

Abstract
Niemann-Pick type C (NP-C) disease is a fatal genetic lipidosis for which there is no Food and Drug Administration (FDA)-approved therapy. Vorinostat, an FDA-approved inhibitor of histone deacetylases, ameliorates lysosomal lipid accumulation in cultured NP-C patient fibroblasts. To assess the therapeutic potential of histone deacetylase inhibition, we pursued these in vitro observations in two murine models of NP-C disease. Npc1nmf164 mice, which express a missense mutation in the Npc1 gene, were treated intraperitoneally, from weaning, with the maximum tolerated dose of vorinostat (150 mg/kg, 5 days/week). Disease progression was measured via gene expression, liver function and pathology, serum and tissue lipid levels, body weight, and life span. Transcriptome analyses of treated livers indicated multiple changes consistent with reversal of liver dysfunction that typifies NP-C disease. Significant improvements in liver pathology and function were achieved by this treatment regimen; however, NPC1 protein maturation and levels, disease progression, weight loss, and animal morbidity were not detectably altered. Vorinostat concentrations were >200 μm in the plasma compartment of treated animals but were almost 100-fold lower in brain tissue. Apolipoprotein B metabolism and the expression of key components of lipid homeostasis in primary hepatocytes from null (Npc1−/−) and missense (Npc1nmf164) mutant mice were altered by vorinostat treatment, consistent with a response by these cells independent of the status of the Npc1 locus. These results suggest that HDAC inhibitors have utility to treat visceral NP-C disease. However, it is clear that improved blood-brain barrier penetration will be required to alleviate the neurological symptoms of human NP-C disease.
Keywords: cholesterol, cholesterol regulation, histone deacetylase inhibitor (HDAC inhibitor) (HDI), lipid trafficking, lysosomal storage disease, neurodegenerative disease, Saccharomyces cerevisiae, sphingolipid, yeast
Introduction
Histone deacetylase (HDAC)5 inhibitors were initially identified as treatments for a variety of proliferative diseases, including T-cell lymphoma and other cancers (1). The mechanism of efficacy primarily reflects changes in the transcriptional program of rapidly growing cells and, in some cases, results in altered expression of 2–10% of the genome (2, 3). Subsequently, intervention at the histone acetylation/deacetylation axis of gene expression has been successfully applied to many unrelated diseases (4). Neurological disorders such as Alzheimer's disease, Huntington's disease, Parkinson's disease, amyotrophic lateral sclerosis, and Friedrich's ataxia have all been found to respond to HDAC inhibitors (4–6). The primary mechanism by which HDAC inhibitors influence these diseases remains undetermined and may well be multivariate. In some disorders (e.g. cystic fibrosis), the treatment is associated with alterations at the proteostatic level due in part to activation of chaperone-mediated refolding pathways (7).
Niemann-Pick type C (NP-C) disease is a rare lysosomal storage disorder typified by defective subcellular transport of cholesterol and sphingolipids as well as premature demise due to central nervous system (largely cerebellar) impairment (8). Autosomal recessive mutations in the NPC1 and NPC2 genes confer 95 and 5% of NP-C disease cases, respectively. Both genes are strikingly conserved throughout evolution (from yeast to humans), although their precise functions remain elusive (9, 10). We have proposed that variation in the activity of additional genes influences NP-C disease severity, and these loci represent novel therapeutic interventions (11). To identify such NP-C disease modification pathways, we have used a model system-based approach in the yeast Saccharomyces cerevisiae. Our “exacerbate-reverse” strategy successfully identified 13 genetic loci whose individual absence conferred complete inviability to the yeast deletion of the NPC1 ortholog conditional upon sterol loading. This approach identified the status of histone acetylation as a key component of lipid accumulation in yeast and in cultured human fibroblasts that are deficient in the NP-C pathways (11). We and others have demonstrated that histone deacetylase inhibition by a variety of pharmacological agents normalizes lipid homeostasis in rapidly growing immortalized cell lines derived from patients with NP-C disease (11–13). These studies have progressed to the extent that the FDA-approved HDAC inhibitor vorinostat (suberoylanilide hydroxamic acid, Zolinza®) is the focus of a clinical trial for safety and tolerability in adult NP-C patients (ClinicalTrials.gov number NCT02124083).
Multitissue lipid accumulation and neurodegeneration define NP-C disease. The aberrant subcellular lipid storage is clearly associated with transcriptional dysregulation of numerous genes, including sterol regulatory element-binding protein targets, such as HMG-CoA reductase (HMGCR) and the low density lipoprotein receptor (LDLR) (14, 15). Any candidate intervention that can restore lipid homeostasis in cultured cells, however, must also be tested for an impact on NP-C disease pathology in an in vivo model. This is particularly critical given the challenge of predicting whether results in cell culture will be conserved in human trials (16). Here we describe the impact of vorinostat in a murine model of NP-C disease caused by a missense mutation in the Npc1 gene. We show that this FDA-approved HDAC inhibitor normalizes hepatic transcriptional regulation of cholesterol, corrects apolipoprotein B homeostasis, and improves liver function and pathology, thus raising the possibility that this intervention may treat visceral NP-C disease. With the regimens used here, however, there was no accompanying improvement in mortality or morbidity, probably due to the poor bioavailability of this drug to brain tissues. Overall, these studies establish HDAC inhibition as an attractive approach for treatment of NP-C disease while emphasizing the need for candidate inhibitors to overcome the blood-brain barrier.
Results
NPC1 Missense Variants Can Be Refolded
In the Npc1nmf164 mouse, NP-C disease arises from a missense mutation at codon 1005 in Npc1 that resembles several NPC1 missense mutations that cause NP-C disease in humans (17). Specifically, the murine aspartic acid-to-glycine NPC1D1005G variant is contextually similar to the NPC1S1004L, NPC1P1007A, and NPC1P1007L variants associated with the juvenile form of human NP-C disease. The Npc1nmf164 mouse model presents with a less severe form of the disease than the Npc1−/− murine model but nevertheless exhibits an abbreviated life span, progressive weight loss, neurological impairment, and cellular lipid accumulation compared with control littermates (17).
In contrast to the Npc1−/− mutant mouse model, Npc1nmf164 mice exhibit low but detectable levels of presumably functionally impaired NPC1 protein (17). To specifically determine whether this phenotype is due to misfolding and premature degradation of the NPC1D1005G variant, fibroblasts derived from the Npc1nmf164 mouse were treated with glycerol, a chemical chaperone known to stabilize misfolded proteins (18). Glycerol increased expression levels of the NPC1D1005G protein compared with untreated cells (Fig. 1) and in addition promoted its maturation to a higher molecular weight form. The properties of the NPC1D1005G protein were then compared with those of NPC1I1061T, a previously described misfolded NPC1 variant associated with the most common form of human NP-C disease (18, 19). NPC1-deficient human osteosarcoma U2OS cells expressing the NPC1I1061T protein were also treated with glycerol. Expression levels of the NPC1I1061T protein were also increased with glycerol treatment (Fig. 1), consistent with previous studies (18). To elucidate the possible contributions of lysosomal and proteasomal degradation to levels of the NPC1D1005G protein, the Npc1nmf164 fibroblasts were treated with 3-methyladenine (an autophagy inhibitor) and two inhibitors of the proteasome, MG132 and lactacystin. Each of these treatments increased expression and maturation of the NPC1D1005G protein (Fig. 1), implicating lysosomal and proteasomal pathways in the degradation of misfolded forms of the NPC1D1005G protein.
FIGURE 1.
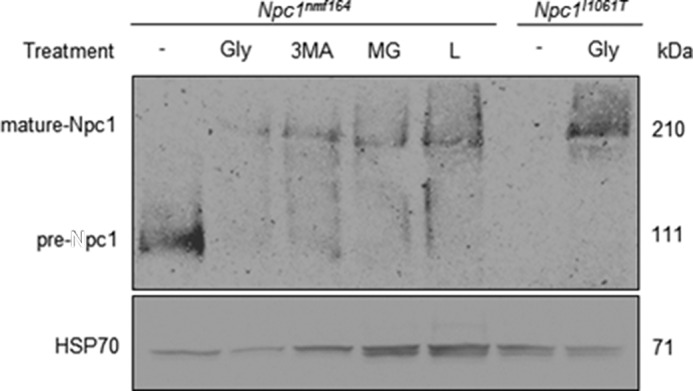
Refolding of NPC1D1005G mutant protein. Representative image of 30 μg of total cell lysates extracted from Npc1nmf164 fibroblasts treated with 5% glycerol (Gly), 10 μm 3-methyladenine (3MA), 10 μm MG132 (MG), and 10 μm lactacystin (L) compared with U2OS cells expressing the NPC1I1061T mutation in the presence and absence of 5% glycerol. Lysates were denatured at 70 °C for 10 min, electrophoresed through a 3–8% Tris acetate gel, and transferred onto a Protran membrane that was incubated with NPC1 or HSP70 antibody and envisaged with chemiluminescence.
Vorinostat Administration in a Misfolded Protein Mouse Model of NP-C Disease
To investigate the role of vorinostat as an NP-C therapeutic in vivo, we first assessed toxicity of this HDAC inhibitor in the Npc1nmf164 NP-C murine model following i.p. administration in a vehicle of 45% PEG 400 + 10% DMSO (hereafter called PEG/DMSO). This protocol was used previously to deliver vorinostat across the blood-brain barrier and inhibit the metastasis of triple-negative breast cancer to the brain (20). 21-day-old, asymptomatic Npc1nmf164 homozygous mutant and WT mice were injected daily with 50, 100, 150, or 200 mg/kg vorinostat for 7 days. All mice were viable after 7 days; however, we observed significant weight loss in mice treated with 200 mg/kg vorinostat. This is in agreement with 200 mg/kg being toxic in control mice as well as in a mouse model of Huntington's disease (5). Using the maximum tolerated dose and treatment regimen, we determined whether HDAC inhibition with vorinostat can impact NP-C disease progression. Control (WT) and Npc1nmf164 mice were treated with 150 mg/kg/day vorinostat or the PEG/DMSO vehicle, 5 times/week from P21, an age where lipid accumulation already exists in the liver and brain (17). Treatment continued until P60, at which point disease symptoms (i.e. weight loss, motor function deficits) are detectable in untreated Npc1nmf164 animals (17).
Transcriptional Modulation in Response to Vorinostat
The lysosomal sequestration of free cholesterol and other bioactive lipids in NPC1-deficient animals has striking and chronologically early consequences in terms of gene regulation. Multiple components of lipid homeostasis and inflammation are transcriptionally up-regulated, and this dysregulation is rapidly normalized by treatment with the lipid chelator, 2-hydroxypropyl-β-cyclodextrin (14, 15). To test whether these pathways respond to vorinostat in the liver of Npc1nmf164 mice as well as to comprehensively explore the impact of vorinostat on hepatic gene expression in these animals, we performed RNA-Seq analysis on RNA isolated from the livers of vorinostat and vehicle-treated Npc1nmf164 mice. The transcripts were mapped to the mouse mm10 genome assembly, normalized, and quantified. Approximately 16–18 million reads/sample were obtained. Expression levels of 14,516 genes were measured, a number comparable with the 14,326 genes measured previously in the murine liver transcriptome (21). Expression of 844 genes (5.9% of the transcriptome) was significantly regulated (>0.6 log-fold change, p < 0.05) by vorinostat treatment, and of these 844 genes, 603 (71.4%) were up-regulated and 241 (28.6%) were down-regulated (Fig. 2, a and b, supplemental Table S1). These genes were classified within an extensive array of Gene Ontology processes (Fig. 2a). Notable cellular aspects and processes relevant to NP-C disease and HDAC inhibition include lipid metabolism, inflammation, and histone-mediated gene expression (Fig. 2c).
FIGURE 2.

Vorinostat administration from P21 to P60 alters the liver transcriptome of Npc1nmf164 mice. a, quantification of the diversity of biological processes regulated by vorinostat as assigned using PANTHER (19). b, heat map depicting the relative repression or induction of 844 transcripts that were differentially expressed in the liver by ≥0.6 log2 ratio -fold change (p < 0.05) between the vehicle- and vorinostat-treated Npc1nmf164 mice (n = 7, both groups). c, functional gene annotation clustering performed on 10 genes identified in cellular functions associated with inhibition of histone deacetylation, 19 genes with cellular functions associated with lipid metabolism, and 16 genes with cellular functions associated with inflammation. The scale bar indicates proportional read counts, with red and white tones representing down-regulated and up-regulated genes, respectively; this scale bar applies to b and c.
Critically, the vorinostat-responsive genes include those known to be modulated by HDAC inhibitors in previous studies. For example, the GATA-binding transcription factor Gata1, a hallmark vorinostat-responsive gene (2, 3, 22), was up-regulated 3.6 log2-fold (p = 0.0001) in vorinostat-treated Npc1nmf164 mice compared with PEG/DMSO-treated Npc1nmf164 mice (Fig. 2c). As observed previously (23, 24), early growth response-1 (Egr1) and amyloid β precursor-like protein 1 (Aplp1) were elevated 1.2 log2-fold (p = 5.67 × 10−4) and 2.56 log2-fold (p = 1.95 × 10−19), respectively, after vorinostat treatment (Fig. 2c). Transcription of the aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL, also referred to as BMAL1) is repressed by HDAC3 (25) and was elevated 2.59 log2-fold (p = 1.18 × 10−7) after treatment with vorinostat (Fig. 2c). Likewise, we determined that vorinostat reduced transcription of the minichromosome maintenance protein-2 (MCM2) by 0.67 log2-fold (p = 0.0015) and the TATA-box-binding protein TAF6L by 0.56 log2-fold (p = 0.004) compared with PEG/DMSO-treated mice (Fig. 2c), consistent with the previously observed down-regulation of these genes by HDAC inhibitors (26, 27). Additionally, the cyclin D1 (Ccnd1) gene has been shown to decrease in expression by HDAC inhibition (28); here vorinostat reduced Ccnd1 expression 1.34 log2-fold (p = 1.8 × 10−6) (Fig. 2c). It is clear from these responses that a variety of HDAC isoforms, and thus their transcriptional targets, were affected by the treatment protocol in place, as would be anticipated for the impact of a global HDAC inhibitor like vorinostat.
To determine the therapeutic potential of vorinostat, we investigated the overlap of vorinostat-mediated gene expression and candidate biomarkers previously shown to be transcriptionally dysregulated in NP-C disease. Lyz1 (lysozyme 1) transcript levels are up-regulated in the liver and brain of the Npc1−/− mouse (29), and plasma lysozyme activity is increased in the Npc1−/− mouse (30). Vorinostat treatment reduced hepatic expression of Lyz1 by 1.1 log2-fold (p = 4.37 × 10−4) in Npc1nmf164 mice compared with vehicle control (Fig. 2c). Likewise, the sterol regulatory element-binding factor (SREBF1) is a transcription factor that regulates cholesterol synthesis and is up-regulated in NPC1-deficient hepatocytes (31), and Srebf1 expression was reduced with the vorinostat treatment 0.64 log2-fold (p = 0.02) compared with vehicle-treated Npc1nmf164 mice (Fig. 2c). In addition, the genes encoding oxysterol-binding protein OSBPL3 and the perilipin PLIN4 were up-regulated in independent studies of the liver of the Npc1−/− mouse (29, 32), and we show here that the vorinostat treatment reduced expression of Osbpl3 and Plin4 by 1.4 log2-fold (p = 2.86 × 10−7) and 1.3 log2-fold (p = 1.44 × 10−5), respectively, in Npc1nmf164 mice (Fig. 2c).
Similarly, several transcriptional pathways associated with inflammation (a characteristic of chronic NP-C disease) were normalized by treatment of the Npc1nmf164 mouse with vorinostat. For example, expression levels of four genes (metalloproteinase matrix-15 (Mmp15), CD276 antigen (Cd276), and histocompatibility antigens H2-Ab1 and H2-Eb1) that were consistently shown to be up-regulated in Npc1−/− mice (29, 32) were down-regulated in vorinostat-treated Npc1nmf164 mice compared with vehicle-treated Npc1nmf164 mice (Fig. 2c). Expression levels of three additional pro-inflammatory CD antigen genes (Cd8a, Cd79b, and Cd74) were similarly reduced by vorinostat (Fig. 2c). Two S100a genes (S100a8 and S100a9) that are targets of NFkβ and critical components of the immune response to liver damage (33) were up-regulated with vorinostat treatment (Fig. 2c). The cathelicidin antimicrobial peptide gene (Camp) is required for inflammatory response regulation; expression of CAMP was up-regulated in two studies of the Npc1−/− mouse (29, 32) and down-regulated in the Npc1nmf164 mice treated with vorinostat. These results suggest that vorinostat activates an anti-inflammatory response as a consequence of NP-C disease in the Npc1nmf164 mouse.
Transcriptional Modulation of Cholesterol Metabolism
We used the Web Gene Ontology Enrichment Analysis Tool Kit (34) to identify pathways statistically overrepresented within the 844 vorinostat-regulated genes in the liver transcriptome of Npc1nmf164 mice. In addition to amino acid metabolism (p = 0.0323), four pathways associated with an anti-inflammatory response were enriched: GPCRs class A rhodopsin-like (p = 0.0008), GPCRs non-odorant (p = 0.0026), GPCRs peptide (p = 0.0323), and chemokine signaling pathway (p = 0.0082). Of particular relevance to NP-C disease, the enrichment analysis revealed that genes critical to the biosynthesis of cholesterol were down-regulated (p = 0.0014) (Fig. 2c). qRT-PCR analysis for seven key genes (Hmgcs1, Hmgcr, Mvk, Mvd, Idi1, Lss, and Cyp51) was conducted on sets of three randomly selected mice from the RNA-Seq analysis (Fig. 3, a–g). Vehicle- and vorinostat-treated WT mice were included to determine whether vorinostat regulates these cholesterol biosynthesis genes independent of the status of the NPC1 pathway.
FIGURE 3.

Vorinostat normalizes gene expression of key components of cholesterol homeostasis. 80 ng of RNA from the indicated animals were assessed for gene expression at P60 using qRT-PCR relative to GAPDH (PCR primers are described under “Experimental Procedures”). Vehicle-treated WT mice were assigned a baseline expression of 1. a–g, cholesterol biosynthesis genes that were significantly enriched in RNA-Seq analysis (Hmgcs, Hmgcr, Mvk, Mvd, Idi1, Lss, and Cyp51). h, Ldlr. All groups are n = 3 with data shown as mean ± S.D. (error bars) *, p < 0.05, Student's t test.
Our analyses suggest that the cholesterol biosynthetic pathway is significantly up-regulated in the Npc1nmf164 model, presumably in response to the lysosomal accumulation of cholesterol in association with reduced free cholesterol in regulatory pools such as the ER. Indeed, expression levels of Hmgcs1, Hmgcr, Mvk, Mvd, Idi1, Lss, and Cyp51 were significantly up-regulated in the mutant mice relative to WT (Fig. 3, a–g), ranging from an increase of 2-fold to an increase of 5.1-fold. Impressively, vorinostat treatment significantly decreased the expression of all seven genes toward WT levels, with -fold change ranging from 1.6- to 2.8-fold. Moreover, these changes were only identified in Npc1nmf164 mice; expression levels of these seven genes in WT mice were not affected by vorinostat treatment. In addition, we measured expression of Ldlr, a gene that is strongly up-regulated in NP-C disease (14, 15); LDLR was up-regulated 1.9-fold in vehicle-treated Npc1nmf164 compared with vehicle-treated WT mice (p = 0.014), and down-regulated 1.7-fold in vorinostat-treated Npc1nmf164 compared with vehicle-treated Npc1nmf164 mice (p = 0.044) (Fig. 3h). Collectively, these results have three major implications. First, cholesterol homeostatic genes are elevated in Npc1nmf164 mice at P60. Second, vorinostat does not regulate expression of cholesterol metabolism genes in WT mice. Third, and most importantly, upon treatment with vorinostat, eight genes critical to cholesterol homeostasis and NP-C disease were down-regulated to WT levels.
Lipid Metabolism Is Fundamental to the Vorinostat Interactome
To identify the core genes in the vorinostat interactome, we used Phenolyzer (35) to visualize gene-gene interactions, protein-protein interactions, and transcriptional regulation among the 844 genes in the vorinostat interactome (Fig. 2a). This analysis distinguished Cyp27b1, Hmgcr, Pnpla3, and Chka as the core hubs in the vorinostat interactome, each with >20 interactions within the interactome (Fig. 4). Cyp27b1 encodes a member of the cytochrome P450 superfamily of enzymes that hydroxylates 25-hydroxyvitamin D3 and regulates the level of biologically active vitamin D derived from cholesterol. Interestingly, vitamin D deficiency is associated with toxic lipid accumulation in chronic liver disease (36). Hmgcr encodes the rate-limiting enzyme in cholesterol biosynthesis that is consistently up-regulated in NP-C disease (14, 15), and we show here that vorinostat treatment restores this dysregulation to normal levels (Fig. 3b). Pnpla3 encodes a patatin-like phospholipase that is a genetic risk factor for lipid-mediated liver disease wherein overexpression confers hepatic steatosis (37). Chka encodes choline kinase α, one of two mammalian enzymes that catalyze the phosphorylation of choline to phosphocholine in the biosynthesis of the major membrane phospholipid, phosphatidylcholine (38). The hubs of the vorinostat interactome are all associated with lipid metabolism; specifically, our identification of Hmgcr as a mediator of vorinostat-mediated down-regulation of cholesterol biosynthesis as well as a key hub in the vorinostat interactome indicates that regulation of cholesterol biosynthesis is fundamental to the impact of vorinostat on NP-C disease.
FIGURE 4.

Lipid metabolism is fundamental to the vorinostat interactome. The 844 genes in the vorinostat interactome were processed through Phenolyzer (35) to visualize the 50 most interactive genes based on gene-gene interactions, protein-protein interactions, and transcriptional regulation within the interactome. This analysis distinguished four lipid metabolism genes (Cyp27b1, Hmgcr, Pnpla3, and Chka) as the core hubs (indicated in blue) in the vorinostat interactome, each with more than 20 interactions within the interactome. The size of the nodes represents the number of interactions.
Vorinostat Improves Liver Health
To determine whether the transcriptional regulation of cholesterol metabolism by vorinostat has functional consequences, we measured serum levels of alanine aminotransferase (ALT), an enzyme that is elevated in NP-C disease as a consequence of liver damage (39). Serum ALT levels were significantly higher (73.9%) in the PEG/DMSO-treated Npc1nmf164 mice compared with PEG/DMSO-treated control mice. Vorinostat treatment significantly reduced serum ALT levels in Npc1nmf164 mice by 25.7% (Fig. 5a). Furthermore, vorinostat treatment significantly reduced serum total cholesterol in Npc1nmf164 mice by 22%, independent of HDL cholesterol or triglyceride concentrations (Fig. 5, b–d).
FIGURE 5.
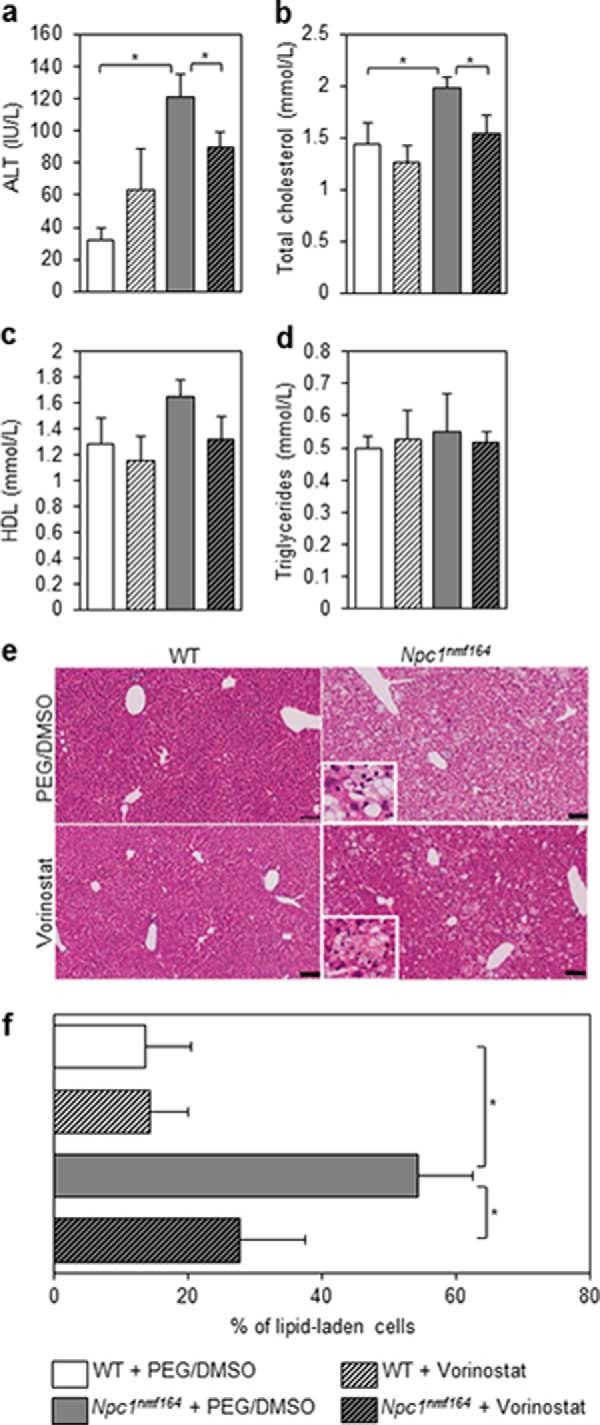
Vorinostat treatment improves liver function in Npc1nmf164 mice. Animals were treated with either vehicle (PEG/DMSO) or 150 mg/kg vorinostat from P21 to P60, at which point serum was harvested from sacrificed animals. Liver function was evaluated via levels of ALT expressed as international units/liter (IU/L; a). Total cholesterol, HDL cholesterol, and triglycerides (b–d) were measured and are expressed as mmol/liter. All data are shown as mean ± S.E. (error bars) for WT + PEG/DMSO (n = 5), WT + vorinostat (n = 5), Npc1nmf164 + PEG/DMSO (n = 4), and Npc1nmf164 + vorinostat (n = 5). *, p < 0.05, Student's t test. e, fixed liver tissue was paraffin-embedded, and 5-μm sections were stained with H&E. Representative images are shown. The prevalence of lipid-laden cells was expressed as a fraction of total cells per field (∼3000) and quantified using SlidePath tissue image analysis (Leica Biosystems, version 2.0) software at ×20 magnification. Data are shown in f as mean ± S.D. (error bars) from 10–15 random fields/animal with 3 animals/treatment group. *, p < 0.001, Student's t test. The inset panels represent a ×80 magnification of the field of view, focusing on the lipid laden cells.
Thin section H&E staining of fixed liver tissue revealed the marked accumulation of lipid-laden cells in untreated Npc1nmf164 mice at P60 (Fig. 5e), characteristic of lipid accumulation at this stage of the disease in this mouse model (17). Treatment of Npc1nmf164 mice with vorinostat convincingly and significantly reduced the prevalence of lipid-laden cells by 48.9% (p < 0.001) and elevated the presence of healthy hepatocytes, relative to vehicle controls (Fig. 5e). Taken together, the transcriptional normalization of hepatic cholesterol homeostatic genes and the reductions in serum cholesterol and ALT levels (liver function) in conjunction with improved liver histopathology indicate a marked recovery from visceral NP-C disease due to vorinostat treatment.
To determine whether the vorinostat-mediated changes in liver heath correlate with changes in total lipid accumulation in vivo, we quantified levels of free cholesterol, sphingomyelin, sphingosine, glucosylceramide, and lactosylceramide using GC/MS and LC/MS. These lipids are biochemical hallmarks of late stage NP-C disease in the liver (8). Because the status of these lipids has not been reported for the Npc1nmf164 mouse at P60, we first determined that these lipids were significantly increased in vehicle-treated Npc1nmf164 mice compared with vehicle-treated WT mice (Fig. 6, a–e). In accordance with the Npc1−/− and Npc1I1061T mouse models of NP-C disease (14, 19, 40), the Npc1nmf164 mice exhibit increased levels of these lipids relative to WT mice (Fig. 6, a–e). Vorinostat did not detectably alter lipid levels in WT or Npc1nmf164 mice (Fig. 6, a–e). To further investigate a hallmark lipid using an independent method, we assessed free cholesterol levels using fluorescent microscopy. Filipin, a fluorescent stain for unesterified cholesterol, is a key tool used in the diagnosis of NP-C patients (8). As expected, we observed increased filipin fluorescence in vehicle-treated Npc1nmf164 mice compared with vehicle-treated WT mice (Fig. 6, f and g). However, filipin fluorescence did not indicate any change in free cholesterol levels after vorinostat treatment. These results indicate that vorinostat did not detectably reduce lipid accumulation in the liver using this treatment regime from P21 to P60. Together, these results of reduced ALT levels, reduced number of lipid-laden cells, and increased number of healthy hepatocytes coincident with no apparent reduction in total mass of unesterified cholesterol suggest that vorinostat reproportions lipids, a phenomenon that would be consistent with partitioning of unesterified cholesterol in Kupffer cells by cyclodextrin (41).
FIGURE 6.

Cholesterol and sphingolipid accumulation in the liver of vorinostat-treated mice. WT and Npc1nmf164 mice were treated with either vehicle (PEG/DMSO) or 150 mg/kg vorinostat from P21 to P60, at which time livers were harvested from sacrificed animals. a–e, free cholesterol and sphingolipids were measured using LC/MS. All groups are n = 5 with data shown as mean ± S.D. (error bars). *, p < 0.05, Student's t test. f–g, unesterified cholesterol was visualized using filipin and quantified using densitometry software (ImageJ). All groups are n = 3 with data shown as mean ± S.D. *, p < 0.05, Student's t test.
Vorinostat Treatment Does Not Delay Weight Loss or Extend Life Span
To determine the effect of vorinostat on the life span of the Npc1nmf164 mice, we monitored the weight of vorinostat- and PEG/DMSO-treated mice during an extended treatment period from P21 to P90 using the regimen described above (150 mg/kg/day vorinostat in PEG/DMSO vehicle, 5 times/week). In agreement with our prior toxicity study, vorinostat-treated WT mice continued to gain weight beyond 10 weeks (Fig. 7a) and were not neurologically impaired compared with vehicle-treated mice. Relative to vehicle-treated WT mice, vehicle-treated Npc1nmf164 mice significantly lost weight between 10 and 13 weeks (p = 0.0002), a result consistent with prior characterization of the Npc1nmf164 mice (17). There was no delay in the weight loss of vorinostat-treated Npc1nmf164 mice at any point during the treatment (Fig. 7a); treated and untreated animals lost weight and exhibited ataxia to similar levels from 9 to 10 weeks until death. Therapies that have delayed weight loss have also extended life span (14, 42); thus, the absence of change with vorinostat treatment would predict that this intervention protocol does not extend life span. Indeed, vorinostat-treated Npc1nmf164 mice survived 94 ± 7 days (n = 8), a time range that is not different than the 94 ± 4-day (n = 5) life span of untreated Npc1nmf164 mice.
FIGURE 7.

Vorinostat does not delay weight loss or penetrate the blood-brain barrier of Npc1nmf164 mice. a, animals were treated with PEG/DMSO or 150 mg/kg vorinostat between P21 and P90 (3–13 weeks) and weighed weekly. Data are shown as mean ± S.D. (error bars) for WT + PEG/DMSO (n = 5), WT + PEG/DMSO (n = 5), Npc1nmf164 + PEG/DMSO (n = 5), and Npc1nmf164 + vorinostat (n = 6). *, p < 0.05, two-way analysis of variance with Bonferroni post hoc test. b and c, vorinostat levels in the brain are <5% of plasma levels. 150 mg/kg vorinostat was administered in Npc1nmf164 mice at P21, and mean vorinostat levels were quantified in the plasma and brain at four time points after injection (0.5, 1, 2, and 4 h).
Penetration of the Blood-Brain Barrier by Vorinostat
Despite its beneficial effect on the liver, the lack of effect of vorinostat treatment on weight and life span of the Npc1nmf164 mouse suggests that the drug fails to reach a concentration in the brain required to reverse the overall impact of the disease. Previously, a concentration of 5–10 μm vorinostat was described as sufficient to limit lipid accumulation in cultured NP-C patient fibroblasts (11–13). To determine the drug concentration achieved in the brain, we quantified vorinostat in plasma and brain of treated Npc1nmf164 mice at P21 (Fig. 7b). The levels of vorinostat in the plasma at 0.5, 1, 2, and 4 h after injection were 222.29, 17.39, 3.01, and 0.97 μm. In marked contrast, the levels of vorinostat in the brain in the same animals were 3.16, 0.23, and 0.12 μm, at 0.5, 1, and 2 h after injection, respectively, and were below the minimum detection level of the assay at 4 h (Fig. 7c). These results indicate a half-life of vorinostat that is <60 min. Our findings of rapid clearance and <5% brain penetration, compared with plasma, in Npc1nmf164 mice are in accordance with observations in normal mice and in a murine model of Huntington's disease (43, 44).
Improved Liver Function Is Independent of Npc1 Expression
To investigate the mechanism by which vorinostat reduced ALT levels, we measured Npc1 mRNA and protein levels to test the hypothesis that vorinostat elevated expression of the NPC1D1005G variant in the Npc1nmf164 mouse. To determine whether vorinostat up-regulated Npc1 mRNA expression, we measured mRNA levels of NPC1 by quantitative real-time PCR (Fig. 8a). Expression of Npc1 was not affected in either genotype with vorinostat treatment, although Npc1 mRNA expression was modestly elevated in vehicle-treated Npc1nmf164 compared with vehicle-treated WT mice (p = 0.066). Quantification of Western blotting analysis of liver protein extracts from treated and untreated animals (n = 7/group) indicated that the vehicle-treated Npc1nmf164 mouse expressed 19% of the amount of Npc1 expressed by vehicle-treated WT mice (p = 5.76 × 10−7; Fig. 8, b and c), consistent with previous reports that the Npc1nmf164 mouse expressed 10–15% of WT levels of NPC1 (17). However, our analysis indicated equivalent steady state levels of NPC1 protein in vorinostat-treated liver and vehicle control-treated livers. The mRNA levels of Npc2 were 4.7-fold higher (p = 0.016) in vehicle-treated Npc1nmf164 compared with vehicle-treated WT mice, a result that has been reported previously for the Npc1−/− null and the Npc1pf/pf missense mutant mouse models of NP-C disease (45, 46). However, there was no significant change in expression of Npc2 in vorinostat-treated Npc1nmf164 compared with vehicle-treated Npc1nmf164 mice (Fig. 8d). These results suggest that the improvements in lipid metabolism and liver function occur independently of the concentration of the NPC1D1005G protein and are not a consequence of vorinostat regulating the NPC1/NPC2 pathways at protein or mRNA levels.
FIGURE 8.

Liver recovery due to vorinostat treatment is independent of changes in expression of Npc1 or Npc2. Animals were treated with either vehicle (PEG/DMSO) or 150 mg/kg vorinostat from P21 to P60. a, liver RNA from the indicated animals was assessed for Npc1 gene expression by qRT-PCR relative to Gapdh. All groups are n = 3 with data shown as mean ± S.D. (error bars). b, representative image of 30 μg of hepatic protein extracted from vehicle- and vorinostat-treated animals that was denatured at 4 °C for 30 min, subjected to electrophoresis, transferred onto PVDF membrane, and incubated with NPC1 antibody or actin antibody. c, NPC1 and actin bands were quantified relative to actin using densitometry software (ImageJ). All groups are n = 7 with data shown as mean ± S.E. *, p < 0.05, Student's t test. d, liver RNA from the indicated animals was assessed for NPC2 gene expression by qRT-PCR relative to GAPDH. All groups are n = 3 with data shown as mean ± S.D. (error bars). *, p < 0.05, Student's t test. e, protein from the indicated animals was extracted, treated with glycosidase enzymes, and analyzed via SDS-PAGE/immunoblotting and probed for NPC1 expression.
Vorinostat Does Not Increase Maturation of the NPC1D1005G Protein
To further understand the lack of impact of vorinostat on expression of the NPC1D1005G protein in the liver of Npc1nmf164 mice, we investigated maturation of the NPC1D1005G protein in the secretory pathway via two glycosidase digestion assays that have been used previously to characterize the NPC1I1061T protein (18, 19). First, we used endoglycosidase H (Endo H), an enzyme that removes immature high mannose N-linked glycans from proteins. Second, we used peptide:N-glycosidase F (PNGase F), an enzyme that removes all N-linked glycan residues regardless of the glycan modification. In livers of vehicle-treated WT mice, the NPC1 protein is glycosidase-sensitive represented by a mature species at ∼225 kDa that is reduced to two deglycosylated species at ∼200 and ∼150 kDa in response to the Endo H and PNGase F treatments, respectively (Fig. 8e). These results indicate that the wild-type NPC1 protein is glycosylated in the ER and further matured in the Golgi. Conversely, the NPC1D1005G protein in both vehicle- and vorinostat-treated Npc1nmf164 mice is predominantly a single species at ∼150 kDa in undigested, Endo H-treated, and PNGase F-treated samples, with electrophoretic mobility similar to that of PNGase F-treated WT protein (Fig. 8e). These results indicate that the majority of NPC1D1005G protein in livers of Npc1nmf164 mice lacks glycosylation and does not exit the ER as a result of misfolding. This defect was not corrected by vorinostat treatment.
Vorinostat Normalizes Apolipoprotein B Metabolism in Npc1 Null and Missense Mutant Hepatocytes
Inhibition of histone deacetylation results in differential expression of 2–10% of the genome (2, 3). In disorders such as cystic fibrosis, the success of vorinostat treatment was, at least in part, due to activation of chaperone-mediated refolding pathways (7). Thus far, our data suggest that vorinostat is therapeutic via a mechanism that does not require functional NPC1 (Figs. 3 and 8). To directly test whether residual levels of NPC1 protein impact the response to vorinostat treatment, we compared primary hepatocytes prepared from Npc1nmf164 and Npc1−/− mice. In contrast to the Npc1nmf164 mice that produce low levels of NPC1 protein, the Npc1−/− model expresses a null variant of the Npc1 gene arising from a transposon insertion at the 5′ end of the gene (47). Metabolic labeling of primary hepatocytes with [35S]methionine followed by immunoprecipitation was used to quantify newly synthesized and secreted isoforms of apolipoprotein B (apoB100 and apoB48). These proteins are major exporters of lipid from the liver (48). As described previously (31), the synthesis of apoB100 by hepatocytes derived from Npc1−/− mice was elevated in response to the lipid accumulation underlying NP-C disease. Similarly, secretion of newly synthesized apoB100 was dramatically increased (507.3%, p = 0.006), and apoB48 was marginally increased (18.8%, p = 0.0007) in Npc1−/− hepatocytes compared with control hepatocytes from WT littermates (Fig. 9, a–c). Despite the absence of the NPC1 protein in these cell lines, the ex vivo treatment of 10 μm vorinostat strikingly reduced secretion of apoB100 by 76% (p = 0.01) to control levels (Fig. 9, a–c). Interestingly, secretion of apoB48 was slightly elevated in vorinostat-treated Npc1−/− hepatocytes compared with vehicle-treated Npc1−/− hepatocytes (23.5%, p = 0.046). ApoB metabolism was also aberrant in primary hepatocytes from Npc1nmf164 mice expressing the NPC1D1005G missense mutant. Unlike the null mutant hepatocytes, secretion levels of apoB100 and apoB48 were significantly reduced by 57.2% (p = 0.04) and 69.6% (p = 0.002), respectively, in vehicle-treated Npc1nmf164 hepatocytes compared with vehicle-treated control hepatocytes (Fig. 9, d–f). Similar to the response of Npc1−/− hepatocytes to vorinostat, secretion of newly synthesized apoB48 levels in media was significantly increased (413.7%, p = 0.0008) in vorinostat-treated Npc1nmf164 hepatocytes compared with vehicle-treated mutant cells (Fig. 9, d–f). Clearly, vorinostat modulated the homeostasis of apoB in both null Npc1−/− and missense Npc1nmf164 hepatocytes, further demonstrating that the mechanism of action of this drug is independent of NPC1.
FIGURE 9.
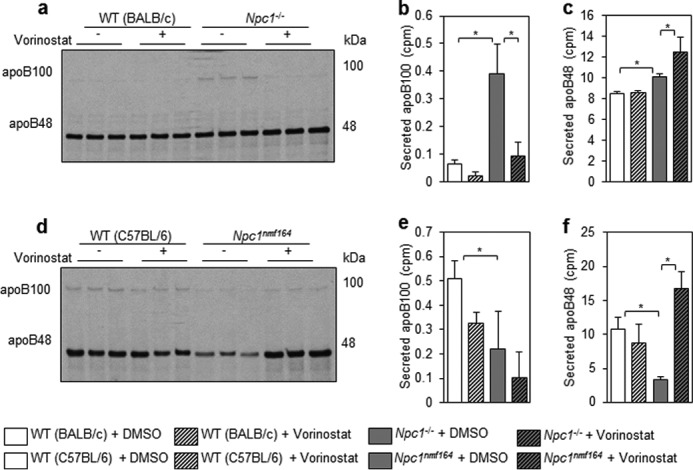
Vorinostat modulates apoB metabolism in Npc1nmf164 and Npc1−/− hepatocytes. Hepatocytes were isolated from livers of Npc1nmf164 and Npc1−/− mice, incubated for 24 h in DMEM + 10% FBS with 10 μm vorinostat or DMSO, and labeled with [35S]methionine in methionine-free DMEM for 2 h. Equal radiolabeled protein was separated on a 4% SDS-polyacrylamide gel to distinguish apoB100 and apoB48 in mutant mice relative to appropriate WT controls. a–c, newly synthesized and secreted apoB in media of Npc1−/− hepatocytes. d–f, newly synthesized and secreted apoB in media of Npc1nmf164 hepatocytes. Data represent mean cpm ± S.D. (error bars) from triplicates. *, p < 0.05, Student's t test.
To further investigate the efficacy of vorinostat in Npc1−/− and Npc1nmf164 primary hepatocytes, we measured expression of several genes integral to lipid metabolism (Npc1, Hmgcr, Abcg1, and Apob) and the cellular response to misfolded proteins (CHOP). The expression of Npc1 was significantly increased by 91% (p = 0.016) in vehicle-treated Npc1nmf164 cells compared with vehicle-treated control hepatocytes, and vorinostat did not impact these expression levels (Fig. 10a), results that are consistent with Npc1 gene expression in the liver of Npc1nmf164 mice compared with controls (Fig. 8a). As expected, expression of Npc1 was significantly reduced (81.4%, p = 0.009) in vehicle-treated Npc1−/− compared with vehicle-treated WT hepatocytes, and interestingly, albeit probably not functional, this was significantly up-regulated following treatment with vorinostat (Fig. 10a). The expression of ApoB was significantly reduced by 50.1% (p = 0.0009) in Npc1nmf164 hepatocytes compared with WT hepatocytes and was markedly elevated by vorinostat treatment beyond control levels in both Npc1nmf164 (p = 0.0007) and Npc1−/− hepatocytes (p = 0.04) (Fig. 10b). The transcription factor, CCAAT/enhancer-binding protein homologous protein (CHOP), an indicator of ER stress that has been associated with disrupted apoB metabolism (49), was significantly increased in Npc1nmf164 hepatocytes (111.6%, p = 0.0006) compared with WT hepatocytes (Fig. 10c). In contrast, the expression of Chop was significantly reduced in Npc1−/− hepatocytes by 40.4% (p = 0.03) compared with control hepatocytes. This dysregulation was reduced further in both mutant cell types by vorinostat treatment (49.9%, p = 0.0003 in Npc1−/− cells; 23.7%, p = 0.001 in Npc1nmf164 cells) (Fig. 10c). These results suggest that vorinostat transcriptionally modulates gene expression in Npc1nmf164 and Npc1−/− hepatocytes independent of the status of the NPC1 locus.
FIGURE 10.
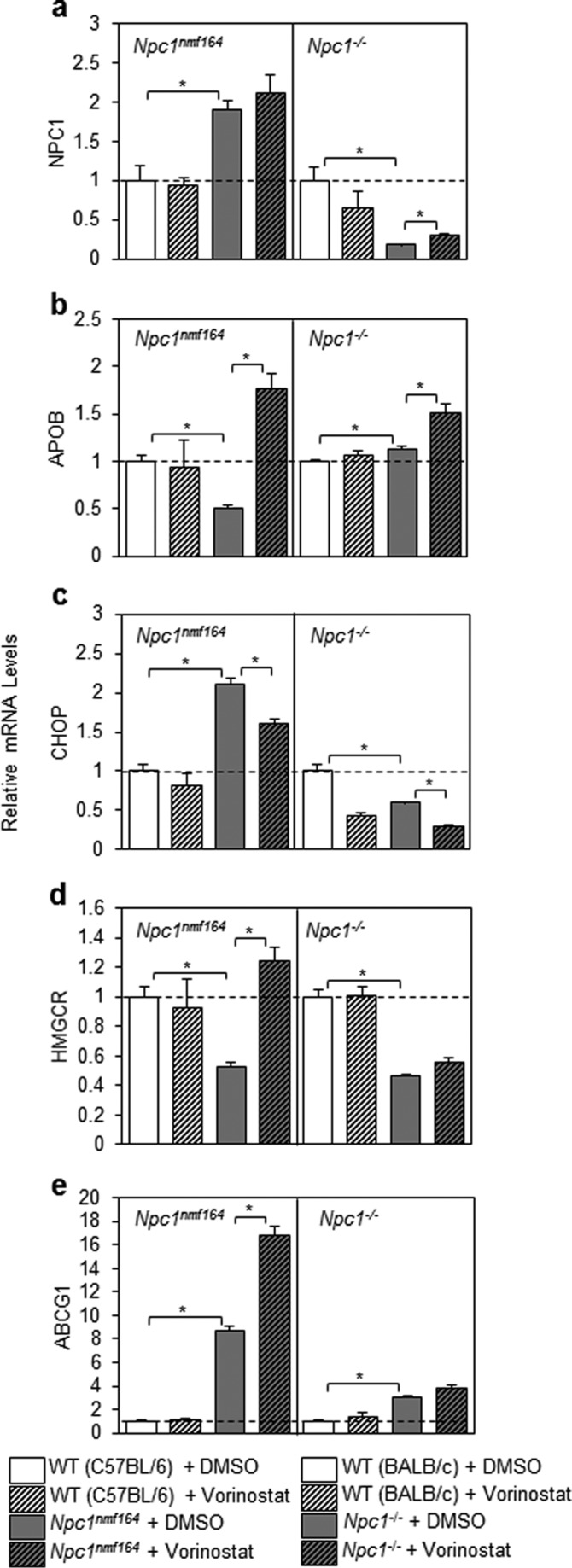
Transcriptional response of Npc1nmf164 and Npc1−/− hepatocytes following treatment with vorinostat. Hepatocytes were isolated from livers of Npc1nmf164 and Npc1−/− mice; incubated for 24 h in DMEM + 10% FBS with 10 μm vorinostat or DMSO; and assessed for Npc1, Apob, Chop, Hmgcr, and Abcg1 gene expression by qRT-PCR relative to cyclophilin (CYCLO) and appropriate WT controls using primers described under “Experimental Procedures.” All groups are n = 3 with data shown as mean ± S.D. (error bars) *, p < 0.05, Student's t test.
In certain instances, the transcriptional response to vorinostat by primary hepatocytes was more striking in the presence of residual NPC1 protein (i.e. in Npc1nmf164 hepatocytes). Relative to controls, expression of Hmgcr was significantly reduced by 47.5% (p = 0.004) in Npc1nmf164 and by 53.4% (p = 0.0003) in Npc1−/− hepatocytes (Fig. 10d), consistent with previously reported levels of processed (nuclear) SREBP2 in Npc1−/− hepatocytes (31). Interestingly, vorinostat significantly increased Hmgcr expression to control levels only in Npc1nmf164 hepatocytes (136.5%, p = 0.002). Similarly, the expression of Abcg1 was significantly increased in Npc1nmf164 and Npc1−/− hepatocytes by 769.7% (p = 3.3 × 10−5) and 206.7% (p = 0.0003), respectively, compared with control hepatocytes (Fig. 10e), results that are in agreement with the previous demonstration of increased expression of Abcg1, a transporter that regulates efflux to HDL, in Npc1−/− hepatocytes (50). Vorinostat treatment significantly increased Abcg1 expression but only in Npc1nmf164 hepatocytes (92.7%, p = 0.001), a mechanism consistent with observations on the anti-atherogenic effects of HDAC inhibition (51).
Discussion
The identification of FDA-approved drugs to treat NP-C disease remains elusive; in nearly 2 decades of effort since the molecular isolation of the NPC1 gene (52), only 2-hydroxypropyl-β-cyclodextrin, the excipient of a failed neurosteroid treatment (53), has progressed to a Phase 3 clinical trial. As an alternate strategy, we applied unbiased, genome-wide, genetic screens in yeast and identified histone acetylation as a negative modifier of the NP-C disease pathway (11). Subsequently, we and others translated this finding by demonstrating the efficacy of HDAC inhibitors to reduce lipid accumulation in NP-C patient fibroblasts (11–13). We hypothesized that this therapeutic effect would be conserved in vivo and that the pathway to regulatory approval of vorinostat for this orphan disease would be relatively rapid and unencumbered. In the present study, we tested the impact of intraperitoneal injection of vorinostat in the Npc1nmf164 mouse model of NP-C disease. We carefully reviewed toxicity and administration of vorinostat and used a concentration and dosing regimen that was the maximum tolerated dose in this animal. This regimen exceeded that used to treat murine models of cancer (the equivalent of a human dose of 400 mg/day). We were successful in that vorinostat treatment was therapeutic for visceral NP-C disease, as evidenced by normalized liver lipid homeostasis at the transcriptional level and marked improvement of liver pathology and function. However, aspects of disease progression such as morbidity, mortality, and neurodegeneration were not impacted, due, at least in part, to ineffective brain penetration.
We interpret these findings as a very promising step toward applying HDAC inhibitors as therapeutics for this disease; however, the manner by which HDAC inhibitors may act remains to be determined. HDAC inhibitors impact expression of the genome in a global fashion, with immense capacity to simultaneously alter numerous metabolic pathways. Indeed, our results show that vorinostat altered the expression of ∼6% of the liver transcriptome of Npc1nmf164 mice, a result consistent with previous reports of vorinostat regulating 2–10% of the genome (2, 3). Most notably, we detected transcriptional regulation of genes involved in cholesterol homeostasis that correlate with reversal of NP-C disease. The Hmgcs1, Hmgcr, Mvk, Mvd, Idi1, Lss, Cyp51, and Ldlr genes were repressed by vorinostat only in the Npc1nmf164 mice, reflecting the successful treatment of hepatic NP-C disease as well as a therapeutic mechanism that is independent of Npc1. Interestingly, HDAC inhibitors have also been shown to repress expression of cholesterol homeostatic genes in U18666A-treated SH-SY5Y human neuroblastoma cells (54). HDAC inhibitors can further impact expression of the genome by directing recovery of misfolded mutated proteins via the induction of chaperone pathways (7). In Npc1nmf164 mice, the NPC1D1005G variant protein persisted as an unglycosylated species that could be chased through the secretion pathway into a fully glycosylated mature species either by a refolding agent, such as glycerol, or by protection from degradation. However, our studies demonstrate that this is not the predominant mechanism by which vorinostat impacts NP-C disease in these animals.
Despite the overall transcriptional responsiveness of the Npc1nmf164 missense mutant mouse, the vorinostat treatment protocol used here did not detectably alter hepatic expression of Npc1 at either mRNA or protein levels or in terms of its maturation through the secretion pathway. Moreover, when we compared apoB homeostasis in primary hepatocytes from both missense (Npc1nmf164) and null (Npc1−/−) mice, we concluded that vorinostat normalized both genotypes, relative to their own age-, gender-, and background-matched controls. Interestingly, apoB levels were significantly increased in Npc1−/− mice in the BALB/c background and significantly decreased in Npc1nmf164 mice in the C57BL/6 background, differences that may be consequences of the strain background and/or the Npc1 mutation. There is precedence for the impact of mouse background in the context of Npc1 missense mutations (e.g. different lipidomic profiles in NPC1I1061T mice in the BALB/c and C57BL/6 backgrounds) (19). Similarly, there is precedence for varying effects of the ∼300 disease-causing mutations in NPC1 on the clinical progression of NP-C disease (8, 55–57). Regarding the efficacy of vorinostat, the extent of misfolding and feasibility of refolding will be critical to decipher for different mutations in NPC1. Our results suggest that vorinostat would be therapeutic in the liver of all NP-C patients regardless of mutation because the mechanism underlying improvement was predominantly independent of Npc1 expression.
It is clear that the bioavailability and stability of vorinostat represent a significant but, we believe, surmountable hurdle to further progress. We determined that i.p. administration and a PEG/DMSO vehicle result in vorinostat levels at <5 μm in the brain, 30 min after injection, a result in stark contrast to the 60 μm concentration in plasma at the same juncture. Concentrations of 5 μm are necessary to reduce lipid accumulation with 24-h treatment in cell culture (11–13). Given the 12-min half-life of vorinostat in rats and dogs (58) compared with the effectively infinite half-life in cell culture, it is plausible that >5 μm is necessary to reduce lipid accumulation in the murine brain. Interestingly, using the PEG/DMSO vehicle and the 150 mg/kg dose in this study, i.p. administered vorinostat reduced the tumor size in the brain of a mouse model of brain cancer (20), a result that was attributed to a disrupted blood-brain barrier in brain cancer models (44). It is widely accepted that the murine and feline models of NP-C disease do not have a compromised blood-brain barrier (59, 60). The blood-brain barrier could be circumvented by a variety of strategies. Rescue of neurodegenerative symptoms in mouse models of Huntington's disease have been accomplished by dissolving vorinostat in 2-hydroxypropyl-β-cyclodextrin (5, 43). This is interesting, because although 2-hydroxypropyl-β-cyclodextrin is a compound not known to cross the blood-brain barrier, perhaps it improves blood-brain barrier penetration of vorinostat in the Huntington's disease studies. Intriguingly, 2-hydroxypropyl-β-cyclodextrin alone, when administered directly into the CNS, is itself a promising therapeutic to treat NP-C disease (14, 42) and is currently being evaluated in a clinical trial. Indeed, Alam et al. (61) recently demonstrated a striking synergy (∼2-fold life extension, relative to 2-hydroxypropyl-β-cyclodextrin monotherapy) when treating Npc1nmf164 mice (in a BALB/c background) by combining high concentrations of vorinostat with therapeutic doses of 2-hydroxypropyl-β-cyclodextrin in a PEG/DMSO vehicle. Although the confounding effect of using already therapeutic doses of 2-hydroxypropyl-β-cyclodextrin and possibly PEG/DMSO remains to be determined, it is apparent that the pharmacokinetics of vorinostat can be improved.
In summary, NP-C disease is a fatal pediatric neurodegenerative disease due to lysosomal accumulation of cholesterol and sphingolipids. Currently, there is no effective FDA-approved therapy to treat NP-C disease. Here we have translated prior results in a yeast model of NP-C disease and NP-C patient fibroblasts (11) to the Npc1nmf164 mouse model of NP-C disease, wherein we report that a marked improvement in liver function was accompanied by normalized expression of key hepatic homeostatic genes in vorinostat-treated mice. With these data, we provide proof of principle that HDAC inhibition has the potential to be therapeutic in vivo in an animal model of NP-C disease. Indeed, the potential of vorinostat to ameliorate peripheral NPC disease in adults is currently the object of a Phase 1/2 clinical trial (ClinicalTrials.gov number NCT02124083).
Our results indicate that vorinostat treatment limits the aberrant up-regulation of cholesterol biosynthesis and uptake that arises from lysosomal sequestration of cholesterol in NP-C disease. Consequently, this markedly improves the visceral pathophysiology of this disease. With improved blood-brain barrier penetration, we propose that vorinostat and other HDAC inhibitors may prove useful for treatment of the neurological aspects of NP-C disease.
Experimental Procedures
Animals
Animal husbandry and experiments were approved by the Columbia University and Victoria University animal ethics committees. Animals were exposed to alternating 12-h periods of dark and light and were fed a standard diet, ad libitum, until dissection. Npc1nmf164 mutant and WT littermates (C57BL/6 background) were generated by crossing heterozygous Npc1nmf164 males and females. Npc1 genotyping was conducted by restriction digests of PCR amplicons as described (17). Mice were i.p. administered with vorinostat (LC Laboratories) in 10% DMSO and 45% PEG 400, as described previously (20), from P21 to P60 (for all in vivo experiments except the body weight/life span experiments, which were continued to P90). Disease progression was monitored in terms of weekly weight gain from P21 to P90. For all experiments except the pharmacokinetic assay, Npc1nmf164 mutant mice and WT mice were anesthetized 6 h after the last injection with an i.p. administered injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), serum was collected, and animals were transcardially perfused with 0.9% saline solution. Following perfusion, half of the cerebrum and liver were removed and immediately frozen at −80 °C for genetic and biochemical analysis with the other half fixed in 4% paraformaldehyde for histological analysis.
Primary Hepatocytes and Cell Culture
Primary hepatocytes were isolated from age- and background-matched control, Npc1nmf164 (C57BL/6 background), and Npc1−/− (BALB/c background, generously provided by X. Huang and S. Walkley) mice (62). Filtered, washed cells were plated into collagen-coated 6-well plates at a density of 500,000 viable cells/well in DMEM + 10% FBS. Fibroblasts derived from Npc1nmf164 mutant mice and wild-type littermates or NPC1-deficient human osteosarcoma U2OS cells expressing the NPC1I1061T protein were generated and maintained as described previously (63).
ApoB Metabolism
For steady-state apoB labeling and immunoprecipitation, primary hepatocytes were incubated in methionine-free DMEM for 1 h and then labeled with [35S]methionine in methionine-free DMEM for 2 h (64). Cells were collected in 200 μl of lysis buffer (62.5 mm sucrose, 0.5% sodium deoxycholate, 0.5% Triton X-100, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm benzamidine, 5 mm EDTA, 100 units/ml aprotinin, 50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 10 mm HEPES, pH 8.0) and boiled with sample buffer for 5 min. Trichloroacetic acid total precipitable 35S counts of the cell lysate were determined and equal radiolabeled intracellular and medium protein was processed by immunoprecipitation (anti-lipoprotein B; Calbiochem) and separated on 4% SDS-polyacrylamide gels. The gels were dried under vacuum at 65 °C for 2 h and exposed to X-ray films to visualize apoB100 and apoB48.
RNA-Seq Analysis
Total RNA was extracted from livers of Npc1nmf164 vehicle control and vorinostat-treated mice (7 animals/group) using the PureLink RNA minikit (Life Technologies) and processed for RNA-Seq analyses at the Australian Genome Research Facility. RNA-Seq libraries were prepared using the TruSeq RNA version 2 kit (Illumina) and quantified using Bioanalyzer (Agilent). The prepared libraries were sequenced using an Illumina HiSeq 2000 (100-bp single end reads). The reads were mapped to the Mus musculus genome (Build version mm10) using TopHat (version 2.0.13). Transcripts were assembled utilizing reference-based annotation with Cufflinks (version 2.2.1). Gene expression was normalized and quantified using trimmed mean of M values. Differential gene expression analysis of vorinostat-treated mice relative to vehicle-treated mice was calculated using log2 ratio (-fold change) and statistically evaluated using edgeR. The data set has been deposited in the NCBI GEO database. Gene ontology processes of differentially expressed genes were classified using PANTHER (protein analysis through evolutionary relationships) and MGD (Mouse Genome Database) (65, 66). The web-based Gene Set Analysis Toolkit (34) was used to conduct enrichment analyses of functions and pathways. Phenolyzer (35) was used to identify core components of the vorinostat interactome.
qRT-PCR
Gene expression was measured using qRT-PCR. For validation of RNA-Seq results, 80 ng of total RNA was converted to mRNA using the Power SYBR Green RNA-to-CT kit (Applied Biosystems) and quantified using SYBR Green chemistry and a CFX Connect real-time PCR detection system (Bio-Rad). For characterization of primary hepatocytes, RNA was isolated from cells with TRIzol (Invitrogen) and was used as a template in qRT-PCR using an Applied Biosystems 7900HT sequence detection system. The mRNA levels were calculated by the 2−ΔCT method (67) using the following 5′ to 3′ primer sequences: Abcg1 (forward, GCTGTGCGTTTTGTGCTGTT; reverse, TGCAGCTCCAATCAGTAGTCCTAA) (14); ApoB (forward, CTGGGCTCCAGCATTCTA; reverse, TCACCAGTCATTTCTGCCTTTG) (68); Chop (forward, CACCACACCTGAAAGCAGAAC; reverse, GGTGAAAGGCAGGGACTCA) (69); Cyclo (forward, TGGAGAGCACCAAGACAGACA; reverse, TGCCGGAGTCGACAATGAT) (70); Cyp51 (forward, CCTGGATGGAGGTTTTACCC; reverse, TCTCTCGATGGGCTCTATCC); Gapdh (forward, TGGCGCCTGTGTCTGTGT; reverse, TCGGGTCTGATTTCTGTTTCTTC) (29); Hmgcr (forward, CTTGTGGAATGCCTTGTGATTG; reverse, AGCCGAAGCAGCACATGAT) (14); Hmgcs (forward, GCCGTGAACTGGGTCGAA; reverse, GCATATATAGCAATGTCTCCTGCAA) (14); Idi1 (forward, AAAGCCGAGTTGGGAATACCC; reverse, ACCATCAGATTGGGCCTTGTA); Ldlr (forward, GAGGAACTGGCGGCTGAA; reverse, GTGCTGGATGGGGAGGTCT) (14); Lss (forward, GCGGCTGTGCGATGCT; reverse, AGGTAGCGAACCCGCCA); Mvd (forward, CCGGTCAACATCGCAGTTATC; reverse, TTGTGGTCGTTTTTAGCTGGT); Mvk (forward, GGTGTGGTCGGAACTTCCC; reverse, CCTTGAGCGGGTTGGAGAC); Npc1 (forward, AACCGTGACACTGCAGGACAT; reverse, CTCATAATGGTGCAGTTCTTGTTGT) (46); and Npc2 (forward, TGGCGCCTGTGTCTGTGT; reverse, TCGGGTCTGATTTCTGTTTCTTC) (46). Primers without references were designed using Primer-BLAST (71).
Immunoblotting and Deglycosylation
Frozen liver was homogenized in radioimmune precipitation assay buffer with the addition of protease inhibitors (Roche Applied Science) and centrifuged at 16,000 × g. Protein concentration was measured using the BCA protein assay (Bio-Rad). For deglycosylation, protein extracts were combined with glycoprotein denaturation buffer (5% SDS, 0.4 m DTT), denatured at 65 °C for 10 min, treated with either GlycoBuffer 3 (New England Biolabs) and 1000 units of Endo H (New England Biolabs) or GlycoBuffer 2 (New England Biolabs) with 1% Nonidet P-40 and 1000 units of PNGase F (New England Biolabs), and incubated at 37 °C for 7 h. Protein was denatured at 4 °C for 30 min, loaded onto a 7.5% BisTris gel, subjected to electrophoresis, transferred onto 0.2 μm PVDF membranes using a semidry system (Bio-Rad), and incubated with NPC1 antibody (Abcam ab134113 or Abcam ab36983) or actin antibody (Sigma). Membranes were probed with goat anti-mouse or goat anti-rabbit secondary antibodies (GE Healthcare), visualized using ECLplus (GE Healthcare) with a fluorescent image analyzer (Fuji FLA-5100), and quantified using densitometry software (ImageJ).
NPC1 Protein Refolding
Cells were incubated in the presence and absence of glycerol, 3-methyladenine, MG132, or lactacystin for 20 h at 37 °C, lysed in 100 mm sodium phosphate, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% IGEPAL CA-630 for 15 min at 4 °C, and centrifuged for 5 min at 14,000 rpm at 4 °C. Total cell lysates were denatured at 70 °C for 10 min, subjected to SDS-PAGE through a 3–8% Tris acetate precast gel (Invitrogen), transferred to nitrocellulose membrane (Protran), and probed using primary antibodies against NPC1 (Abcam ab134113) or Hsp70 (Millipore) and a secondary HRP-conjugated rabbit IgG (GE Healthcare). Proteins were visualized by chemiluminescence using SuperSignal West Dura substrate (Thermo Scientific) on a FluorChemQ imager (ProteinSimple) using AlphaView software (Alpha Innotech).
Pharmacokinetics
Vorinostat (150 mg/kg) was administered as described above with one injection in Npc1nmf164 mice. Plasma and brain were collected at 30, 60, 120, and 240 min after the injection. Vorinostat levels were measured using LC-MS/MS as described previously (43).
Serum Analysis
Serum measurements of ALT total cholesterol, HDL cholesterol, LDL cholesterol, and triglyceride were assessed at a commercial laboratory (New Zealand Veterinary Pathology) as part of a general metabolic profile.
Histology
Fixed liver tissue was paraffin-embedded, and 5-μm sections were stained with H&E by the Columbia University Medical Center Molecular Pathology Core Facility. The prevalence of lipid-laden cells was expressed as a fraction of total cells per field and quantified using SlidePath tissue image analysis (Leica Biosystems, version 2.0) software at ×20 magnification.
Lipids
Homogenized frozen tissue (half liver lobe) was used to prepare lipids by a modified Bligh and Dyer extraction procedure (72). Free cholesterol, sphingomyelin, sphingosine, glucosylceramide, and lactosylceramide were measured using LC/MS as described previously (73, 74) modified for use with a Shimadzu 8040 LC/MS system, an APCI source with MRM detection, and a Waters Acquity BEH C18 1.7-μm, 2.1 × 150-mm column. Standards (Avanti Polar Lipids) were used for quantification, and all measurements were normalized to liver weight. Subcellular accumulation of free cholesterol in liver sections was visualized using fluorescence of filipin as described previously (42) and quantified using densitometry software (ImageJ).
Author Contributions
A. B. M., N. H., and S. L. S. conceived, coordinated, and conducted the study and wrote the paper. R. T. S. and R. A. M. contributed to the toxicity study and the development of the drug administration protocol used for in vivo experiments. F. W. C. and Y. A. I. provided technical assistance for the experiment in Fig. 1. D. S. S. provided technical assistance and contributed to the preparation of Fig. 2. K. L. and S. J. B. provided technical assistance for the experiment shown in Fig. 6, a–e. D. S. O. designed, performed, and analyzed the experiments shown in Fig. 7, c and d. K. H. performed the experiment shown in Fig. 8e. A. H.-O. and H. N. G. provided technical assistance for the experiments in Figs. 9 and 10. N. D. and J. J. R. provided technical assistance for the experiment in Fig. 10. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful for the support of Rodney Rothstein and Michael Shelanski in the final stages of this study.
This work was supported by grants from the Ara Parseghian Medical Research Foundation (to S. L. S. and J. J. R.) and Dana's Angels Research Trust (to S. L. S.) and by National Institutes of Health Grants DK54320 (to S. L. S.), DK082712 (to Y. A. I.), NS092653 (to D. S. O.), and HL55638 (to H. N. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table 1.
- HDAC
- histone deacetylase
- NP-C
- Niemann-Pick type C
- FDA
- Food and Drug Administration
- P
- postnatal day
- RNA-Seq
- RNA sequencing
- qRT-PCR
- quantitative RT-PCR
- ER
- endoplasmic reticulum
- ALT
- alanine aminotransferase
- Endo H
- endoglycosidase H
- PNGase F
- peptide:N-glycosidase F
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Dokmanovic M., Clarke C., and Marks P. A. (2007) Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 5, 981–989 [DOI] [PubMed] [Google Scholar]
- 2. Richon V. M., Sandhoff T. W., Rifkind R. A., and Marks P. A. (2000) Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. U.S.A. 97, 10014–10019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitsiades C. S., Mitsiades N. S., McMullan C. J., Poulaki V., Shringarpure R., Hideshima T., Akiyama M., Chauhan D., Munshi N., Gu X., Bailey C., Joseph M., Libermann T. A., Richon V. M., Marks P. A., and Anderson K. C. (2004) Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl. Acad. Sci. U.S.A. 101, 540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Falkenberg K. J., and Johnstone R. W. (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug. Discov. 13, 673–691 [DOI] [PubMed] [Google Scholar]
- 5. Hockly E., Richon V. M., Woodman B., Smith D. L., Zhou X., Rosa E., Sathasivam K., Ghazi-Noori S., Mahal A., Lowden P. A., Steffan J. S., Marsh J. L., Thompson L. M., Lewis C. M., Marks P. A., and Bates G. P. (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2041–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herman D., Jenssen K., Burnett R., Soragni E., Perlman S. L., and Gottesfeld J. M. (2006) Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol. 2, 551–558 [DOI] [PubMed] [Google Scholar]
- 7. Hutt D. M., Herman D., Rodrigues A. P., Noel S., Pilewski J. M., Matteson J., Hoch B., Kellner W., Kelly J. W., Schmidt A., Thomas P. J., Matsumura Y., Skach W. R., Gentzsch M., Riordan J. R., et al. (2010) Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 6, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vanier M. T. (2010) Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Munkacsi A. B., Porto A. F., and Sturley S. L. (2007) Niemann-Pick type C disease proteins: orphan transporters or membrane rheostats? Future Lipidol. 2, 357–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopez M. E., and Scott M. P. (2013) Genetic dissection of a cell-autonomous neurodegenerative disorder: lessons learned from mouse models of Niemann-Pick disease type C. Dis. Model Mech. 6, 1089–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munkacsi A. B., Chen F. W., Brinkman M. A., Higaki K., Gutiérrez G. D., Chaudhari J., Layer J. V., Tong A., Bard M., Boone C., Ioannou Y. A., and Sturley S. L. (2011) An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 286, 23842–23851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pipalia N. H., Cosner C. C., Huang A., Chatterjee A., Bourbon P., Farley N., Helquist P., Wiest O., and Maxfield F. R. (2011) Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 108, 5620–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wehrmann Z. T., Hulett T. W., Huegel K. L., Vaughan K. T., Wiest O., Helquist P., and Goodson H. (2012) Quantitative comparison of the efficacy of various compounds in lowering intracellular cholesterol levels in Niemann-Pick type C fibroblasts. PLoS One 7, e48561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu B., Turley S. D., Burns D. K., Miller A. M., Repa J. J., and Dietschy J. M. (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. U.S.A. 106, 2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor A. M., Liu B., Mari Y., Liu B., and Repa J. J. (2012) Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream. J. Lipid Res. 53, 2331–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wienkers L. C., and Heath T. G. (2005) Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug. Discov. 4, 825–833 [DOI] [PubMed] [Google Scholar]
- 17. Maue R. A., Burgess R. W., Wang B., Wooley C. M., Seburn K. L., Vanier M. T., Rogers M. A., Chang C. C., Chang T. Y., Harris B. T., Graber D. J., Penatti C. A., Porter D. M., Szwergold B. S., Henderson L. P., Totenhagen J. W., Trouard T. P., Borbon I. A., and Erickson R. P. (2012) A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 21, 730–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gelsthorpe M. E., Baumann N., Millard E., Gale S. E., Langmade S. J., Schaffer J. E., and Ory D. S. (2008) Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 283, 8229–8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Praggastis M., Tortelli B., Zhang J., Fujiwara H., Sidhu R., Chacko A., Chen Z., Chung C., Lieberman A. P., Sikora J., Davidson C., Walkley S. U., Pipalia N. H., Maxfield F. R., Schaffer J. E., and Ory D. S. (2015) A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 35, 8091–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmieri D., Lockman P. R., Thomas F. C., Hua E., Herring J., Hargrave E., Johnson M., Flores N., Qian Y., Vega-Valle E., Taskar K. S., Rudraraju V., Mittapalli R. K., Gaasch J. A., Bohn K. A., et al. (2009) Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin. Cancer Res. 15, 6148–6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu Y., Ping J., Chen H., Jiao L., Zheng S., Han Z. G., Hao P., and Huang J. (2010) A comparative analysis of liver transcriptome suggests divergent liver function among human, mouse and rat. Genomics 96, 281–289 [DOI] [PubMed] [Google Scholar]
- 22. Marks P. A., and Breslow R. (2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 [DOI] [PubMed] [Google Scholar]
- 23. Su L., Cheng H., Sampaio A. V., Nielsen T. O., and Underhill T. M. (2010) EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor. Oncogene 29, 4352–4361 [DOI] [PubMed] [Google Scholar]
- 24. Claerhout S., Lim J. Y., Choi W., Park Y. Y., Kim K., Kim S. B., Lee J. S., Mills G. B., and Cho J. Y. (2011) Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS One 6, e24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yin L., and Lazar M. A. (2005) The orphan nuclear receptor Rev-erbα recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 19, 1452–1459 [DOI] [PubMed] [Google Scholar]
- 26. Liu Y., He G., Wang Y., Guan X., Pang X., and Zhang B. (2013) MCM-2 is a therapeutic target of Trichostatin A in colon cancer cells. Toxicol. Lett. 221, 23–30 [DOI] [PubMed] [Google Scholar]
- 27. Halsall J. A., Turan N., Wiersma M., and Turner B. M. (2015) Cells adapt to the epigenomic disruption caused by histone deacetylase inhibitors through a coordinated, chromatin-mediated transcriptional response. Epigenetics Chromatin 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stimson L., and La Thangue N. B. (2009) Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett. 280, 177–183 [DOI] [PubMed] [Google Scholar]
- 29. Alam M. S., Getz M., Safeukui I., Yi S., Tamez P., Shin J., Velázquez P., and Haldar K. (2012) Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS One 7, e48273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alam M. S., Getz M., Yi S., Kurkewich J., Safeukui I., and Haldar K. (2014) Plasma signature of neurological disease in the monogenetic disorder Niemann-Pick Type C. J. Biol. Chem. 289, 8051–8066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kulinski A., and Vance J. E. (2007) Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J. Biol. Chem. 282, 1627–1637 [DOI] [PubMed] [Google Scholar]
- 32. Cluzeau C. V., Watkins-Chow D. E., Fu R., Borate B., Yanjanin N., Dail M. K., Davidson C. D., Walkley S. U., Ory D. S., Wassif C. A., Pavan W. J., and Porter F. D. (2012) Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 21, 3632–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Németh J., Stein I., Haag D., Riehl A., Longerich T., Horwitz E., Breuhahn K., Gebhardt C., Schirmacher P., Hahn M., Ben-Neriah Y., Pikarsky E., Angel P., and Hess J. (2009) S100A8 and S100A9 are novel nuclear factor κB target genes during malignant progression of murine and human liver carcinogenesis. Hepatology 50, 1251–1262 [DOI] [PubMed] [Google Scholar]
- 34. Wang J., Duncan D., Shi Z., and Zhang B. (2013) WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41, W77–W83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H., Robinson P. N., and Wang K. (2015) Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 12, 841–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han Y. P., Kong M., Zheng S., Ren Y., Zhu L., Shi H., and Duan Z. (2013) Vitamin D in liver diseases: from mechanisms to clinical trials. J. Gastroenterol. Hepatol. 28, 49–55 [DOI] [PubMed] [Google Scholar]
- 37. Li J. Z., Huang Y., Karaman R., Ivanova P. T., Brown H. A., Roddy T., Castro-Perez J., Cohen J. C., and Hobbs H. H. (2012) Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Invest. 122, 4130–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vance D. E., and Vance J. E. (2009) Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J. Lipid Res. 50, S132–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sayre N. L., Rimkunas V. M., Graham M. J., Crooke R. M., and Liscum L. (2010) Recovery from liver disease in a Niemann-Pick type C mouse model. J. Lipid Res. 51, 2372–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fan M., Sidhu R., Fujiwara H., Tortelli B., Zhang J., Davidson C., Walkley S. U., Bagel J. H., Vite C., Yanjanin N. M., Porter F. D., Schaffer J. E., and Ory D. S. (2013) Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling. J. Lipid Res. 54, 2800–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davidson C. D., Fishman Y. I., Puskás I., Szemán J., Sohajda T., McCauliff L. A., Sikora J., Storch J., Vanier M. T., Szente L., Walkley S. U., and Dobrenis K. (2016) Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol. 3, 366–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., and Walkley S. U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4, e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mielcarek M., Benn C. L., Franklin S. A., Smith D. L., Woodman B., Marks P. A., and Bates G. P. (2011) SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease. PLoS One 6, e27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hanson J. E., La H., Plise E., Chen Y. H., Ding X., Hanania T., Sabath E. V., Alexandrov V., Brunner D., Leahy E., Steiner P., Liu L., Scearce-Levie K., and Zhou Q. (2013) SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS One 8, e69964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aqul A., Liu B., Ramirez C. M., Pieper A. A., Estill S. J., Burns D. K., Liu B., Repa J. J., Turley S. D., and Dietschy J. M. (2011) Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31, 9404–9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xie X., Brown M. S., Shelton J. M., Richardson J. A., Goldstein J. L., and Liang G. (2011) Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 15330–15335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., Pentchev P. G., and Pavan W. J. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235 [DOI] [PubMed] [Google Scholar]
- 48. Ota T., Gayet C., and Ginsberg H. N. (2008) Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J. Clin. Invest. 118, 316–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Conlon D. M., Thomas T., Fedotova T., Hernandez-Ono A., Di Paolo G., Chan R. B., Ruggles K., Gibeley S., Liu J., and Ginsberg H. N. (2016) Inhibition of apolipoprotein B synthesis stimulates endoplasmic reticulum autophagy that prevents steatosis. J. Clin. Invest. 126, 3852–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang M. D., Franklin V., Sundaram M., Kiss R. S., Ho K., Gallant M., and Marcel Y. L. (2007) Differential regulation of ATP binding cassette protein A1 expression and ApoA-I lipidation by Niemann-Pick type C1 in murine hepatocytes and macrophages. J. Biol. Chem. 282, 22525–22533 [DOI] [PubMed] [Google Scholar]
- 51. Van den Bossche J., Neele A. E., Hoeksema M. A., de Heij F., Boshuizen M. C., van der Velden S., de Boer V. C., Reedquist K. A., and de Winther M. P. (2014) Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem. Biophys. Res. Commun. 455, 396–402 [DOI] [PubMed] [Google Scholar]
- 52. Carstea E. D., Morris J. A., Coleman K. G., Loftus S. K., Zhang D., Cummings C., Gu J., Rosenfeld M. A., Pavan W. J., Krizman D. B., Nagle J., Polymeropoulos M. H., Sturley S. L., Ioannou Y. A., Higgins M. E., et al. (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277, 228–231 [DOI] [PubMed] [Google Scholar]
- 53. Griffin L. D., Gong W., Verot L., and Mellon S. H. (2004) Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 10, 704–711 [DOI] [PubMed] [Google Scholar]
- 54. Nunes M. J., Moutinho M., Gama M. J., Rodrigues C. M., and Rodrigues E. (2013) Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS One 8, e53394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Garver W. S., Francis G. A., Jelinek D., Shepherd G., Flynn J., Castro G., Walsh Vockley C., Coppock D. L., Pettit K. M., Heidenreich R. A., and Meaney F. J. (2007) The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am. J. Med. Genet. A 143A, 1204–1211 [DOI] [PubMed] [Google Scholar]
- 56. Runz H., Dolle D., Schlitter A. M., and Zschocke J. (2008) NPC-db, a Niemann-Pick type C disease gene variation database. Hum. Mutat. 29, 345–350 [DOI] [PubMed] [Google Scholar]
- 57. Stampfer M., Theiss S., Amraoui Y., Jiang X., Keller S., Ory D. S., Mengel E., Fischer C., and Runz H. (2013) Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J. Rare Dis. 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sandhu P., Andrews P. A., Baker M. P., Koeplinger K. A., Soli E. D., Miller T., and Baillie T. A. (2007) Disposition of vorinostat, a novel histone deacetylase inhibitor and anticancer agent, in preclinical species. Drug Metab. Lett. 1, 153–161 [DOI] [PubMed] [Google Scholar]
- 59. Pontikis C. C., Davidson C. D., Walkley S. U., Platt F. M., and Begley D. J. (2013) Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J. Inherit. Metab. Dis. 36, 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vite C. H., Bagel J. H., Swain G. P., Prociuk M., Sikora T. U., Stein V. M., O'Donnell P., Ruane T., Ward S., Crooks A., Li S., Mauldin E., Stellar S., De Meulder M., Kao M. L., et al. (2015) Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 7, 276ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alam M. S., Getz M., and Haldar K. (2016) Chronic administration of an HDAC inhibitor treats both neurological and systemic Niemann-Pick type C disease in a mouse model. Sci. Transl. Med. 8, 326ra23. [DOI] [PubMed] [Google Scholar]
- 62. Smedsrod B., and Pertøft H. (1985) Preparation of pure hepatocytes and reticuloendothelial cells in high yield from a single rat liver by means of Percoll centrifugation and selective adherence. J. Leukoc. Biol. 38, 213–230 [DOI] [PubMed] [Google Scholar]
- 63. Chen F. W., Gordon R. E., and Ioannou Y. A. (2005) NPC1 late endosomes contain elevated levels of non-esterified (“free”) fatty acids and an abnormally glycosylated form of the NPC2 protein. Biochem. J. 390, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dixon J. L., Furukawa S., and Ginsberg H. N. (1991) Oleate stimulates secretion of apolipoprotein B-containing lipoproteins from Hep G2 cells by inhibiting early intracellular degradation of apolipoprotein B. J. Biol. Chem. 266, 5080–5086 [PubMed] [Google Scholar]
- 65. Mi H., Muruganujan A., Casagrande J. T., and Thomas P. D. (2013) Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eppig J. T., Blake J. A., Bult C. J., Kadin J. A., Richardson J. E., and Mouse Genome Database Group (2015) The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 43, D726–D736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schmittgen T. D., and Livak K. J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 68. Tian J., Goldstein J. L., and Brown M. S. (2016) Insulin induction of SREBP-1c in rodent liver requires LXRα-C/EBPβ complex. Proc. Natl. Acad. Sci. U.S.A. 113, 8182–8187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chuang J. C., Cha J. Y., Garmey J. C., Mirmira R. G., and Repa J. J. (2008) Research resource: nuclear hormone receptor expression in the endocrine pancreas. Mol. Endocrinol. 22, 2353–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cha J. Y., and Repa J. J. (2007) The liver X receptor (LXR) and hepatic lipogenesis: the carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 282, 743–751 [DOI] [PubMed] [Google Scholar]
- 71. Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., and Madden T. L. (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bligh E. G., and Dyer W. J. (1959) (1959) A rapid method of total lipid extraction and purification. Can J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 73. Butovich I. A. (2009) Cholesteryl esters as a depot for very long chain fatty acids in human meibum. J. Lipid Res. 50, 501–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shaner R. L., Allegood J. C., Park H., Wang E., Kelly S., Haynes C. A., Sullards M. C., and Merrill A. H. Jr. (2009) Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 50, 1692–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.