

Abstract
Alterations in mitochondrial function contribute to diabetic cardiomyopathy. We have previously shown that heart mitochondrial proteins are hyperacetylated in OVE26 mice, a transgenic model of type 1 diabetes. However, the universality of this modification and its functional consequences are not well established. In this study, we demonstrate that Akita type 1 diabetic mice exhibit hyperacetylation. Functionally, isolated Akita heart mitochondria have significantly impaired maximal (state 3) respiration with physiological pyruvate (0.1 mm) but not with 1.0 mm pyruvate. In contrast, pyruvate dehydrogenase activity is significantly decreased regardless of the pyruvate concentration. We found that there is a 70% decrease in the rate of pyruvate transport in Akita heart mitochondria but no decrease in the mitochondrial pyruvate carriers 1 and 2 (MPC1 and MPC2). The potential role of hyperacetylation in mediating this impaired pyruvate uptake was examined. The treatment of control mitochondria with the acetylating agent acetic anhydride inhibits pyruvate uptake and pyruvate-supported respiration in a similar manner to the pyruvate transport inhibitor α-cyano-4-hydroxycinnamate. A mass spectrometry selective reactive monitoring assay was developed and used to determine that acetylation of lysines 19 and 26 of MPC2 is enhanced in Akita heart mitochondria. Expression of a double acetylation mimic of MPC2 (K19Q/K26Q) in H9c2 cells was sufficient to decrease the maximal cellular oxygen consumption rate. This study supports the conclusion that deficient pyruvate transport activity, mediated in part by acetylation of MPC2, is a contributor to metabolic inflexibility in the diabetic heart.
Keywords: acetylation, bioenergetics, diabetes, heart, mitochondria, mitochondrial transport, pyruvate dehydrogenase complex (PDC), mitochondrial pyruvate carrier
Introduction
Metabolic flexibility is essential for cardiac function. The heart normally generates 70% of its energy from fatty acids and ∼30% from carbohydrates (1, 2). However, with diabetes, the heart relies almost exclusively on fatty acid oxidation. This promotes diabetic cardiomyopathy via mechanisms that include lipotoxicity and loss of mitochondrial function.
Both type 1 and type 2 diabetes are associated with damaged mitochondria that have an impaired capacity to perform oxidative phosphorylation. However, changes in mitochondrial metabolic flexibility occur prior to overt defects in respiratory capacity. For example, we previously reported that heart mitochondria from a transgenic type 1 diabetic mouse model (OVE26 mice) have normal state 3 respiration rates when fatty acids, but not other oxidizable substrates, are supplied as the energy source (3). It is important to identify the mechanisms that underlie this bioenergetic abnormality to promote cardiac health.
The molecular mechanisms that induce metabolic inflexibility are complex. Pyruvate dehydrogenase (PDH)3 is a key regulatory point for determining the fate of pyruvate generated by glycolysis. We and others have shown that PDH is inactivated by hyperphosphorylation in the diabetic heart (3, 4). Other changes to mitochondrial function may be mediated by increased acetylation of mitochondrial protein lysines (3, 5). This reversible post-translational modification is implicated in regulating intermediary metabolism (1). Thus, overabundant acetylation may impede normal regulatory processes and contribute to metabolic inflexibility.
In the present work, we examine changes in mitochondrial function in Akita mouse hearts, a type 1 diabetic model that develops mitochondrial dysfunction (6). We identify a decrease in pyruvate supported respiration, pyruvate dehydrogenase activity, and pyruvate transport. Concurrently, there is hyperacetylation of mitochondrial proteins. We present evidence that acetylation inhibits pyruvate uptake. Using an MS-based selected reactive monitoring technique, we demonstrate that acetylation of the mitochondrial pyruvate carrier 2 (MPC2) is significantly increased in Akita heart mitochondria.
Results
Akita Heart Mitochondria Gave Decreased Pyruvate-supported Respiration, Pyruvate Dehydrogenase Activity, and Pyruvate Uptake
Experiments were performed to determine the relative capacity of Akita heart mitochondria to support mitochondrial oxidative phosphorylation using either pyruvate or palmitoylcarnitine (PC). As shown in Fig. 1, A and B, wild type and Akita mitochondria have comparable state 3 respiration rates with PC as the oxidizable substrate. State 4 respiration rates were also unchanged (not shown). This indicates that mitochondrial integrity is preserved in diabetic animals and that electron transport chain defects are not present at a magnitude that impairs the maximal rate of oxygen consumption. In contrast to PC, state 3 respiration was significantly impaired (26% decrease) when a physiological concentration of pyruvate (0.1 mm) was used. However, this decrease in respiration was largely attenuated (9% decrease; nonsignificant) when the concentration of pyruvate was increased to 1.0 mm in the assay.
FIGURE 1.

Akita heart mitochondria have impaired pyruvate supported respiration, PDH activity, and pyruvate transport. A, mitochondria were isolated from control and Akita hearts. Respiration was measured by a fiber optic oxygen measurement system with 10 mm malate and either 30 μm PC or the indicated amounts of pyruvate. State 3 was initiated by the addition of 0.5 mm ADP. Representative oxygen traces are shown. B, state 3 respiration rates were quantified and are shown either as specific activities (left) or as the percentage of Akita relative to control rates compared on a day-by-day basis (right; n = 5–6). C, mitochondria were incubated with the indicated amounts of pyruvate for 2.0 min at room temperature. 0.5 mm ADP was added as indicated, and samples were incubated an additional minute. PDH activity was then measured as described under “Experimental Procedures” (n = 4). D, pyruvate uptake was measured in isolated mitochondria as described under “Experimental Procedures” (n = 3). E, MPC1 and MPC2 levels were measured by Western blotting (WB) analysis as described under “Experimental Procedures” (n = 5). The MPC1 and MPC2 Western blots reveal single bands at the expected molecular masses and are cropped for clarity. LA, lipoic acid. Experimental points are from unique mitochondrial preparations, and error bars are the standard deviation. *, p < 0.05, unpaired Student's t test; NS, not significant.
The decrease in pyruvate-supported respiration may be mediated, in part, by decreased PDH activity. PDH activity was measured in intact mitochondria preincubated with either 0.1 or 1.0 mm pyruvate and in the presence or absence of ADP. Preincubation of mitochondria with pyruvate and ADP initiates state 3 respiration and induces maximal PDH activity (7). After the indicated incubations, mitochondria were solubilized, and PDH activity was measured spectrophotometrically. The maximal rate of PDH activity was established in wild type mitochondria preincubated with 1.0 mm pyruvate and ADP (Fig. 1C). Under these conditions, PDH activity was 49% less in Akita heart mitochondria (Fig. 1B). In mitochondria preincubated with 0.1 mm pyruvate and ADP, PDH activity was 59% less in Akita mice than in wild types. This demonstrates that the magnitude of the decrease in PDH activity is not necessarily indicative of total respiratory loss under similar assay conditions (Fig. 1A).
An essential factor for maximal respiratory capacity is the transport of substrates into the mitochondrial matrix. Indeed, pyruvate transport is the rate-limiting factor in cardiac mitochondrial pyruvate oxidation (for a review, see Ref. 8). We therefore assayed pyruvate transport in wild type and Akita heart mitochondria by determining the α-cyano-4-hydroxycinnamate (CHC)-sensitive accumulation of [2-14C]pyruvate. As shown in Fig. 1D, the rate of pyruvate uptake in Akita heart mitochondria was 60% less than wild type controls. However, this decreased pyruvate uptake was not mediated by a decreased amount of MPC1 and MPC2 (Fig. 1E). This suggests that pyruvate transport activity may be decreased via another means, such as by post-translational modifications.
The Prevalence of Cardiac Mitochondrial Protein Acetylation Reflects Preference for Fatty Acid Oxidation
We have previously reported that OVE26 type 1 diabetic mice have hyperacetylation of cardiac mitochondrial proteins (3). We sought to determine whether the increase of this post-translational modification is common to other type 1 diabetic rodent models and, furthermore, whether this may contribute to impaired pyruvate-supported respiration. As shown in Fig. 2, A and B, Akita heart mitochondria have a nearly 2-fold increase in acetylation compared with wild type controls. Likewise, protein acetylation is also significantly increased in STZ diabetic mice heart mitochondria (1.5-fold increase). Thus, increased mitochondrial protein acetylation is a common occurrence in multiple models of chronic type 1 diabetes.
FIGURE 2.

Mitochondrial proteins are hyperacetylated in the hearts of diabetic mice and hypoacetylated in PPARα−/− knock-out mice. The levels of acetylation are a predictor of whether pyruvate or palmitoylcarnitine produces maximal state 3 respiratory rates. A, mitochondria were analyzed by Western blotting (WB) using an acetylated lysine (AcK) antibody. An antibody against lipoic acid (LA) bound to the E2 subunit of PDH was used as a loading control (32) as its levels did not significantly differ between experimental groups. The samples shown are from a single blot and the same exposure. The image was subsequently cropped for clarity of presentation. B, acetylation densitometry was determined on the entire lane of Akita (n = 7), STZ (n = 5), and PPARα−/− (n = 6) mitochondria and expressed relative to the appropriate age-matched control. C, state 3 respiration rates were determined with either PC and malate or pyruvate and malate for wild type (n = 4), Akita (n = 5), and PPARα−/− (n = 4) heart mitochondria. The lines connect measurements from the same preparation of mitochondria. D, the ratio of PC-supported state 3 to pyruvate (Pyr)-supported state 3 rates was quantified. Experimental points are from unique mitochondrial preparations, and error bars are the standard deviation. *, p < 0.05, paired Student's t test.
The increased occurrence of mitochondrial protein acetylation in diabetic samples reflects an increased reliance on fatty acid oxidation. When comparing the maximal rates of state 3 respiration in control heart mitochondria with either PC or pyruvate, the PC-supported rate is on average 33% faster (Fig. 2, C and D). In Akita heart mitochondria, PC-supported respiration is 79% faster (Fig. 2, C and D). In contrast, mice that lack the PPARα transcriptional factor, which regulates the levels of several fatty acid oxidation enzymes (9), have a significant increase in the rate of state 3 respiration with pyruvate relative to PC (Fig. 2, C and D). Furthermore, mitochondria from PPARα−/− mouse hearts have a corresponding decrease (21%) in mitochondrial protein acetylation (Fig. 2, A and B). These results support that the preferential usage of fatty acids is accompanied by increased acetylation, and inversely less reliance on fatty acid utilization results in less mitochondrial acetylation.
Acetylation Decreases Pyruvate Oxidation
We next sought to determine whether hyperacetylation of mitochondrial proteins might contribute to the observed deficits in pyruvate-supported respiration and pyruvate uptake. Control and Akita heart mitochondria were treated with the acetylating agent acetic anhydride (Ac2O), which preferentially modifies the primary amine of lysines at neutral pH (3, 10). As shown in Fig. 3A, control mitochondria treated with low concentrations of Ac2O exhibit a remarkably similar complement of acetylated proteins as are found endogenously in Akita heart mitochondria. This result is also similar to what we have previously reported in OVE26 heart mitochondria (3).
FIGURE 3.

Acetylation of control heart mitochondria with acetic anhydride mimics the respiratory profile of Akita heart mitochondria. A, control and Akita heart mitochondria were treated with the indicated amounts of Ac2O and then subjected to Western blotting (WB) analysis using an acetyl-lysine (AcK) antibody. Lipoic acid (LA) content was probed as a loading control as its levels were not affected by Ac2O treatment. The samples shown are from a single blot and the same exposure. The image was subsequently cropped for clarity of presentation. B, state 3 respiration rates were measured 2 min after addition of 10 μm Ac2O as described under “Experimental Procedures” (n = 4). C, mitochondria (n = 5) were treated with 10 μm Ac2O as indicated. After 2.0 min, PDH activity was measured as described under “Experimental Procedures.” D, mitochondria (n = 5) were treated with 10 μm Ac2O as indicated. After 2.0 min, the radiolabeled pyruvate mixture was added, and pyruvate transport was measured as described under “Experimental Procedures.” Experimental points are from unique mitochondrial preparations, and error bars are the standard deviation. *, p < 0.05, unpaired Student's t test.
Experiments were next performed to demonstrate that the acetylation of control mitochondria in vitro mimics the deficits in pyruvate-supported respiration observed in Akita mice. Treatment of control heart mitochondria with 10 μm Ac2O significantly inhibited state 3 respiration in the presence of 0.1 mm pyruvate (34% decrease). However, this effect was attenuated in mitochondria respiring with 1.0 mm pyruvate (13% decrease; Fig. 3B). In contrast, Ac2O treatment caused a slight but significant drop (16% decrease) in PC-supported state 3 respiration (Fig. 3B) but was independent of the PC concentration (not shown). Thus, pyruvate-supported respiration is more sensitive than PC-supported respiration to acetylation-mediated inhibition.
Previous reports have shown that acetylation of the PDH complex inhibits its enzymatic activity (11–13). We therefore tested whether Ac2O treatment of control mitochondria was inhibiting pyruvate-supported respiration by decreasing PDH activity. Treatment of control heart mitochondria with Ac2O had no effect on basal or maximal PDH activity regardless of the pyruvate concentration (Fig. 3C). This suggests that another aspect of pyruvate oxidation, such as substrate transport, may be inhibited by acetylation. Control mitochondria were therefore treated with Ac2O, and pyruvate uptake was measured. As shown in Fig. 3C, Ac2O (10 μm) inhibits the rate of pyruvate uptake by 44%. This supports that protein acetylation may represent a means for decreasing pyruvate transport and the overall rate of mitochondrial pyruvate oxidation.
We hypothesized that if protein acetylation acts as a pyruvate transporter inhibitor then the effects of Ac2O on mitochondrial respiration should be similar to the classic pyruvate transport inhibitor CHC. We noted that CHC inhibits pyruvate supported state 3 respiration in a dose-dependent manner in coupled, control heart mitochondria (Fig. 4A). However, the sensitivity to CHC-mediated inhibition is significantly enhanced in uncoupled mitochondria (Fig. 4, B and C). For example, 1.0 μm CHC inhibits ADP- and FCCP-supported respiration by 15 and 50%, respectively (Fig. 4C). This enhanced inhibition is likely due to the drop in membrane potential that helps drive pyruvate uptake and not mediated by changes in either basal or maximal PDH activities. The maximal PDH activities are similar in control heart mitochondria treated with either ADP or FCCP and were unaffected by 1.0 μm CHC (Fig. 4D).
FIGURE 4.

The classic pyruvate transport inhibitor CHC preferentially inhibits uncoupled state 3 respiration. A, state 3 respiration rates were measured in control heart mitochondria in the presence of 1.0 mm malate, 0.1 mm pyruvate, and the indicated amounts of CHC (n = 4). B, representative oxygen consumption traces of control mitochondria in the presence of 1.0 mm malate, 0.1 mm pyruvate, and 1.0 μm CHC as indicated are shown. State 3 respiration was induced by either the addition of ADP (top) or the uncoupler FCCP (bottom). C, the magnitude of state 3 inhibition induced by 1.0 μm CHC was quantified in coupled and uncoupled mitochondria (n = 6). D, PDH activity was measured in control and CHC-treated mitochondria basally and following stimulation of maximal respiration with either ADP or FCCP. PDH activity was measured as described under “Experimental Procedures” (n = 3–5). *, p < 0.05, unpaired Student's t test. Error bars are the standard deviation.
We next examined the effect of Ac2O on coupled and uncoupled pyruvate-supported respiration in control heart mitochondria. Ac2O (10 μm), like CHC, preferentially inhibits uncoupled state 3 respiration (Fig. 5, A and B). This result supports that, in vitro, acetylation inhibits pyruvate-supported respiration in a manner similar to CHC.
FIGURE 5.

Acetylation preferentially inhibits uncoupled mitochondrial respiration. A decrease in the ratio of uncoupled to coupled respiration is indicative of decreased pyruvate uptake. A, control heart mitochondria were treated with 10 μm Ac2O as indicated, and maximal pyruvate-supported respiration was measured upon addition of ADP (coupled; top) or FCCP (uncoupled; bottom). B, the magnitude of state 3 inhibition induced by 10 μm Ac2O was quantified in coupled and uncoupled mitochondria (n = 4). *, p < 0.05, paired Student's t test. C, the ratio of uncoupled to coupled state 3 respiration was quantified in control heart mitochondria, control heart mitochondria treated with 1.0 μm CHC, control heart mitochondria treated with 10 μm Ac2O Ac2O, or Akita heart mitochondria (n > 4). *, p < 0.05, unpaired Student's t test. Error bars are the standard deviation.
Our results indicate that the pyruvate-supported uncoupled/coupled respiration ratio acts as a functional indicator for detecting decreased pyruvate uptake. For example, this ratio is ∼0.78 in control mitochondria and decreases to 0.37 in CHC (1.0 μm)-treated mitochondria (Fig. 5C). Likewise, the ratio is also significantly decreased in Ac2O (10 μm)-treated control mitochondria to 0.49. Thus, hyperacetylation inhibits mitochondrial respiration in a manner similar to CHC. Finally, we found that the uncoupled/coupled respiration ratio is also significantly less in Akita mice (0.55) than in controls (Fig. 5C). This supports that decreased pyruvate uptake is a phenomenon in diabetic heart mitochondria and that it may be mediated by hyperacetylation.
MPC2 Acetylation Is Increased in Diabetic Heart Mitochondria
Pyruvate is transported into the mitochondria via the mitochondrial pyruvate carrier, which is composed of the MPC1 and MPC2 proteins. Neither subunit is decreased in Akita heart mitochondria (Fig. 1E), and our biochemical characterization supports a role for acetylation in decreasing pyruvate uptake. We therefore focused our ensuing experiments on developing a selected reaction monitoring (SRM) assay to detect the acetylation of MPC subunits in Akita heart mitochondria.
The SRM assay was developed by enriching MPCs from control heart mitochondria by immunoprecipitation and then acetylating the proteins with Ac2O prior to in-gel digestion and MS analysis. This approach allowed us to determine the retention times and CID spectra of specific acetylated peptides that may occur in vivo. This experiment resulted in the identification of MPC2 peptides containing acetylation sites at Lys-19, Lys-26, Lys-27, and Lys-122. Although MPC1 was successfully enriched (not shown), subsequent MS analysis failed to resolve acetylated peptides (see “Discussion”). Ensuing experiments performed to identify acetylation of lysines in vivo thus focused on MPC2 peptides.
MPC2 was equally enriched by immunoprecipitation in control and Akita heart mitochondria (Fig. 6A). The enriched proteins were subjected to in-gel digestion and SRM analysis. We detected acetylation of Lys-19 and Lys-26 but not Lys-27 and Lys-122. Furthermore, the in vivo abundance of acetylation was significantly increased at Lys-19 (40%) and Lys-26 (80%) in Akita heart mitochondria relative to wild types (Fig. 6B). Collectively, our results demonstrate that Akita heart mitochondria have decreased pyruvate-supported respiration and pyruvate transport and hyperacetylation of MPC2.
FIGURE 6.
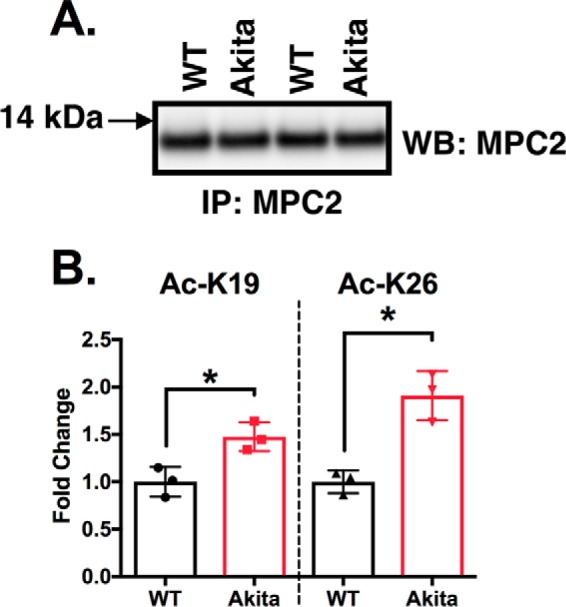
Acetylation of MPC2 at Lys-19 and Lys-26 is significantly increased in Akita heart mitochondria. A, representative Western blots (WB), on duplicate samples, of MPC2 immunoprecipitated (IP) from wild type or Akita heart mitochondria. B, a selected reaction monitoring technique demonstrated that lysines 19 and 26 are significantly more acetylated in Akita heart mitochondria than in wild types (n = 3). *, p < 0.05, unpaired Student's t test. Error bars are the standard deviation.
Acetylation Mimetics of Lys-19 and Lys-26 Decrease Maximal Mitochondrial Respiration
The functional significance of Lys-19 and Lys-26 acetylation was next evaluated by Seahorse XF analysis of oxygen consumption rates (OCRs). We first established that mitochondrial pyruvate transport activity can affect the OCR in H9c2 cells, a commonly used rat ventricular cell line that has a metabolic profile similar to primary cardiomyocytes (14). For the assay, cells were incubated with pyruvate as the lone nutrient, and the OCR was measured in the presence or absence of CHC. As shown in Fig. 7, A and B, CHC significantly decreased the maximal OCR induced by the addition of FCCP. This establishes that the maximal OCR can be used as a proxy to evaluate mitochondrial pyruvate transport activity in H9c2 cells.
FIGURE 7.

The double acetylation mimetic of Lys-19 and Lys-26 (K19Q/K26Q) decreases the pyruvate-dependent cellular oxygen consumption rate. A, H9c2 cells were placed in medium containing pyruvate as the sole nutrient source, and the OCR was measured as described under “Experimental Procedures.” CHC (0.1 mm) was added just prior to the beginning of the experiment as indicated. OCR measurements taken were basal (1), post-oligomycin (2), post-FCCP (3), and post-antimycin A (4). B, the maximal OCR was calculated as the post-FCCP OCR minus the post-oligomycin OCR (n = 4). C, wild type Myc-MPC2 and RR and QQ Myc-tagged MPC2 were expressed in H9c2 cells. A representative Western blot (WB) is shown indicating protein expression levels. D and E, the effects of exogenously expressed Myc-MPC2 on OCR (D) and maximal (Max) OCR (E) were evaluated. F, a schematic representation of the proposed transmembrane domains and topology of MPC2 (adapted from Ref. 36) and relative positions of Lys-19 and Lys-26. IMM, inner mitochondrial membrane; IMS, intermembrane space. n = 4; *, p < 0.05, unpaired Student's t test. Error bars are the standard deviation.
We next generated MPC2 mutants to determine whether modifications of Lys-19 and Lys-26 can affect the rate of pyruvate transport. Glutamine approximates an acetylated lysine structurally and functionally, whereas arginine approximates a lysine that cannot be acetylated (15). We expressed Myc-tagged MPC2 that was either wild type, a K19R/K26R (RR) double mutant, or a K19Q/K26Q (QQ) double mutant. The Myc tag was used to discern expressed proteins from endogenous MPC2. As shown in Fig. 7C, transfection of wild type or RR Myc-MPC2 resulted in similar protein levels. However, identical transfection conditions consistently resulted in less of the QQ double mutant. This reduced protein content was dependent upon both lysines being modified as the K19Q and K26Q single mutants were expressed at levels similar to wild type Myc-MPC2 (data not shown). Furthermore, we noted that none of the Myc-MPC2 constructs affected the endogenous levels of MPC2 (Fig. 6E). These results suggest modification of Lys-19 and Lys-26 in tandem may affect MPC2 structure or stability.
We next evaluated whether expression of mutant MPC2 proteins can affect the cellular OCR. As shown in Fig. 7, D and E, wild type and RR Myc-MPC2 had no significant effects on basal or maximal OCR. In contrast, a significant decrease in basal and maximal OCRs was observed with expression of the QQ mutant. This decrease was dependent upon the double mutation as no significant changes in OCR were observed with either the single K19Q or K26Q mutation (data not shown). Collectively, these results support that modification of Lys-19 and Lys-26, as seen in the diabetic heart, can affect MPC2 stability and function.
Discussion
Mitochondrial deficits are an important contribution to diabetic cardiomyopathy (16). However, changes to mitochondrial function precede irreversible organelle damage. As we show here, Akita heart mitochondria display deficits in pyruvate- but not palmitoylcarnitine-supported respiration. This metabolic inflexibility is in agreement with our previous work in OVE26 heart mitochondria (3). Also like OVE26 animals, we show that Akita heart mitochondria have hyperacetylation of mitochondrial proteins. Our results here support acetylation as a means of decreasing metabolic flexibility via a decrease in mitochondrial pyruvate uptake.
This work establishes that hyperacetylation is a common occurrence in both Akita and STZ diabetic animals. We have previously shown that hyperacetylation is also present in OVE26 type 1 diabetic mice and in wild type mice fed a high fat diet (17). However, the functional significance of these post-translational modifications is uncertain. We provide evidence that the increase or decrease of this general protein modification is indicative of substrate preference. Less acetylation corresponds to lower palmitoylcarnitine respiration rates relative to pyruvate. Reciprocally, more acetylation corresponds to increased palmitoylcarnitine rates relative to pyruvate (Fig. 2). Thus, both hyper- and hypoacetylation may be signs of metabolic inflexibility and, consequentially, mitochondrial abnormalities.
PDH is a primary site of metabolic regulation, and decreased cardiac PDH activity is a well established consequence of diabetes (1, 18, 19). This loss of PDH activity may be mediated by overactivation of pyruvate dehydrogenase kinases or decreased PDH phosphatase activities. Consistent with this idea, we demonstrate that, in Akita heart mitochondria, PDH activity is decreased under basal conditions and fails to reach maximal rates of activity comparable with wild types under state 3 respiration conditions. A possible contributor to decreased PDH activity is the increased occurrence of mitochondrial acetylation. It has been previously shown that PDH is acetylated and that this decreases its activity (11–13). However, the occurrence of this modification to PDH subunits in the diabetic heart is not currently known and will be important for future investigation.
Our results demonstrate that the changes in metabolic flexibility in the Akita mouse heart are multifaceted. In addition to the decreased PDH activity, a deficit in pyruvate transport also contributes to metabolic inflexibility in the diabetic heart. This conclusion is supported by our observations that pyruvate-supported respiration can be increased in Akita heart mitochondria by increasing the amount of pyruvate and that the rate of pyruvate transport is decreased. The mitochondrial pyruvate transporter is composed of two components, MPC1 and MPC2 (20, 21). Although exhibiting decreased pyruvate transport activity, Akita heart mitochondria have levels of MPC proteins comparable with wild types (Fig. 1E). Both of the MPCs contain acetylated lysines, previously identified through unbiased proteomic screens (22–24). However, the occurrence of these modifications in the diabetic heart and their effects on pyruvate transport activity have not been previously determined. We thus hypothesized that acetylation may affect pyruvate transport activity. Furthermore, this modification may work in concert with other factors in the Akita model, such as a possible decrease in membrane potential, to decrease pyruvate oxidation.
A recent report demonstrated changes in MPC1 activity based on site-directed mutagenesis of putative acetylation sites (25). Although that work did not directly measure pyruvate transport, the data support the notion that acetylation decreases MPC activity. In our work, we did not identify acetylation of MPC1 in Akita hearts. However, this may be due to limitations of the SRM technique we used. By this established approach (17), we first acetylate lysine residues via chemical modification before the in-gel digestion process and subsequent MS analysis. However, acetylation of lysine residues may either impair proteolytic cleavage of the protein or MS detection. Nevertheless, our SRM approach can be further refined and tailored to other target proteins to determine the in vivo occurrence and significance of acetylation under normal and pathological states.
Two lysines were found hyperacetylated in Akita heart mitochondria. These sites, Lys-19 and Lys-26 of MPC2, are conserved in humans. The structures of MPCs have not been previously solved. However, based on homology to related transporters, MPC2 is predicted to have the orientation presented in Fig. 7E based upon homology to the bacterial semi-SWEET glucose transporter (26, 27). Our site-directed mutagenesis studies demonstrated that modification of these sites to glutamines, but not arginines, decreased protein content. Furthermore, this double mutant decreased pyruvate-supported OCR even in the presence of endogenous levels of MPC2. One explanation is that the QQ double mutant acts as a dominant negative regulator of MPC2 activity. However, mutation of these sites to glutamines individually had no effect on expression or the cellular OCR. Collectively, these results suggest that these lysines are important both functionally and perhaps for protein stability and that acetylation may play a role in regulating pyruvate transport activity.
The two lysines Lys-19 and Lys-26 are proposed to be exposed to the intermembrane space, thereby sequestering these residues away from SIRT3, the primary mitochondrial deacetylase (28). Thus, acetylation of these residues may not be as readily reversible as other proteins in the mitochondrial matrix. The question also remains as to how these sites become hyperacetylated because they are not exposed to the high acetyl-CoA concentration of the mitochondrial matrix. Future work that directly addresses the structural nature of these proteins will help to address these issues.
Experimental Procedures
Animal Models
Akita mice were purchased from The Jackson Laboratory (stock number 003548) and bred at the Oklahoma Medical Research Foundation animal facility. Akita mice contain a spontaneous mutation in the Ins2 locus and develop type 1 diabetes and diabetic cardiomyopathy (29, 30). Both the control and Akita mice are on the C57BL/6J background that contains the nicotinamide nucleotide transhydrogenase mutation. Hearts from STZ diabetic mice (24 weeks diabetic; 32 weeks old) and PPARα−/− mice (32 weeks old) were obtained from the University of Oklahoma Health Sciences Center animal core.
Isolation of Cardiac Mitochondria
Male Akita or age-matched C57 mice were euthanized by cervical dislocation. Their chest cavities were immediately opened, and their hearts were perfused with 5 ml of ice-cold buffer containing 210 mm mannitol, 70 mm sucrose, 1.0 mm EDTA, and 5.0 mm MOPS, pH 7.4 (buffer A), via injection into the left ventricle. Hearts were excised and placed into 5 ml of buffer A and then minced with scissors. This was followed by five passes with a motor-driven Potter-Elvehjem tissue grinder. The homogenate was spun at 500 × g for 5 min at 4 °C, and the supernatant was collected, passed through a cheesecloth, and spun again at 5000 × g for 10 min. The resulting mitochondrial pellet was resuspended in ∼60 μl of buffer A, and the protein concentration was determined by the BCA (bicinchoninic acid) method (Thermo Scientific) using BSA as a standard.
Mitochondrial Respiration Measurements
Mitochondria were diluted to 0.25 mg/ml in 210 mm mannitol, 70 mm sucrose, 5.0 mm KH2PO4, 0.5 mg/ml BSA, and 10 mm MOPS, pH 7.4 (buffer B), containing the indicated oxidizable substrates. For chemically induced acetylation experiments, mitochondria (0.25 mg/ml) were treated with the indicated amounts of Ac2O in buffer B with 0.5 mg/ml BSA for 2.0 min at 20 °C. Respiration was measured at 20 °C by using a fiber optic oxygen measurement system (Instech) that utilizes the fluorescence lifetime technique. State 3 respiration was initiated by the addition of ADP (0.5 mm) or FCCP (30 μm). The starting amount of molecular oxygen in the 0.4-ml electrode chamber was based on the assumption that 265 nmol of molecular oxygen are dissolved per ml at atmospheric pressure and 20 °C.
Pyruvate Dehydrogenase Assay
Mitochondria were diluted to 0.25 mg/ml in buffer B with the indicated substrates and Ac2O. To obtain maximal PDH activity, 0.5 mm ADP or FCCP (25 μm) was also added to mitochondria. After a 2.0-min incubation, mitochondria were diluted into a buffer containing 025% Triton X-100, 5.0 mm MgCl2, and 20 mm MOPS, pH 7.2, to a final concentration of 0.025 mg/ml. PDH activity was measured spectrophotometrically by the increase in NADH absorbance at 340 nm upon addition of 2.5 mm pyruvate, 0.1 mm CoA, 0.2 mm thiamine pyrophosphate, and 1.0 mm NAD+ (3). The increase in absorbance was linear for over a 5.0-min period.
Pyruvate Transport Assay
Pyruvate transport was measured in isolated mitochondria (adapted from Ref. 31). Isolated mitochondria were diluted to 0.25 mg/ml in 0.4 ml of buffer B. To determine the affect of acetylation, samples were treated with Ac2O (10 μm) for 2.0 min as indicated. A mixture of 10 mm pyruvate (1.0 mm [2-14C]pyruvate and 9.0 mm unlabeled pyruvate) and 100 mm malate was prepared fresh. The pyruvate/malate mixture was added to the mitochondria at a 1:100 dilution. Samples were incubated for 2.5 min at 20 °C and then centrifuged for 2.0 min at 16,000 × g. The supernatant was discarded, and the pellets were resuspended in 0.2 ml of liquid scintillation mixture (Ultima Gold, PerkinElmer Life Sciences). The resuspended pellet was transferred to a scintillation vial with a final volume of 3.0 ml of scintillation mixture, and counts were measured. Negative control samples were prepared by adding 20 μm CHC for 1.0 min prior to the addition of the pyruvate/malate mixture. The values reported represent experimental counts minus the negative control counts. A standard curve, determined with known amounts of [2-14C] pyruvate, was used to calculate specific activities. The uptake of pyruvate was determined to be linear for over 5.0 min (data not shown).
Western Blotting Analysis
Samples (20 μg/well) were separated by SDS-PAGE (NuPAGE 4–12% denaturing gels, Thermo Scientific) with an MES running buffer and then transferred onto nitrocellulose. Acetyl-lysine, MPC1, and MPC2 antibodies were purchased from Cell Signaling Technology. An antibody against lipoic acid (32), covalently bound to the E2 subunit of PDH, was used as a loading control in mitochondria because its content did not change under any experimental condition. Actin-HRP (Santa Cruz Biotechnology) was used as the loading control for H9c2 samples. Western blots were imaged with an Alpha Infotech Fluorchem HD2 imaging system, and band densities were determined with NIH ImageJ software.
MS Analysis
A method for detecting MPC2 acetylation in vivo was developed in a manner similar to our previous report (17). MPC2 was immunoprecipitated by dilution of 0.3 mg of mitochondrial protein into 0.3 ml radioimmune precipitation assay buffer (150 mm NaCl, 0.1% Nonidet P-40, and 100 mm NaPO4, pH 7.4) and addition of 10 μl of MPC2 antibody. Samples were incubated with mixing overnight at 4 °C. Samples were then incubated with 40 μl of Protein A/G beads (Pierce) with mixing at 4 °C. Beads were then washed three times, and bound proteins eluted with 0.1 m glycine, pH 2.5. For identification of acetylation sites, immunoprecipitated proteins were treated with acetic anhydride (0.5 mm) for 10 min prior to glycine elution. The eluted proteins were diluted in Laemmli buffer and separated by SDS-PAGE, and subsequently the gel was fixed and stained. The MPC bands at ∼12–15 kDa were cut from the gel, reduced, alkylated, and digested with trypsin.
All LC-tandem mass spectrometry experiments used a Thermo Scientific Q-Exactive Plus mass spectrometer with an Ultimate 3000 nanoflow HPLC system. The HPLC used a 10-cm × 75-μm capillary column packed with Phenomenex Aeris 3.6-μm Peptide XB-C18 stationary phase. Samples were injected in 1% acetic acid and eluted with a linear gradient of acetonitrile in water with 0.1% formic acid. The acetylation sites were mapped using data-dependent analysis acquiring full scan spectra at R = 140,000 and CID spectra at R = 17,500. These data were used to map the acetylation sites by searching the mouse RefSeq database with Proteome Discoverer. All identified sites were verified by manual interpretation of the corresponding CID spectra. Unmodified MPC2 peptides were also detected in the data-dependent analyses. The targeted quantitative experiments used parallel reaction monitoring (PRM). Parent ions were selected with a 1 m/z window and full scan CID spectra were acquired at R = 17,500. The parallel reaction monitoring data were processed to generate chromatograms of the various peptides of interest using the product ions seen in the respective CID spectra and the observed chromatographic retention time. Calculations used the geometric mean of the abundance of three unmodified peptides (VELLLPK, YSLVIIPK, and LRPLYNHPAGPR) for normalization based on our previously reported native reference peptide method (33, 34).
Tissue Culture, Constructs, and Transfection
H9c2(2-1) cells (ATCC, CRL-1446) were maintained in Dulbecco's modified Eagle's medium (DMEM) + GlutaMAX (Gibco, 10567-014) supplemented with 10% fetal bovine serum (Gibco, A3160401) and penicillin/streptomycin (Gibco,15140-122). Point mutations were engineered into the mouse cDNA of MPC2-Myc (OriGene, MR200690) using site-directed mutagenesis to generate K19R, K19Q, K26R, and K26Q and K19R/K26R and K19Q/K26Q double mutations. Plasmid constructs were prepared with a Miniprep kit (Thermo Fisher, K0503) and sequenced to confirm mutations. H9c2 cells were transfected using Lipofectamine 2000 (Invitrogen, 11668-027) according to the manufacturer's recommendations. Briefly, 3 μl of Lipofectamine 2000 was diluted into 150 μl of Opti-MEM (Gibco, 31985-070), and 0.5 μg of plasmid DNA was added to 150 μl of Opti-MEM. After a 5-min incubation, solutions were combined and incubated for 20 min before dropwise addition to cells. Medium was removed, fresh medium was applied 4 h after transfection, and cells were processed 36 h after transfection. Expression levels of MPC2-Myc were verified by Western blotting analysis.
Seahorse XF Analysis
Pyruvate supported respiration was measured in control and transfected cells at 37 °C using a Seahorse XFe24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA) (35). H9c2 cells were plated at 25,000 cells/well into XF24 microplates in DMEM containing 10% FBS. 36 h after transfection, medium was removed and replaced with unbuffered DMEM supplemented with 1.0 mm pyruvate and then cultured for 30 min at 37 °C in an air-equilibrated incubator. The OCR measurements were performed using cycles comprising 3-min mixing, 2-min wait, and 2-min measurement. Injections of oligomycin (1.0 μm), FCCP (1.0 μm), and antimycin A (1.5 μm) were made at the indicated time points. Experimental treatments were performed on 4 wells of each plate as biological replicates.
Statistical Analysis
Data are presented as mean ± S.E. Data were analyzed using either a paired or unpaired two-tailed Student's t test as indicated. p values of <0.05 were considered significant.
Author Contributions
S. S. V. performed the mitochondria isolation experiments and analyzed the data. J. R. G. and S. M. assisted with acetylation experiments. S. M. performed the Seahorse XF analysis. C. A. E. performed the site-directed mutagenesis of MPC2 and cell culture. C. S. K. performed aspects of the mass spectrometry analysis. M. K. designed and performed mass spectrometry experiments and analyzed the data. L. B. B. assisted with presentation of data and its interpretation. K. M. H. conceived the idea for the project, assisted with experimental design and data interpretation, and wrote the manuscript.
Acknowledgment
The Q-Exactive Plus system used in this study was purchased by the Oklahoma City Veterans Affairs Medical Center with a grant from the Development of Veterans Affairs to Dr. Arlan Richardson.
This work was supported in part by American Heart Association Grant 14GRNT20510031; Institutional Development Award P20GM104934 from the NIGMS, National Institutes of Health; and NHLBI, National Institutes of Health Grant R01HL125625. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PDH
- pyruvate dehydrogenase
- CHC
- α-cyano-4-hydroxycinnamate
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- MPC
- mitochondrial pyruvate carrier
- PC
- palmitoylcarnitine
- STZ
- streptozotocin
- PPARα
- peroxisome proliferator-activated receptor
- Ac2O
- acetic anhydride
- SRM
- selected reaction monitoring
- CID
- collision-induced dissociation
- OCR
- oxygen consumption rate
- RR
- K19R/K26R
- K19Q/K26Q
- R
- resolution.
References
- 1. Fukushima A., and Lopaschuk G. D. (2016) Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 1860, 1525–1534 [DOI] [PubMed] [Google Scholar]
- 2. Griffin T. M., Humphries K. M., Kinter M., Lim H. Y., and Szweda L. I. (2016) Nutrient sensing and utilization: getting to the heart of metabolic flexibility. Biochimie 124, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vadvalkar S. S., Baily C. N., Matsuzaki S., West M., Tesiram Y. A., and Humphries K. M. (2013) Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochem. J. 449, 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang B., Wu P., Popov K. M., and Harris R. A. (2003) Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 52, 1371–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baeza J., Smallegan M. J., and Denu J. M. (2016) Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem. Sci. 41, 231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bugger H., and Abel E. D. (2009) Rodent models of diabetic cardiomyopathy. Dis. Model. Mech. 2, 454–466 [DOI] [PubMed] [Google Scholar]
- 7. Crewe C., Kinter M., and Szweda L. I. (2013) Rapid inhibition of pyruvate dehydrogenase: an initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. PLoS One 8, e77280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schell J. C., and Rutter J. (2013) The long and winding road to the mitochondrial pyruvate carrier. Cancer Metab. 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Finck B. N. (2007) The PPAR regulatory system in cardiac physiology and disease. Cardiovasc. Res. 73, 269–277 [DOI] [PubMed] [Google Scholar]
- 10. Buechler J. A., Vedvick T. A., and Taylor S. S. (1989) Differential labeling of the catalytic subunit of cAMP-dependent protein kinase with acetic anhydride: substrate-induced conformational changes. Biochemistry 28, 3018–3024 [DOI] [PubMed] [Google Scholar]
- 11. Jing E., O'Neill B. T., Rardin M. J., Kleinridders A., Ilkeyeva O. R., Ussar S., Bain J. R., Lee K. Y., Verdin E. M., Newgard C. B., Gibson B. W., and Kahn C. R. (2013) Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan J., Shan C., Kang H. B., Elf S., Xie J., Tucker M., Gu T. L., Aguiar M., Lonning S., Chen H., Mohammadi M., Britton L. M., Garcia B. A., Alečković M., Kang Y., et al. (2014) Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ozden O., Park S. H., Wagner B. A., Yong Song H., Zhu Y., Vassilopoulos A., Jung B., Buettner G. R., and Gius D. (2014) SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic. Biol. Med. 76, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuznetsov A. V., Javadov S., Sickinger S., Frotschnig S., and Grimm M. (2015) H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta 1853, 276–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tao R., Coleman M. C., Pennington J. D., Ozden O., Park S. H., Jiang H., Kim H. S., Flynn C. R., Hill S., Hayes McDonald W., Olivier A. K., Spitz D. R., and Gius D. (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bugger H., and Abel E. D. (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57, 660–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandes J., Weddle A., Kinter C. S., Humphries K. M., Mather T., Szweda L. I., and Kinter M. (2015) Lysine acetylation activates mitochondrial aconitase in the heart. Biochemistry 54, 4008–4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu P., Sato J., Zhao Y., Jaskiewicz J., Popov K. M., and Harris R. A. (1998) Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 329, 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schummer C. M., Werner U., Tennagels N., Schmoll D., Haschke G., Juretschke H. P., Patel M. S., Gerl M., Kramer W., and Herling A. W. (2008) Dysregulated pyruvate dehydrogenase complex in Zucker diabetic fatty rats. Am. J. Physiol. Endocrinol. Metab. 294, E88–E96 [DOI] [PubMed] [Google Scholar]
- 20. Bricker D. K., Taylor E. B., Schell J. C., Orsak T., Boutron A., Chen Y. C., Cox J. E., Cardon C. M., Van Vranken J. G., Dephoure N., Redin C., Boudina S., Gygi S. P., Brivet M., Thummel C. S., et al. (2012) A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herzig S., Raemy E., Montessuit S., Veuthey J. L., Zamboni N., Westermann B., Kunji E. R., and Martinou J. C. (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96 [DOI] [PubMed] [Google Scholar]
- 22. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., and Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., and Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., et al. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liang L., Li Q., Huang L., Li D., and Li X. (2015) Sirt3 binds to and deacetylates mitochondrial pyruvate carrier 1 to enhance its activity. Biochem. Biophys. Res. Commun. 468, 807–812 [DOI] [PubMed] [Google Scholar]
- 26. Xu Y., Tao Y., Cheung L. S., Fan C., Chen L. Q., Xu S., Perry K., Frommer W. B., and Feng L. (2014) Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature 515, 448–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vanderperre B., Bender T., Kunji E. R., and Martinou J. C. (2015) Mitochondrial pyruvate import and its effects on homeostasis. Curr. Opin. Cell Biol. 33, 35–41 [DOI] [PubMed] [Google Scholar]
- 28. Anderson K. A., and Hirschey M. D. (2012) Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 52, 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J., Takeuchi T., Tanaka S., Kubo S. K., Kayo T., Lu D., Takata K., Koizumi A., and Izumi T. (1999) A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. J. Clin. Investig. 103, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Basu R., Oudit G. Y., Wang X., Zhang L., Ussher J. R., Lopaschuk G. D., and Kassiri Z. (2009) Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am. J. Physiol. Heart Circ. Physiol. 297, H2096–H2108 [DOI] [PubMed] [Google Scholar]
- 31. Halestrap A. P., and Denton R. M. (1975) The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by α-cyano-4-hydroxycinnamate and related compounds. Biochem. J. 148, 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Humphries K. M., and Szweda L. I. (1998) Selective inactivation of α-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry 37, 15835–15841 [DOI] [PubMed] [Google Scholar]
- 33. Ruse C. I., Willard B., Jin J. P., Haas T., Kinter M., and Bond M. (2002) Quantitative dynamics of site-specific protein phosphorylation determined using liquid chromatography electrospray ionization mass spectrometry. Anal. Chem. 74, 1658–1664 [DOI] [PubMed] [Google Scholar]
- 34. Willard B. B., Ruse C. I., Keightley J. A., Bond M., and Kinter M. (2003) Site-specific quantitation of protein nitration using liquid chromatography/tandem mass spectrometry. Anal. Chem. 75, 2370–2376 [DOI] [PubMed] [Google Scholar]
- 35. Gerencser A. A., Neilson A., Choi S. W., Edman U., Yadava N., Oh R. J., Ferrick D. A., Nicholls D. G., and Brand M. D. (2009) Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal. Chem. 81, 6868–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCommis K. S., and Finck B. N. (2015) Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem. J. 466, 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]