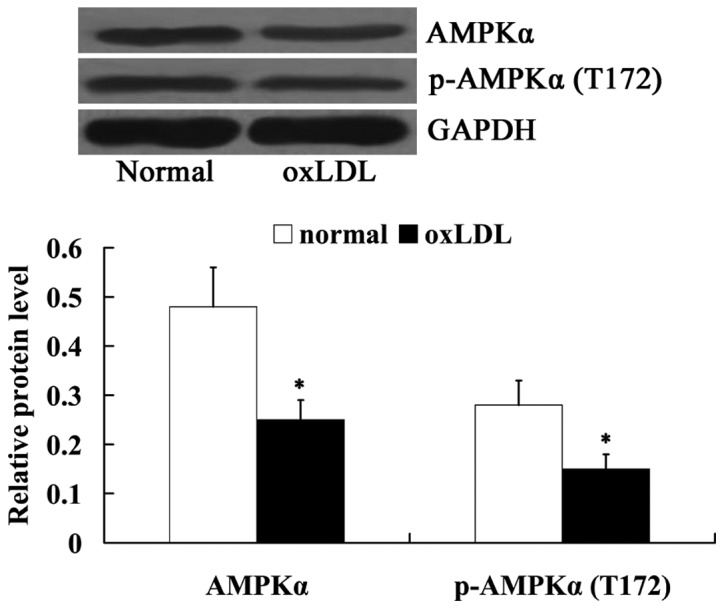
oxLDL inhibited AMPKα and p-AMPKα in human umbilical vein endothelial cells. *P<0.05 vs. normal group. OxLDL, oxidized low density lipoprotein; p-AMPKα, phosphorylated-AMP-activated protein kinase α.
Lp-PLA2 suppression protected against endothelial dysfunction in oxLDL-exposed HUVECs
To investigate the effects of Lp-PLA2 on endothelial dysfunction, Lp-PLA2 protein was suppressed in oxLDL-exposed HUVECs by incubation with the Lp-PLA2 inhibitor SB-435495, then cell viability, endothelial vasorelaxation capacity and the secretion of adhesion molecules were investigated.
As expected, the results from the western blot analysis demonstrated that the expression of Lp-PLA2 protein was significantly inhibited by SB-435495 in oxLDL-exposed HUVECs (P<0.01; Fig. 5).
Figure 5.
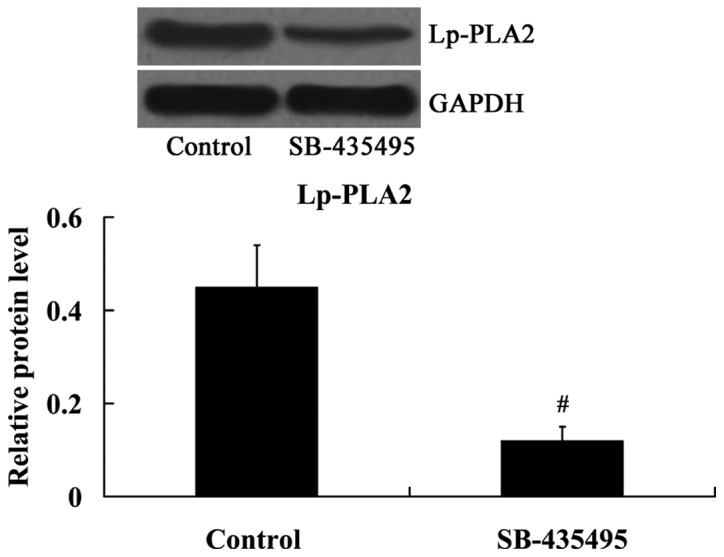
Expression levels of Lp-PLA2 in oxLDL-exposed human umbilical vein endothelial cells following treatment with SB-435495. #P<0.01 vs. control group. OxLDL, oxidized low density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2.
The results of the MTT assay revealed that cell viability was significantly increased following treatment with SB-435495 (P<0.05; Fig. 6A). Western blot analysis results demonstrated that NO expression levels were significantly increased, while ET-1 expression was significantly decreased in the oxLDL-exposed HUVECs following treatment with SB-435495 (P<0.05; Fig. 6B). The expression of adhesion molecules ICAM-1 and PECAM-1 was significantly decreased as a result of treatment with SB-435495 (P<0.05; Fig. 6C).
Figure 6.
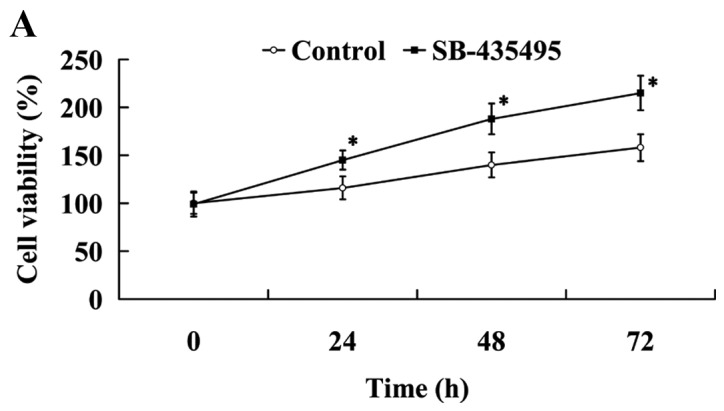

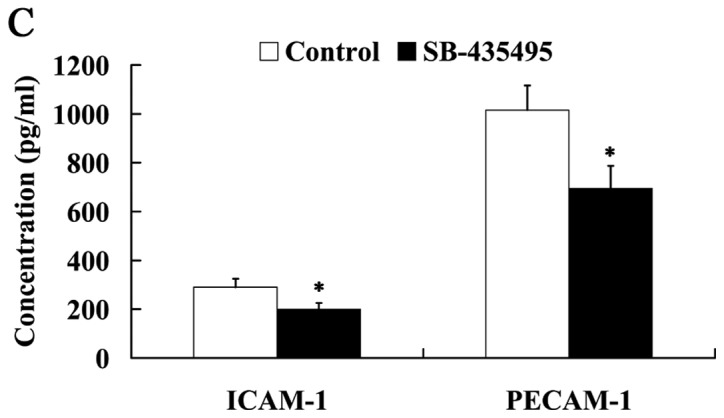
Lp-PLA2 suppression protected against endothelial dysfunction in oxLDL-exposed human umbilical vein endothelial cells. Effect of Lp-PLA2 suppression on (A) cell viability, (B) NO and ET-1 and (C) ICAM-1 and PECAM-1 expression levels. *P<0.05 vs. control group. OxLDL, oxidized low density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2; NO, nitric oxide; ET-1, endothelin 1; ICAM-1, intercellular adhesion molecule 1; PECAM-1, platelet/endothelial cell adhesion molecule 1.
Lp-PLA2 suppression activated AMPKα in oxLDL-exposed HUVECs
Subsequently, the expression levels of AMPKα and phosphorylated-AMPKα (T172) in oxLDL-exposed HUVECs were detected following incubation with SB-435495. It was found that Lp-PLA2 suppression led to increased expression levels of AMPKα and phosphorylated-AMPKα (T172) in oxLDL-exposed HUVECs (P<0.05; Fig. 7).
Figure 7.
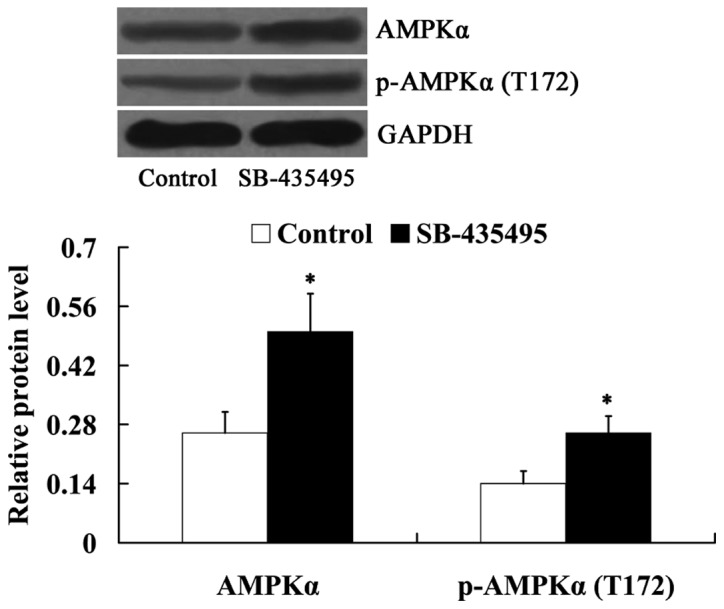
Lp-PLA2 suppression activated AMPKα in oxLDL-exposed human umbilical vein endothelial cells. *P<0.05 vs. control group. OxLDL, oxidized low density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2; p-AMPKα, phosphorylated-AMP-activated protein kinase alpha.
AMPK mediates the effects of Lp-PLA2 on endothelial dysfunction in oxLDL-exposed HUVECs
In order to determine whether AMPK mediates the effects of Lp-PLA2 on endothelial dysfunction, cells were treated with compound C to suppress AMPK expression. As demonstrated in Fig. 8, compared with the SB-435495 group, the relative protein levels of AMPKα and phosphorylated-AMPKα (T172) were significantly decreased in the SB-435495 + compound C group (P<0.01 and P<0.05, respectively).
Figure 8.
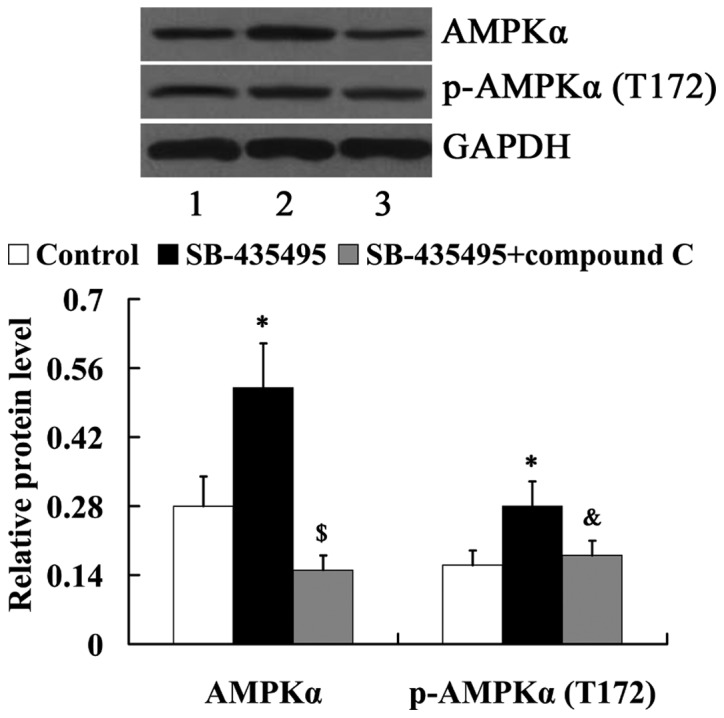
Expression of AMPKα and p-AMPKα in oxLDL-exposed human umbilical vein endothelial cells following treatment with SB-435495 and compound C. Lane 1, control; lane 2, SB-435495; lane 3, SB-435495 + compound C. *P<0.05 vs. control group; &P<0.05 and $P<0.01 vs. SB-435495 group. OxLDL, oxidized low density lipoprotein; p-AMPKα, phosphorylated-AMP-activated protein kinase alpha.
The MTT results showed that Lp-PLA2 suppression led to significantly increased cell viability compared with the control group (P<0.05); however, this effect was reversed by treatment with compound C (P<0.05; Fig. 9A). Western blot analysis results revealed that the significantly altered expression of NO and ET-1 by Lp-PLA2 suppression in oxLDL-exposed HUVECs was attenuated by compound C (P<0.05; Fig. 9B). In addition, it was found that the relative protein expression levels of ICAM-1 and PECAM-1 were significantly increased in the SB-435495 + compound C group, compared with those in the SB-435495 group (P<0.01; Fig. 9C).
Figure 9.
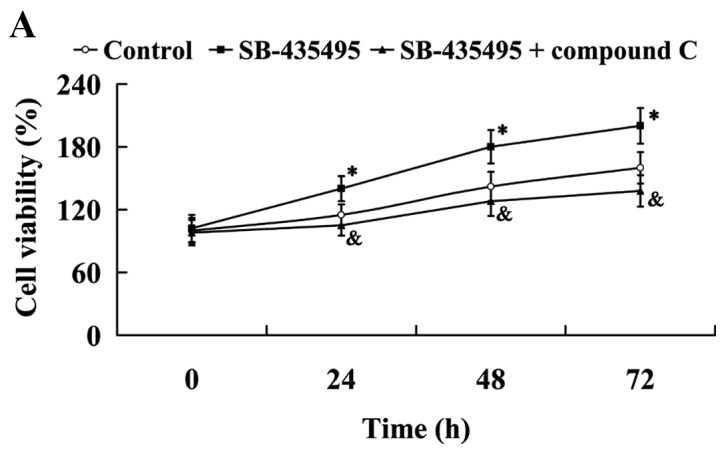
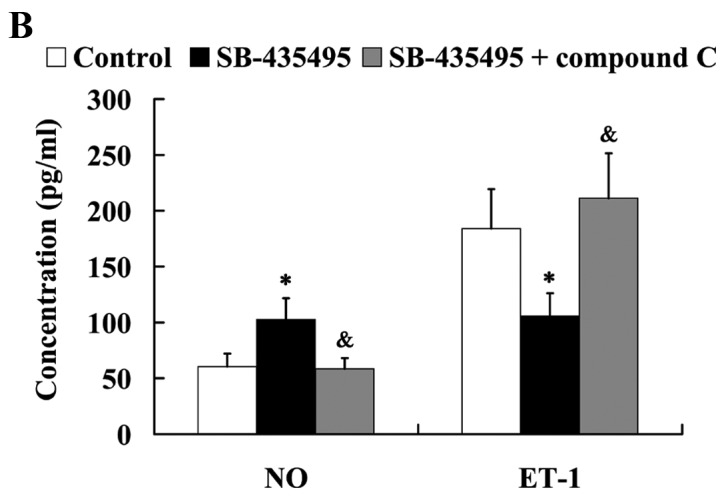
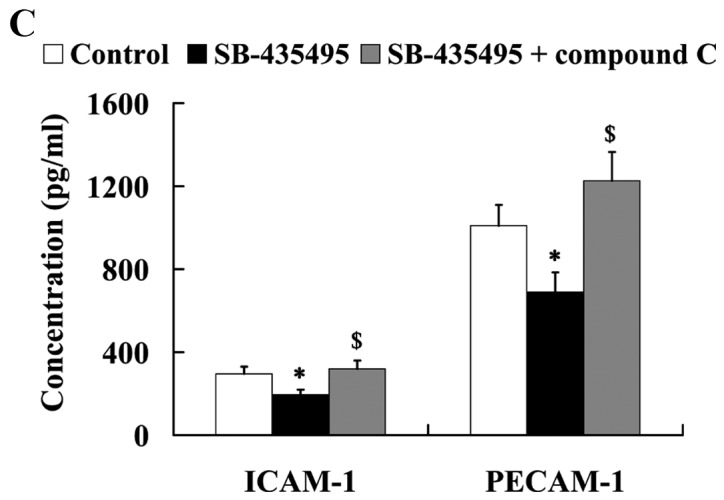
AMPK mediates the effects of Lp-PLA2 on endothelial dysfunction in oxLDL-exposed HUVECs. (A) Cell viability, (B) expression levels of NO and ET-1 and (C) ICAM-1 and PECAM-1 in oxLDL-exposed HUVECs following treatment with SB-435495 + compound C. *P<0.05 vs. control group; &P<0.05 and $P<0.01 vs. SB-435495 group. OxLDL, oxidized low density lipoprotein; HUVECs, human umbilical vein endothelial cells; Lp-PLA2, lipoprotein-associated phospholipase A2; AMPKα, AMP-activated protein kinase alpha; NO, nitric oxide; ET-1, endothelin 1; ICAM-1, intercellular adhesion molecule 1; PECAM-1, platelet/endothelial cell adhesion molecule 1.
Discussion
In the present study, a large number of CAD samples were collected, including those from patients with SAP, UAP, ACS and AMI. Atherosclerosis is the most common cause of stenosis of the heart arteries and, as such, of angina pectoris, including SAP and UAP (15). The instability of atherosclerotic plaques forms the pathological basis of ACS (16). Plaques can become unstable, rupture and promote the formation of blood clots that may occlude the artery, leading to AMI (17,18). The current study demonstrated that plasma Lp-PLA2 expression levels were significantly upregulated in patients with CAD, and this result was consistent with a previous report (19). Subsequently, the current study investigated the role of Lp-PLA2 in endothelial dysfunction in atherosclerosis.
oxLDL is an important factor in the initiation and development of atherosclerosis (20). In the present study, an in vitro cell model of atherosclerosis was established by exposing HUVECs to 200 µg/ml oxLDL for 24 h. Thus, the present study provided the first in vitro evidence that Lp-PLA2 induction results in endothelial dysfunction in experimental atherosclerosis, and that AMPK was involved in mediating the effects of Lp-PLA2.
The endothelium has a central role in vascular disease, to the extent that several clinical tests have been conducted to evaluate the functional properties of normal and activated endothelium (21,22). Endothelial dysfunction is defined as an imbalance between vasodilating and vasoconstricting substances that are produced by or act on the endothelium (23), in addition to the altered anti-inflammatory and anticoagulant properties of the endothelium (6,24). Endothelial dysfunction is associated with numerous vascular diseases, including atherosclerosis, stroke, myocardial ischemia and acute coronary syndrome (6). Clinical assessments of endothelial function have enabled the detection of early disease, quantification of risk, judgment of response to interventions, and a reduction in the occurrence of later adverse events in patients. Endothelial dysfunction is often regarded as a key early event in the development of atherosclerosis (6). NO is the most important vasorelaxation factor released by the endothelium. The decrease in NO production could induce pathological conditions associated with endothelial dysfunction, such as atherosclerosis (25,26). ET-1 is another endothelium-derived regulatory protein that functions as a potent vasoconstrictor, and is a marker of endothelial dysfunction (27). During the process of atherosclerosis, endothelial dysfunction could be induced by the inflammatory cytokines, and consequently led to the induction of cell adhesion molecules (28). ICAM-1 and PECAM-1 are cell adhesion molecules expressed by several cell types, including endothelial cells (29–31). The endothelial expression of ICAM-1 and PECAM-1 is increased in atherosclerotic lesions and is involved in their progression (32,33). In the present study, it was found that cell viability was inhibited by oxLDL treatment. Furthermore, oxLDL treatment led to decreased production of NO and increased secretion of ET-1 in HUVECs. In addition, the expression of cell adhesion molecules, including ICAM-1 and PECAM-1, was induced by oxLDL. Collectively, the aforementioned results demonstrated that oxLDL caused endothelial dysfunction in HUVECs.
Lp-PLA2 is strongly associated with several cardiovascular risk markers and cardiovascular events in the clinic (34,35). A number of drugs and diet components have been demonstrated to demonstrate the vascular protective effect of Lp-PLA2 through influencing the enzyme concentration and activity (35). Lp-PLA2 is an important enzyme that can be found in human and rabbit atherosclerotic plaques (36). A large study that included 172 patients with no significant coronary artery disease (<30% stenosis) reported that circulating Lp-PLA2 levels were elevated in patients with early atherosclerosis (10). Compared with the patients without evidence of atherosclerosis, local net production of Lp-PLA2 is significantly increased in the patients with minimal coronary atherosclerosis (11). A study by Yang et al (10) demonstrated the potential of circulating Lp-PLA2 as an independent predictor of coronary endothelial dysfunction. In addition, local coronary production of Lp-PLA2 is associated with local endothelial function (11). In the present study, in vitro assessments were initially performed to investigate the role of Lp-PLA2 in endothelial dysfunction. Consistent with previous studies (37–39), the expression of Lp-PLA2 was induced by oxLDL. SB-435495, an inhibitor of Lp-PLA2, was used to suppress Lp-PLA2 expression, and it was subsequently identified that oxLDL-induced endothelial dysfunction was reversed by Lp-PLA2 suppression. The aforementioned results indicated that Lp-PLA2 induction could cause endothelial dysfunction in experimental atherosclerosis.
AMPK has been identified as a sensor of cellular energy and cellular redox status (40,41). AMPK activation appears to be a shared molecular target in vascular tissues, and the beneficial effects of certain agents are mediated by the activation of AMPK in endothelial cells (12–14). The phosphorylation of AMPKα at Thr172 is required for AMPK activation (42). AMPK phosphorylation has been demonstrated to reverse the decreases in cell viability caused by oxLDL (43), and the activation of AMPK activity in endothelial cells stimulates NO production (44). AMPK also decreases ET-1 expression and secretion from endothelial cells (45). Pretreatment with an AMPK inhibitor compound C significantly increased basal ET-1 mRNA and protein expression levels. In addition, Cacicedo et al (46) reported that AMPK reduces tumor necrosis factor-induced elevated expression of adhesion molecules in endothelial cells. Cell adhesion molecules such as ICAM-1 and PECAM-1 have been reported to be suppressed by AMPK activation (14). It has also been reported that the knockdown or mutation of Lp-PLA2 could induce apoptosis in macrophages and Cos-7 cells, and a reduction in the activated Akt level was involved in this phenomenon (47). AMPK is the upstream regulator of Akt (48–50). In the present study, the association between Lp-PLA2 and AMPK was initially investigated in an in vitro cell model of atherosclerosis, and whether AMPK mediates the effects of Lp-PLA2 on endothelial dysfunction was also investigated. Western blot analysis results indicated a negative role of Lp-PLA2 in the regulation of AMPK, which was evidenced by the increased expression levels of AMPKα and phosphorylated-AMPKα (Thr172) in oxLDL-exposed HUVECs following treatment with the Lp-PLA2 inhibitor SB-435495. Furthermore, the present study found that compound C, an AMPK inhibitor, was able to reverse the effects of Lp-PLA2 on cell viability, endothelial vasorelaxation capacity and the secretion of adhesion molecules in oxLDL-exposed HUVECs. Thus, these results suggest that AMPK serves an important role in mediating the effects of Lp-PLA2 on endothelial dysfunction.
In conclusion, the present study demonstrated the role of Lp-PLA2 in endothelial dysfunction and the associated mechanism in an in vitro cell model of atherosclerosis. Lp-PLA2 expression was induced in oxLDL-exposed HUVECs. The increased expression levels of Lp-PLA2 promotes endothelial dysfunction by inhibiting cell viability and endothelial vasorelaxation capacity, as well as inducing the secretion of adhesion molecules protein, at least partially through the downregulation of AMPK, thus contributing to the progression of atherosclerosis. The present study provided a novel molecular mechanism underlying the role of Lp-PLA2 in endothelial dysfunction and atherosclerosis, and it may aid the improvement of treatments for atherosclerotic vascular diseases.
Acknowledgements
The present study was supported by the Science and Technology Foundation of Tianjin Municipal Health Bureau (grant no. 2011kz63).
References
- 1.Leys D. Atherothrombosis: A major health burden. Cerebrovasc Dis. 2011;11(Suppl 2):S1–S4. doi: 10.1159/000049137. [DOI] [PubMed] [Google Scholar]
- 2.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*) Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 4.Eskin SG, Ives CL, McIntire LV, Navarro LT. Response of cultured endothelial cells to steady flow. Microvasc Res. 1984;28:87–94. doi: 10.1016/0026-2862(84)90031-1. [DOI] [PubMed] [Google Scholar]
- 5.Langille BL, Adamson SL. Relationship between blood flow direction and endothelial cell orientation at arterial branch sites in rabbits and mice. Circ Res. 1981;48:481–488. doi: 10.1161/01.RES.48.4.481. [DOI] [PubMed] [Google Scholar]
- 6.Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168–175. doi: 10.1161/01.ATV.0000051384.43104.FC. [DOI] [PubMed] [Google Scholar]
- 7.Karabina SA, Ninio E. Plasma PAF-acetylhydrolase: An unfulfilled promise? Biochim Biophys Acta. 2006;1761:1351–1358. doi: 10.1016/j.bbalip.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Venable ME, Zimmerman GA, McIntyre TM, Prescott SM. Platelet-activating factor: A phospholipid autacoids with diverse actions. J Lipid Res. 1993;34:691–702. [PubMed] [Google Scholar]
- 9.Snyder F. Platelet-activating factor and its analogs: Metabolic pathways and related intracellular processes. Biochim Biophys Acta. 1995;1254:231–249. doi: 10.1016/0005-2760(94)00192-2. [DOI] [PubMed] [Google Scholar]
- 10.Yang EH, McConnell JP, Lennon RJ, Barsness GW, Pumper G, Hartman SJ, Rihal CS, Lerman LO, Lerman A. Lipoprotein-associated phospholipase A2 is an independent marker for coronary endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2006;26:106–111. doi: 10.1161/01.ATV.0000191655.87296.ab. [DOI] [PubMed] [Google Scholar]
- 11.Lavi S, McConnell JP, Rihal CS, Prasad A, Mathew V, Lerman LO, Lerman A. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidylcholine in the coronary circulation: Association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation. 2007;115:2715–2721. doi: 10.1161/CIRCULATIONAHA.106.671420. [DOI] [PubMed] [Google Scholar]
- 12.Hardie DG. AMPK and raptor: Matching cell growth to energy supply. Mol Cell. 2008;30:263–265. doi: 10.1016/j.molcel.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Uchida K, Nakanishi N, Hattori Y. Cilostazol activates AMP-activated protein kinase and restores endothelial function in diabetes. Am J Hypertens. 2008;21:451–457. doi: 10.1038/ajh.2008.6. [DOI] [PubMed] [Google Scholar]
- 15.Hombach V, Höher M, Kochs M, Eggeling T, Schmidt A, Höpp HW, Hilger HH. Pathophysiology of unstable angina pectoris-correlations with coronary angioscopic imaging. Eur Heart J. 1988;9(Suppl N):40–45. doi: 10.1093/eurheartj/9.suppl_N.40. [DOI] [PubMed] [Google Scholar]
- 16.Plutzky J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. Am J Cardiol. 2001;88:10K–15K. doi: 10.1016/S0002-9149(01)01924-5. [DOI] [PubMed] [Google Scholar]
- 17.Tsujita K, Kaikita K, Soejima H, Sugiyama S, Ogawa H. Acute coronary syndrome-initiating factors. Nihon Rinsho. 2010;68:607–614. (In Japanese) [PubMed] [Google Scholar]
- 18.Dohi T, Daida H. Change of concept and pathophysiology in acute coronary syndrome. Nihon Rinsho. 2010;68:592–596. (In Japanese) [PubMed] [Google Scholar]
- 19.Khuseyinova N, Imhof A, Rothenbacher D, Trischler G, Kuelb S, Scharnagl H, Maerz W, Brenner H, Koenig W. Association between Lp-PLA2 and coronary artery disease: Focus on its relationship with lipoproteins and markers of inflammation and hemostasis. Atherosclerosis. 2005;182:181–188. doi: 10.1016/j.atherosclerosis.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272:20963–20966. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- 21.Ryu JH, Yu M, Lee S, Ryu DR, Kim SJ, Kang DH, Choi KB. AST-120 improves microvascular endothelial dysfunction in end-stage renal disease patients receiving hemodialysis. Yonsei Med J. 2016;57:942–949. doi: 10.3349/ymj.2016.57.4.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ceriello A, De Nigris V, Pujadas G, La Sala L, Bonfigli AR, Testa R, Uccellatore A, Genovese S. The simultaneous control of hyperglycemia and GLP-1 infusion normalize endothelial function in type 1 diabetes. Diabetes Res Clin Pract. 2016;114:64–68. doi: 10.1016/j.diabres.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 23.Deanfield J, Donald A, Ferri C, Giannattasio C, Halcox J, Halligan S, Lerman A, Mancia G, Oliver JJ, Pessina AC, et al. Endothelial function and dysfunction. Part I: Methodological issues for assessment in the different vascular beds: A statement by the working group on endothelin and endothelial factors of the european society of hypertension. J Hypertens. 2005;23:7–17. doi: 10.1097/00004872-200501000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Gokce N, Keaney JF, Jr, Hunter LM, Watkins MT, Menzoian JO, Vita JA. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: A prospective study. Circulation. 2002;105:1567–1572. doi: 10.1161/01.CIR.0000012543.55874.47. [DOI] [PubMed] [Google Scholar]
- 25.Shah V, Toruner M, Haddad F, Cadelina G, Papapetropoulos A, Choo K, Sessa WC, Groszmann RJ. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology. 1999;117:1222–1228. doi: 10.1016/S0016-5085(99)70408-7. [DOI] [PubMed] [Google Scholar]
- 26.Taguchi K, Matsumoto T, Kobayashi T. G-protein-coupled receptor kinase 2 and endothelial dysfunction: Molecular insights and pathophysiological mechanisms. J Smooth Muscle Res. 2015;51:37–49. doi: 10.1540/jsmr.51.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Böhm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. 2007;76:8–18. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18:4266–4288. doi: 10.2174/138161212802481246. [DOI] [PubMed] [Google Scholar]
- 29.Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. 2009;61:22–32. doi: 10.1016/S1734-1140(09)70004-0. [DOI] [PubMed] [Google Scholar]
- 30.Jackson DE. The unfolding tale of PECAM-1. FEBS Lett. 2003;540:7–14. doi: 10.1016/S0014-5793(03)00224-2. [DOI] [PubMed] [Google Scholar]
- 31.Albelda SM, Muller WA, Buck CA, Newman PJ. Molecular and cellular properties of PECAM-1 (endoCAM/CD31): A novel vascular cell-cell adhesion molecule. J Cell Biol. 1991;114:1059–1068. doi: 10.1083/jcb.114.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2000;191:189–194. doi: 10.1084/jem.191.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zibara K, Chignier E, Covacho C, Poston R, Canard G, Hardy P, McGregor J. Modulation of expression of endothelial intercellular adhesion molecule-1, platelet-endothelial cell adhesion molecule-1, and vascular cell adhesion molecule-1 in aortic arch lesions of apolipoprotein E-deficient compared with wild-type mice. Arterioscler Thromb Vasc Biol. 2000;20:2288–2296. doi: 10.1161/01.ATV.20.10.2288. [DOI] [PubMed] [Google Scholar]
- 34.Silva IT, Mello AP, Damasceno NR. Antioxidant and inflammatory aspects of lipoprotein-associated phospholipase A2 (Lp-PLA2): A review. Lipids Health Dis. 2011;10:170. doi: 10.1186/1476-511X-10-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: Biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25:923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 36.Häkkinen T, Luoma JS, Hiltunen MO, Macphee CH, Milliner KJ, Patel L, Rice SQ, Tew DG, Karkola K, Ylä-Herttuala S. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1999;19:2909–2917. doi: 10.1161/01.ATV.19.12.2909. [DOI] [PubMed] [Google Scholar]
- 37.Wang WY, Li J, Yang D, Xu W, Zha RP, Wang YP. OxLDL stimulates lipoprotein-associated phospholipase A2 expression in THP-1 monocytes via PI3K and p38 MAPK pathways. Cardiovasc Res. 2010;85:845–852. doi: 10.1093/cvr/cvp367. [DOI] [PubMed] [Google Scholar]
- 38.De Keyzer D, Karabina SA, Wei W, Geeraert B, Stengel D, Marsillach J, Camps J, Holvoet P, Ninio E. Increased PAFAH and oxidized lipids are associated with inflammation and atherosclerosis in hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2009;29:2041–2046. doi: 10.1161/ATVBAHA.109.196592. [DOI] [PubMed] [Google Scholar]
- 39.Pasini A Fratta, Stranieri C, Pasini A, Vallerio P, Mozzini C, Solani E, Cominacini M, Cominacini L, Garbin U. Lysophosphatidylcholine and carotid intima-media thickness in young smokers: A role for oxidized LDL-induced expression of PBMC lipoprotein-associated phospholipase A2? PLoS One. 2013;8:e83092. doi: 10.1371/journal.pone.0083092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou MH, Wu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin Exp Pharmacol Physiol. 2000;35:535–545. doi: 10.1111/j.1440-1681.2007.04851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C. Genetics and molecular biology-protein kinase C-zeta as an AMP-activated protein kinase kinase kinase: The protein kinase C-zeta-LKB1-AMP-activated protein kinase pathway. Curr Opin Lipidol. 2008;19:541–542. doi: 10.1097/MOL.0b013e32830f4a44. [DOI] [PubMed] [Google Scholar]
- 42.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 43.Guo H, Chen Y, Liao L, Wu W. Resveratrol protects HUVECs from oxidized-LDL induced oxidative damage by autophagy upregulation via the AMPK/SIRT1 pathway. Cardiovasc Drugs Ther. 2013;27:189–198. doi: 10.1007/s10557-013-6442-4. [DOI] [PubMed] [Google Scholar]
- 44.Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- 45.Reiter CE, Kim JA, Quon MJ. Green tea polyphenol epigallocatechin gallate reduces endothelin-1 expression and secretion in vascular endothelial cells: Roles for AMP-activated protein kinase, Akt, and FOXO1. Endocrinology. 2010;151:103–114. doi: 10.1210/en.2009-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cacicedo JM, Yagihashi N, Keaney JF, Jr, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324:1204–1209. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- 47.Maeda T, Takeuchi K, Xiaoling P, P Zankov D, Takashima N, Fujiyoshi A, Kadowaki T, Miura K, Ueshima H, Ogita H. Lipoprotein-associated phospholipase A2 regulates macrophage apoptosis via the Akt and caspase-7 pathways. J Atheroscler Thromb. 2014;21:839–853. doi: 10.5551/jat.21386. [DOI] [PubMed] [Google Scholar]
- 48.Ji L, Zhang X, Liu W, Huang Q, Yang W, Fu F, Ma H, Su H, Wang H, Wang J, et al. AMPK-regulated and Akt-dependent enhancement of glucose uptake is essential in ischemic preconditioning-alleviated reperfusion injury. PLoS One. 2013;8:e69910. doi: 10.1371/journal.pone.0069910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leclerc GM, Leclerc GJ, Fu G, Barredo JC. AMPK-induced activation of Akt by AICAR is mediated by IGF-1R dependent and independent mechanisms in acute lymphoblastic leukemia. J Mol Signal. 2010;5:15. doi: 10.1186/1750-2187-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]