

Abstract
Proteins responsible for Pi homeostasis are critical for all life. In Saccharomyces cerevisiae, extracellular [Pi] is “sensed” by the inositol-hexakisphosphate kinase (IP6K) that synthesizes the intracellular inositol pyrophosphate 5-diphosphoinositol 1,2,3,4,6-pentakisphosphate (5-InsP7) as follows: during a period of Pi starvation, there is a decline in cellular [ATP]; the unusually low affinity of IP6Ks for ATP compels 5-InsP7 levels to fall in parallel (Azevedo, C., and Saiardi, A. (2017) Trends. Biochem. Sci. 42, 219–231. Hitherto, such Pi sensing has not been documented in metazoans. Here, using a human intestinal epithelial cell line (HCT116), we show that levels of both 5-InsP7 and ATP decrease upon [Pi] starvation and subsequently recover during Pi replenishment. However, a separate inositol pyrophosphate, 1,5-bisdiphosphoinositol 2,3,4,6-tetrakisphosphate (InsP8), reacts more dramatically (i.e. with a wider dynamic range and greater sensitivity). To understand this novel InsP8 response, we characterized kinetic properties of the bifunctional 5-InsP7 kinase/InsP8 phosphatase activities of full-length diphosphoinositol pentakisphosphate kinases (PPIP5Ks). These data fulfil previously published criteria for any bifunctional kinase/phosphatase to exhibit concentration robustness, permitting levels of the kinase product (InsP8 in this case) to fluctuate independently of varying precursor (i.e. 5-InsP7) pool size. Moreover, we report that InsP8 phosphatase activities of PPIP5Ks are strongly inhibited by Pi (40–90% within the 0–1 mm range). For PPIP5K2, Pi sensing by InsP8 is amplified by a 2-fold activation of 5-InsP7 kinase activity by Pi within the 0–5 mm range. Overall, our data reveal mechanisms that can contribute to specificity in inositol pyrophosphate signaling, regulating InsP8 turnover independently of 5-InsP7, in response to fluctuations in extracellular supply of a key nutrient.
Keywords: inositol phosphate, metabolism, phosphatase, signal transduction, signaling
Introduction
Phosphate has multiple functions that direct the survival of all living organisms: in its organic form, Pi is a component of genomic material, it serves as an energy currency, and it is ubiquitous in cell signaling. Thus, Pi homeostasis is essential to life, but the mechanisms by which this occurs in humans and other metazoans are largely unknown (1, 2). Most of the previous work in this field of research has focused on yeast models (3–5). In particular, recent studies with Saccharomyces cerevisiae have revealed a new function in Pi homeostasis for inositol pyrophosphates (5). The latter are soluble, intracellular signals that contain multiple phosphates and diphosphates; up to seven (InsP7)4 or eight (InsP8) phosphates in total are crammed around a six-carbon inositol ring (see Refs. 6–8 and Fig. 1). In S. cerevisiae, levels of one inositol pyrophosphate, 5-InsP7, track perturbations to Pi homeostasis (5).
FIGURE 1.

Inositol pyrophosphate metabolism. The schematic describes all known mammalian enzyme classes that interconvert InsP6 with InsP8. Note that current thinking (28, 29) has the major route from InsP6 to InsP8 in mammalian cells progressing through 5-InsP7 rather than 1-InsP7.
This Pi-sensing activity of 5-InsP7 appears to reflect it being synthesized by a kinase class (kcs1 in yeast; IP6Ks in metazoans) that exhibits an unusually low affinity for ATP (9, 10). Consequently, cellular levels of 5-InsP7 in yeast decrease in response to the drop in [ATP] that accompanies extracellular [Pi] depletion (5, 11). Furthermore, these ATP-driven changes in 5-InsP7 levels appear to comprise a dynamic signaling response because 5-InsP7 regulates proteins that maintain Pi homeostasis through interactions with their SPX domains (5). However, it is not known to what extent this signaling response is applicable to metazoan cells, which lack orthologs of many of the yeast genes that function in Pi sensing and Pi homeostasis (2).
In the current study, we have searched for links between Pi homeostasis and inositol pyrophosphates in a human model system: the HCT116 intestinal epithelial cell line. This choice reflects the physiological relevance of both small and large fluctuations in [Pi] within the gastrointestinal tract (12). One of our goals has been to investigate whether there are changes in extracellular [Pi] that might cause intracellular [ATP] and [5-InsP7] to co-vary, which as mentioned above is considered to be an IP6K-dependent phenomenon.
Perhaps as a consequence of 5-InsP7 being the most abundant of the inositol pyrophosphates, it has been the focus of much of the literature in this field (6, 8, 13, 14). In the current study, we also study a different inositol pyrophosphate, InsP8 (Fig. 1). We describe some new features to InsP8 turnover that solidify its own, independent cell signaling credentials. This information arises out of our focus on the PPIP5Ks (Fig. 2). The latter enzymes are of general interest; in addition to hosting a kinase domain that phosphorylates 5-InsP7 to InsP8, PPIP5Ks posses a separate phosphatase domain that dephosphorylates InsP8 back to 5-InsP7 (15–17). That is, PPIP5Ks interconvert substrates and products in apparent “futile cycles” (Figs. 1 and 2). Kinase/phosphatase and other covalent modification cycles are a nexus for regulatory inputs into metabolic and signaling pathways (18); in fact this phenomenon is considered a core motif in the field of systems biology (19). However, in general, such competing catalytic activities are hosted by separate proteins for the purposes of compartmentalization and for promoting signaling fidelity (20). Only in rare cases have these apparent benefits been selected against in order that the mutually antagonistic catalytic activities co-exist within a single protein (19, 21, 22). The PPIP5K family is one of these exceptions; representatives from humans, yeasts, and plants contain kinase and phosphatase domains (16), indicating that this bifunctionality has survived at least 1.5 billion years of evolutionary pressure (23).
FIGURE 2.

Domain graphic for human PPIP5Ks. Domain graphics are shown for the human PPIP5Ks used in this study (type 1, BC057395.1; type 2, XM_005271938). For PPIP5K1, amino acid residues defining each domain are numbered as in a previous study, which also defined the intrinsically disordered domain (IDR) (49). These boundaries were matched to those of the corresponding domains in PPIP5K2 by sequence alignments using Clustal Omega. The aligned intrinsically disordered domain boundaries in PPIP5K2 are consistent with those independently predicted from the PSIPRED Protein Sequence Analysis Workbench. The percent sequence identities across each of the three domains are also indicated. Also indicated are the nature and the locations of our engineered mutations in the kinase and phosphatase domains.
Among a number of proposed advantages of having competing catalytic activities in a single, bifunctional protein are the following: (a) preventing signaling incoherence that can otherwise arise due to stochastic fluctuations in the degrees of expression of two separate proteins; (b) robustness, i.e. invariance to quantitative changes of the system's components, including substrate concentration; and (c) increased “parametric sensitivity,” that is, a situation in which signaling output is amplified following relatively small changes in the concentration of a particular parameter, such as an enzyme regulator (19, 21). However, the significance of these phenomena in vivo is dictated by the catalytic parameters of the mutually antagonistic domains (19, 21). Hitherto, we lacked this information. The full kinetic profile for PPIP5Ks has not previously been determined in the full-length versions of these enzymes. Moreover, there is no information in the literature describing the existence of a modulator of either the kinase or the phosphatase activity of any mammalian PPIP5K. The current study addresses these important gaps in our understanding of inositol pyrophosphate turnover. We demonstrate that Pi regulates the catalytic activities of the PPIP5Ks. Furthermore, our data indicate that InsP8 and 5-InsP7 each act through separate mechanisms to individually sense extracellular Pi status.
Results and Discussion
The Effects of Extracellular [Pi] Starvation and Replenishment upon Levels of Inositol Pyrophosphates in HCT116 Cells
Previous work with S. cerevisiae (24) has shown that extracellular [Pi] is sensed by the IP6K that synthesizes the intracellular inositol pyrophosphate 5-InsP7. During a period of Pi starvation, there is a decline in cellular [ATP]. The unusually low affinity of IP6Ks for ATP compels 5-InsP7 levels to fall in parallel (5). As noted in a recent review of this field (24), yeasts are the only organisms that have previously been used to study 5-InsP7 turnover in response to fluctuations in extracellular Pi availability. We have addressed this gap in the field by using the HCT116 human intestinal epithelial cell line as a model system. We prelabeled cells with [3H]inositol for several days, and then we investigated the effects of removal and replenishment of extracellular Pi. We used HPLC to assay the intracellular levels of “higher” InsPs, including InsP5, InsP6, and the inositol pyrophosphates.
First (Fig. 3A), we analyzed cells that had been incubated under Pi-replete conditions (i.e. 2 mm [Pi] (25, 26)). As in all metazoan cells, levels of InsP5 and InsP6 are much higher than those of the inositol pyrophosphates (InsP7 and InsP8); thus for clarity, those regions of the chromatographs that include the inositol pyrophosphates are replotted on an expanded y axis (Fig. 3, A, B, C, and D). The depiction of InsP7 as being the 5-isomer is based on previous work with HCT116 cells in which our HPLC procedures were shown to resolve 5-InsP7 from 1-InsP7; the latter comprises <2% of total InsP7 (27). Indeed, it is generally believed that most of the cell's InsP8 is synthesized from 5-InsP7 rather than 1-InsP7 (28, 29).
FIGURE 3.

The effects of Pi starvation and replenishment upon intracellular levels of inositol phosphates, ATP, and Pi in HCT116 cells. [3H]Inositol-labeled HCT116 cells were treated to four different conditions of extracellular Pi availability as represented schematically by the horizontal bar at the top of each panel (filled bar, 2 mm Pi; empty bar, 0 mm Pi): A, 2 mm Pi for 6 h; B, no Pi for 6 h; C, no Pi for 5. 75 h followed by 2 mm Pi for 0.25 h; D, no Pi for 3 h followed by 2 mm Pi for 3 h. Next, cells were quenched, and the levels of the indicated inositol phosphates (InsP5, InsP6, InsP7, and InsP8) were analyzed by HPLC (see “Experimental Procedures”). Data for InsP7 and InsP8 are replotted on an expanded y axis scale. A–D are representative of three biological replicates; scatter plots (error bars represent S.D.) are compiled from all three experiments in E (InsP5), F (InsP6), G (InsP7), and H (InsP8). The identification of InsP7 as the 5-isomer is based on our previous work with HCT116 cells in which our HPLC procedures were shown to resolve 5-InsP7 from 1-InsP7; the latter is a minor isomer that comprises <2% of total InsP7 (27). The graphics below the x axes correspond to those described above that depict the various extracellular [Pi] conditions. Scatter plots (error bars represent S.D.) in I and J show total intracellular Pi (malachite green/molybdate method; n = 8–10) and ATP (n = 4–7), respectively, determined in parallel experiments with non-radiolabeled cells. The mean values for cell Pi content (nmol/mg of protein) from left to right are as follows (error bars represent S.D.): 62 ± 2.3, 28 ± 1.9, 62 ± 2.7, and 53 ± 2.2; these values closely match the Pi levels that were obtained by an independent enzymatic method (60 ± 5.6, 32 ± 2.5, 59 ± 2.9, and 50 ± 2.7; n = 6).
We next investigated the effects of perturbations to Pi homeostasis. We challenged cells with 6 h of Pi starvation (Fig. 3B). This procedure did not impact levels of InsP5 or InsP6 (Fig. 3, A, B, E, and F), but InsP7 levels decreased by ∼70% (Fig. 3, A, B, and G). This is the first demonstration, for any metazoan cell type, that levels of InsP7 are sensitive to a change in extracellular [Pi]. Nevertheless, InsP8 levels fell by 98%, which is a much greater response to the Pi depletion protocol (Fig. 3, A, B, and H). It has not previously been reported that InsP8 levels are influenced by extracellular [Pi] in any organism (e.g. see Ref. 24). In other words, our data describe a new connection between nutrient status and the poise of a cell signaling cascade.
Next, we substituted the last 15 min of the Pi starvation protocol with the readdition of 2 mm Pi. The levels of 5-InsP7 were not influenced by this 15-min period of Pi replenishment (Fig. 3, B, C, and G). In contrast, there was a substantial rescue of the InsP8 peak (Fig. 3, B, C, and H). These data testify to an acute effect of extracellular Pi status upon InsP8 synthesis that is independent of the supply of 5-InsP7 (i.e. the major InsP7 precursor for InsP8; see above). We revisit this point below.
In further experiments, a 3-h period of Pi starvation was followed by 3 h of 2 mm Pi readdition. After this protocol, levels of 5-InsP7 were similar to those found in cells incubated with 2 mm Pi for 6 h (Fig. 3, A, D, and G). In contrast, levels of InsP8 in Pi-starved/Pi-replenished cells were 80% higher than those of control cells (Fig. 3, A, D, and H), suggestive of a longer term adaptive response to a period of Pi starvation. Indeed, a major conclusion to draw from all of these experiments is that InsP8 reacts more dramatically to fluctuations in extracellular [Pi] than does 5-InsP7.
The Effects of Extracellular [Pi] Starvation and Replenishment upon Levels of Intracellular [Pi] and [ATP] in HCT116 Cells
How do intracellular inositol pyrophosphates sense short term fluctuations in extracellular Pi levels? As a first step toward answering that question, we investigated whether intracellular [Pi] tracked the imposed changes in extracellular [Pi]. We found that our 6-h Pi starvation protocol reduced intracellular [Pi] by 55% (Fig. 3I); only 15 min of Pi replenishment was sufficient to completely restore intracellular [Pi] (Fig. 3I). The recovery in InsP8 lags behind that for Pi (Fig. 3, H and I); the rate of change in InsP8 levels depends upon dynamic fluctuations in InsP8 synthesis and metabolism (see below).
How might inositol pyrophosphate levels be modified by changes in intracellular [Pi]? Previous work has indicated that Pi levels influence ATP production (30). Indeed, we found that our Pi starvation protocol reduced [ATP] by 66% (Fig. 3J), i.e. to a value of about 1.5 mm, based on the concentration of ATP normally being around 5 mm (31–33). Thus, the associated drop in 5-InsP7 levels (Fig. 3, A, B, and G) may reflect the unusually low affinity (Km approximately 1 mm) of IP6Ks for ATP (9, 10). Both ATP and 5-InsP7 were at normal levels when a 3-h period of Pi starvation was followed by 3 h of replenishment with 2 mm [Pi] (Fig. 3, G and J).
On the other hand, there is no reason to suspect that synthesis of InsP8 by the PPIP5Ks would be influenced by these changes in [ATP]. In contrast to IP6Ks, the PPIP5Ks exhibit a low Km value for ATP (20 μm (29)), so their kinase activities would be saturated even by the reduced levels of [ATP] caused by 6 h of Pi starvation (Fig. 3J). We therefore searched for other mechanisms by which Pi might regulate InsP8 turnover. There are precedents for Pi itself being a physiologically relevant regulator of both kinases and phosphatases (34, 35). Thus, we decided to study the synthesis and metabolism of InsP8 by the human bifunctional PPIP5Ks in vitro and then investigate whether these catalytic activities are regulated by Pi. To our knowledge, no previous study has characterized the competing kinase and phosphatase activities of full-length PPIP5Ks purified from a human expression system.
Kinetic Analysis of the Bifunctional Human PPIP5Ks: the Interconversion of 5-InsP7 and InsP8
Mammals express two PPIP5Ks, type 1 and type 2 (Fig. 2). HEK cells were used as hosts for the expression of full-length, recombinant wild-type PPIP5K1 and PPIP5K2, each of which were engineered to possess an N-terminal FLAG tag to facilitate purification with anti-FLAG resin. Upon SDS-PAGE, both recombinant proteins eluted at their expected molecular sizes (Fig. 4).
FIGURE 4.
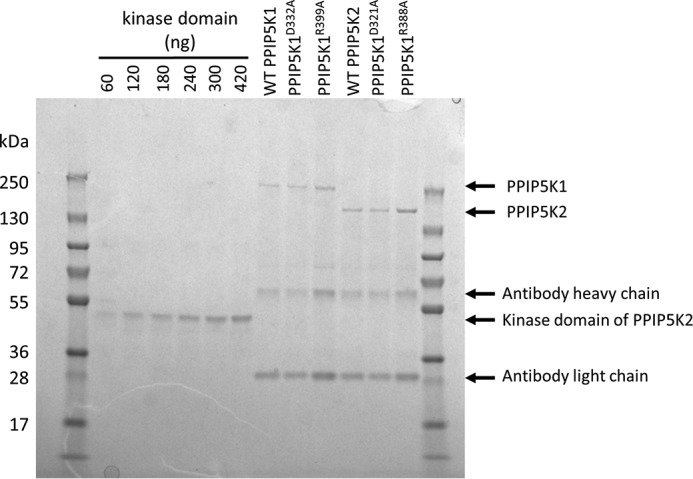
SDS-PAGE of purified human recombinant PPIP5Ks. Shown is the analysis by SDS-PAGE and Coomassie Brilliant Blue staining of 5-μl aliquots of each of the indicated recombinant PPIP5Ks after immunoaffinity purification. There are six lanes toward the left-hand side of the gel that are labeled with the quantities of the recombinant human PPIP5K2 kinase domain (39) that were used as standards to calibrate through densitometry the concentrations of the PPIP5K constructs. The lanes containing PPIP5K1 serve as “no-transfection” controls for PPIP5K2 and vice versa.
We assayed the kinase activities of both PPIP5K1 and PPIP5K2 in incubations with physiologically relevant concentrations of 5-InsP7 (1 μm; see Refs. 36–38) and MgATP (5 mm; see Refs. 31–33). These levels of substrates are up to 15- and 200-fold higher, respectively, than their Km values for kinase activity (15, 29, 38). Thus, our reaction conditions are zero-order (see below for empirical confirmation). In parallel, we assayed the kinase mutants PPIP5K1D332A and PPIP5K2D321A (15, 39); these enzymes did not convert 5-InsP7 to InsP8 (data not shown), indicating that our enzyme preparations are free of any contaminating 5-InsP7 kinase activities.
Analysis of the wild-type kinase activities by HPLC confirmed that the InsP8 product accumulated (Fig. 5, A, B, and C). Nevertheless, when these assays were performed with phosphatase mutants (PPIP5K1R399A and PPIP5K2R388A), a 2.4–4-fold larger net formation of InsP8 was observed (Fig. 5, A, B, and C). Thus, the phosphatase activities constrain but do not overwhelm kinase activities.
FIGURE 5.

HPLC chromatographs showing net 5-InsP7 kinase activities and InsP8 phosphatase activities of wild-type and phosphatase mutant versions of human PPIP5Ks. Kinase assays, performed as described under “Experimental Procedures” for 120 min with 1 μm 5-InsP7, contained the following: no enzyme (open triangles) (A), 90 ng of either wild-type PPIP5K1 (open circles) or PPIP5K1R399A (closed circles) (B), or 90 ng of either wild-type PPIP5K2 (open circles) or PPIP5K2R388A (closed circles) (C). Assays were quenched, neutralized, and then analyzed by HPLC (see “Experimental Procedures”). Phosphatase assays, performed as described under “Experimental Procedures” for 30 min with 1 μm InsP8, contained the following: no enzyme (open triangles) (D), 25 ng of either wild-type PPIP5K1 (open circles) or PPIP5K1R399A (closed circles) (E), or 40 ng of either wild-type PPIP5K2 (open circles) or PPIP5K2R388A (closed circles) (F). Data shown are representative of three to four biological replicates.
We next verified that wild-type PPIP5K1 and PPIP5K2 both dephosphorylate InsP8 to 5-InsP7 (Fig. 5, D, E, and F). We also confirmed that the single site mutations in the phosphatase domain nearly completely impaired InsP8 hydrolysis (Fig. 5, D, E, and F). This observation also usefully confirms that there is negligible contamination of our enzyme preparations with any other InsP8 phosphatase activities. In fact, the only other class of mammalian enzyme known to hydrolyze InsP8 is DIPP (40), and, even if that had been present, it would have dephosphorylated InsP8 to InsP6 without appreciable accumulation of 5-InsP7 (see below).
Clearly, the phosphatase mutants of the PPIP5Ks yield the more accurate rates of the 5-InsP7 kinase reactions (Figs. 5 and 6A). In this respect, we found that PPIP5K2 is 6-fold more active than is PPIP5K1. Our phosphatase mutants were also useful in confirming that zero-order conditions prevail when physiologically relevant concentrations of 5-InsP7 are phosphorylated by PPIP5Ks: reaction rates were not increased when the 5-InsP7 concentration was increased from 1 to 5 μm (representative examples: PPIP5K1R399A, 1.2 and 0.9 nmol/mg of protein/min, respectively; PPIP5K2R388A, 6.4 and 5.8 nmol/mg of protein/min, respectively).
FIGURE 6.
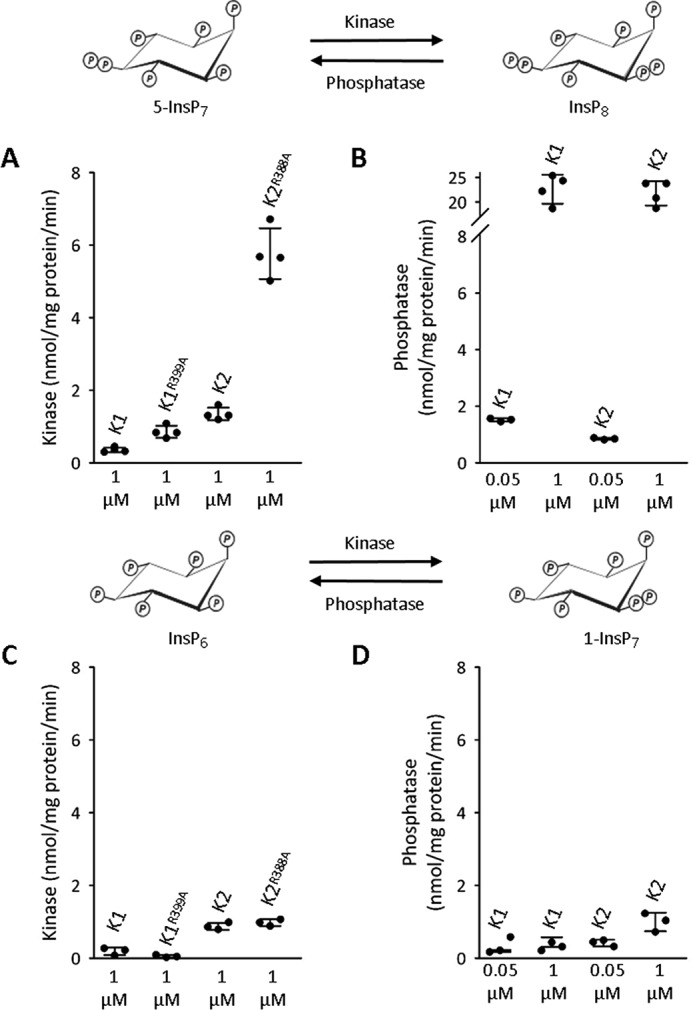
Kinase and phosphatase activities of recombinant PPIP5Ks. All assays were performed as described under “Experimental Procedures” with either wild-type or phosphatase mutant versions of PPIP5Ks (K1, PPIP5K1; K2, PPIP5K2) plus either 1 or 0.05 μm substrate as indicated beneath each scatter plot. A and B show kinase and phosphatase activities toward 5-InsP7 or InsP8, respectively (as indicated by the graphic above the graphs). C and D show kinase and phosphatase activities toward InsP6 or 1-InsP7, respectively (as indicated by the graphic above the graphs). Error bars represent S.D. and are derived from three to four biological replicates.
In the assays of phosphatase activities toward InsP8 described above, we incubated the PPIP5Ks with 0.05 μm InsP8, which is the estimated concentration of this inositol pyrophosphate in HCT116 cells cultured in DMEM/F-12 (27). In this reaction condition, the InsP8 phosphatase activities (0.8–1.5 nmol/mg of protein/min; Fig. 6B) lie in a range that is similar to that for the kinase activities (determined by assaying the phosphatase mutants: 0.7–4.5 nmol/mg of protein/min; Fig. 6A). These same data further indicate that, for PPIP5K2, the 5-InsP7 kinase/InsP8 phosphatase ratio slightly favors the kinase activity. For PPIP5K1, the phosphatase activity is favored. Such subtle differences between the two PPIP5K isoforms may serve some as yet unsuspected physiological purpose.
Our next goal was to determine how the InsP8 phosphatase activities might respond to elevated concentrations of substrate. Therefore, we assayed phosphatase activities against 1 μm InsP8; the reaction rates for both PPIP5Ks are 15- 20-fold higher than those observed with 0.05 μm InsP8 (Fig. 6B). Thus, we conclude that the phosphatase activities in vivo may be modulated by fluctuations to supply of substrate (i.e. InsP8). In contrast, the 5-InsP7 kinase activities proceed under zero-order conditions in vivo (see above and Ref. 29). Mathematical modeling of just such a situation for a kinase/phosphatase bifunctional protein (zero-order for the kinase; first-order for the phosphatase) has established that a biological outcome is a degree of concentration robustness for the kinase product (21), in this case InsP8. In other words, it can be concluded that InsP8 is inherently insensitive to changes in 5-InsP7 levels. This property of PPIP5Ks could promote specificity of inositol pyrophosphate signaling by stabilizing InsP8 levels during periods of stimulus-dependent regulation of 5-InsP7 levels. Conversely, such concentration robustness indicates that the response of InsP8 to changes in Pi homeostasis (Fig. 3) reflects active regulation of InsP8 turnover and not mass action effects due to fluctuations in 5-InsP7 supply.
There are three additional conclusions that can be drawn from our kinetic data (Figs. 5 and 6). First, these results provide the first direct demonstration that the phosphatase activity of mammalian PPIP5Ks has the potential to significantly restrict net kinase activity in the context of the full-length proteins. That is, we conclude that so-called futile cycling by bifunctional PPIP5Ks is a realistic physiological scenario. Second, the activity of the kinase domain in full-length PPIP5K2 is 30-fold below its potential maximal capacity (190 nmol/mg of protein/min; as recorded for the isolated kinase domain expressed in Escherichia coli (29)). This catalytic constraint upon the kinase domain, when expressed as a full-length protein in a human cell type, could reflect covalent modification and/or a conformational constraint enforced by the other protein domains. Irrespective of the mechanism, the submaximal kinase activity in the full-length protein can be viewed as a selective advantage: at steady-state levels of 5-InsP7 (1 μm) and InsP8 (0.05 μm), the rates of the kinase and phosphatase reactions are similar, so in such a situation, the slower these individual rates, the lower the amount of ATP that must be expended to maintain a given cellular content of InsP8. A third conclusion that emerges from the phosphatase and kinase activities of full-length PPIP5Ks being similar is that it is intuitive that it maximizes the sensitivity with which a modulator of either activity could affect net flux through the cycle. This is a topic we return to later (see below).
Kinetic Analysis of the Bifunctional Human PPIP5Ks: the Interconversion of InsP6 and 1-InsP7
PPIP5Ks are also capable of catalyzing a separate cycle of competing kinase and phosphatase activities that interconvert InsP6 with 1-InsP7 (Fig. 1). We next used the wild-type and phosphatase mutant PPIP5K constructs to determine the relative rates of these particular reactions. For the kinase assays (Fig. 6C), we set InsP6 concentrations to 1 μm. Most estimates of cellular levels of InsP6 are 10 μm or more (36–38), but the use of 1 μm increases assay sensitivity while still being more than sufficient to saturate PPIP5Ks with this substrate and obtain Vmax values (15, 29). For the phosphatase assays (Fig. 6D), we initially used a concentration of 0.05 μm 1-InsP7 (because cellular levels of the latter are 2–10% of total InsP7 (27, 28)). The maximum rates of the kinase activities toward InsP6 (0.2–1 nmol/mg of protein/min; Fig. 6C) are 4–6-fold lower than the kinase activities toward 5-InsP7 (Fig. 6A). Furthermore, the rates of the phosphatase activities toward 0.05 μm 1-InsP7 (0.2–0.4 nmol/mg of protein/min; Fig. 6C) are 4-fold lower than the phosphatase activities toward InsP8 (Fig. 6A). That is, kinetic parameters dictate that substrate cycling through InsP6 and 1-InsP7 is quantitatively less significant than is cycling through 5-InsP7 and InsP8. The latter conclusion is directly relevant to current thinking (6, 28) that the major route from InsP6 to InsP8 progresses through 5-InsP7 rather than 1-InsP7.
Although there is no direct evidence that the intracellular concentration of 1-InsP7 in mammalian cells exceeds 0.05 μm (27), we also studied in vitro PPIP5K phosphatase activity toward this substrate at a concentration of 1 μm (Fig. 6D). At this higher substrate concentration, the phosphatase activity was double that against 0.05 μm 1-InsP7 (Fig. 6D), indicating that this activity (like the phosphatase activity toward InsP8; see above) does not operate under zero-order conditions in vivo; instead it is capable of being regulated by substrate supply. Armed with an improved kinetic understanding of the operation of the PPIP5Ks, we next sought information concerning how changes in intracellular Pi might mediate the effects upon InsP8 turnover described above.
Regulation of PPIP5Ks by Inorganic Phosphate in Vitro
We investigated whether Pi might directly modify the catalytic activities of the PPIP5Ks. We found that Pi reduced the rate of InsP8 hydrolysis by our preparations of recombinant PPIP5K1 and PPIP5K2 in a dose-dependent manner (90 and 40% inhibition, respectively, by 1 mm Pi; Fig. 7, A, B, C, and D). At the very least, these data indicate that cytoplasmic Pi will alter the kinase/phosphatase poise of the PPIP5Ks in favor of InsP8 synthesis. Moreover, these data suggest a mechanism by which Pi starvation of HCT116 cells promotes a loss of InsP8 levels (Fig. 3, A, B, and H): the associated depletion of intracellular Pi (Fig. 3I) could relieve its inhibition of InsP8 phosphatase activity. Equally, the restoration of InsP8 levels upon readdition of extracellular Pi (Fig. 3, C, D, and H) may be explained by the replenishment of intracellular Pi (Fig. 3I), which inhibits the PPIP5K phosphatase activities (Fig. 7, B and D). Our data therefore represent the first demonstration of a mechanism for regulating the catalytic activities of PPIP5K in response to an extracellular stimulus (in this case nutrient availability). It is of further interest that DIPP-mediated InsP8 phosphatase activity proceeds without InsP7 accumulation (Fig. 7, E and F), and is not inhibited by Pi (Fig. 7, E and F). That is, Pi is not a general inhibitor of all inositol pyrophosphate phosphatases. Moreover, this observation excludes any residual possibility that contaminating DIPPs contribute to the phosphatase activities of our PPIP5K preparations.
FIGURE 7.

The effects of [Pi] upon the phosphatase activities of PPIP5K1, PPIP5K2, and DIPP1. Phosphatase assays, performed as described under “Experimental Procedures,” contained 1 μm [3H]InsP8, either zero [Pi] (open circles) or 5 mm [Pi] (closed circles), and either 25 ng of PPIP5K1 (A and B), 40 ng of PPIP5K2 (C and D), or 33 ng of DIPP1 (E and F). Assays were quenched, neutralized, and then analyzed by HPLC (see “Experimental Procedures”); representative chromatographs are shown. Insets to B and D depict additional experiments (error bars represent S.E. from three biological replicates) in which the concentration of Pi was varied as shown.
Finally, we found that not only does Pi inhibit PPIP5K2 phosphatase activity (Fig. 7D) but also its kinase activity is activated 2-fold as the concentration of Pi was raised from 0 to 5 mm (Fig. 8). This is the first description of a mechanism by which the kinase activity of any PPIP5K may be regulated. Reciprocal regulation of competing catalytic activities by a single regulator is a particularly sensitive control mechanism. Thus, it is possible that, in vivo, quite small fluctuations in cytoplasmic [Pi] could be amplified into proportionately large changes in net [InsP8] synthesis by cytoplasmic (15) PPIP5Ks. Remarkably, the activation of kinase activity is also specific to PPIP5K2: Pi has no effect upon the kinase activity of PPIP5K1 (Fig. 8). These data underscore a significant difference in the regulation of the activities of the tightly conserved catalytic domains of the two PPIP5Ks.
FIGURE 8.
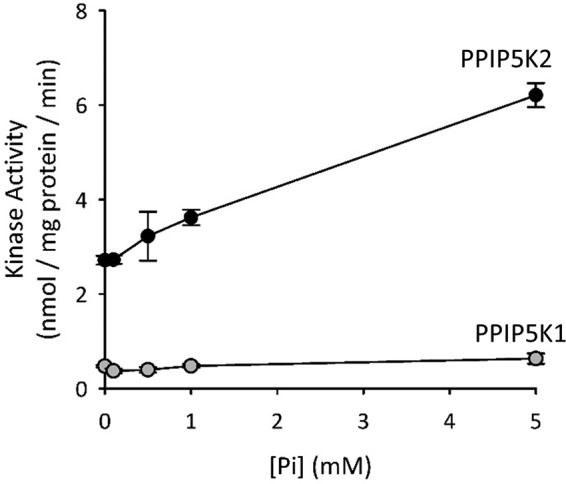
The effects of varying [Pi] (0–5 mm) upon the 5-InsP7 kinase activities of PPIP5K1 and PPIP5K2. Kinase assays, performed as described under “Experimental Procedures,” contained 1 μm 5-[3H]InsP7, the indicated concentrations of Pi, and either PPIP5K1R399A (gray circles) or PPIP5K2R388A (black circles).Data points represent means and error bars represent S.E. from three biological replicates.
Elevated Extracellular [Pi] Specifically Promotes InsP8 Accumulation
Our main experimental paradigm has been to impose a nutritional challenge upon cultured cells: deprive and then restore the levels of Pi found in serum. Other groups have studied the impact upon signal transduction cascades when cells are incubated with “high” [Pi] (5–10 mm) (2, 30). We noted that such dramatic fluctuations in extracellular Pi concentration (up to 10 mm or more) are physiologically relevant for cells that line the gastrointestinal tract (12). We therefore used the HCT116 model to study the effects upon inositol pyrophosphate turnover of elevating extracellular Pi from 1 to 6 mm for periods of up to 1 h. This procedure did not significantly affect levels of either InsP5, InsP6, or 5-InsP7 (Fig. 9, A, B, and C). The absence of an effect upon 5-InsP7 illustrates how the reaction of inositol pyrophosphates to changes in Pi homeostasis may depend upon the nature of the experimental protocol. We propose that levels of 5-InsP7 do not respond to the high [Pi] protocol because [ATP] and hence IP6K activity (see above) are also not altered (Fig. 9D).
FIGURE 9.

The effects of elevated extracellular [Pi] upon phosphate homeostasis and inositol phosphate levels in HCT116 cells. HCT116 cells were labeled with [3H]inositol in medium containing 1 mm Pi and then an additional 5 mm Pi (as a NaH2PO4/Na2HPO4 mixture (pH 7.4)) was added for intervals of up to 60 min. Next, cells were quenched, and the levels of the indicated inositol phosphates (InsP5, InsP6, InsP7, and InsP8) were analyzed by HPLC (see “Experimental Procedures”). A and B show representative HPLC chromatographs for the 0- and 60- min time points; data for InsP7 and InsP8 are replotted on an expanded y axis scale. Data are representative of three biological replicates. C shows time course data (InsP5, InsP6, and InsP7; mean ± S.E., n = 3) from all experiments. D and E show time course data (error bars represent S.E.) for intracellular levels of ATP and Pi (malachite green/molybdate method), respectively, determined in parallel experiments with non-radiolabeled cells (n = 6–7) incubated for the indicated times with 6 mm extracellular [Pi]. The data for cell Pi content (nmol/mg of protein) at 0, 15, 30, 45, and 60 min (error bars represent S.E.; 56 ± 4.3, 59 ± 4.7, 58 ± 3.3, and 60 ± 1.8) closely match the Pi levels that we obtained by an independent enzymatic method (54 ± 3.5, 57 ± 3.7, 56 ± 2.7, and 62 ± 4.3; n = 6–7). F shows time course data for InsP8 (*, p < 0.05; **, p < 0.02; paired t test). G shows representative HPLC chromatographs from the region in which InsP8 elutes (as indicated by the elution profile (open circles) for an [3H]InsP8 standard from a parallel run); cell extracts (closed circles) were derived from [3H]inositol-labeled PPIP5K−/− HCT116 cells (see Ref. 27) that had been incubated for 1 h with either 1 or 6 mm Pi as indicated.
In response to elevated extracellular [Pi], intracellular [Pi] trended higher (by 9–14%; Fig. 9E legend) but not with statistical significance. Nevertheless, InsP8 levels responded dramatically: a 2.3-fold increase within 15 min and a 5-fold elevation after 1 h (Fig. 9, A, B, and F). This response of InsP8 consolidates our discovery (see above) that it is more sensitive to changes in extracellular [Pi] than is 5-InsP7. In view of how acutely and specifically InsP8 responds to high [Pi], we sought further evidence for the participation of PPIP5Ks in this event. We generated an HCT116 cell line in which both PPIP5Ks have been knocked out using CRISPR (27). These cells do not synthesize InsP8 irrespective of the concentration of extracellular [Pi] (Fig. 9G).
Concluding Comments
Until quite recently, signal transduction cascades and metabolic circuits were not recognized to be intimately connected; now it is appreciated that a key aspect of cellular and organismal homeostasis is the acute regulation of cell signaling pathways by the levels of a particular metabolite or nutrient (41). In particular, inositol pyrophosphates have gained considerable attention for their actions that dovetail signaling with metabolism (6, 7, 24, 42). In our study, we add several new aspects to this important research topic by showing that InsP8 levels sense and respond to fluctuations in the extracellular levels of a vital nutrient, Pi.
Our in vitro data describe physiologically relevant mechanisms by which Pi may directly regulate the catalytic activities of the PPIP5Ks: inhibition of InsP8 phosphatase (Fig. 7) and stimulation of 5-InsP7 kinase (Fig. 8). Prior to this study, no regulators of either the kinase or phosphatase activities of mammalian PPIP5Ks had been described. Reciprocal regulation of any covalent modification cycle by a single modulator is a particularly efficient regulatory paradigm. Moreover, it is intuitive that the sensitivity of such a process is enhanced when the two opposing activities proceed at approximately equal rates as is the case for PPIP5K2 under physiologically relevant conditions (Fig. 6, A and B). Thus, there may be scenarios in which proportionately large changes in [InsP8] may be promoted by quite small fluctuations in cytoplasmic [Pi] that are beyond the sensitivity of our assays, which record total intracellular [Pi] (Fig. 9E). A more precise spatiotemporal understanding of the inter-relationships between Pi and InsP8 in vivo requires additional information that is not readily obtained: a quantitative resolution of their separation into different cellular pools (2, 43). Furthermore, differential levels of expression of PPIP5K1 versus PPIP5K2 could contribute to quantitative and qualitative cell type-specific differences in InsP8 responses to extracellular [Pi] because this anion only stimulates the kinase domain of PPIP5K2 and not that in PPIP5K1 (Fig. 8). It will be important to determine how widespread in other cell types is the impact of fluctuations in extracellular [Pi] upon intracellular levels of InsP8.
It is also possible that regulation of PPIP5K activity by Pi is not the only cell signaling mechanism that regulates InsP8 levels, particularly those that might be activated by high extracellular [Pi] (2, 30). This would not be surprising considering the many interactions between inositol pyrophosphate signaling and metabolic homeostasis (6, 7, 24, 42) and the domain complexities of the PPIP5Ks (Fig. 2).
Our new data are also intriguing in light of recent data derived from yeast that show that 5-InsP7 rheostatically regulates mechanisms of Pi homeostasis mediated by SPX domains (5). The only human protein known to contain an SPX domain is XPR1, which transports Pi out of cells (5). In the current study with a human cell type, we show [ATP] and [5-InsP7] to co-vary in their response to Pi starvation. This is the first time such a response has been observed in any metazoan model. In addition, we show that InsP8 senses changes in Pi status by separate mechanisms that exhibit greater sensitivity as compared with 5-InsP7. It could be useful to study whether InsP8 is a homeostatic regulator of XPR1, perhaps adjusting the rate of cellular Pi efflux depending upon organismal Pi status. This would be a new direction for this field, which prior to our study had mainly focused on the biological actions of 5-InsP7 (see Refs. 8 and 14). The possibility of there being other types of InsP8 receptor should not be ignored. Finally, our results raise the possibility that dysregulation of inositol pyrophosphate signaling by genetic or environmental factors could conceivably contribute to mechanisms of toxicity of unbalanced [Pi] homeostasis (1).
Experimental Procedures
Cell Culture
HEK293T cells and HCT116 cells were obtained from ATCC; HCT116 cells in which both PPIP5K1 and PPIP5K2 were knocked out using CRISPR were derived as described previously (27). The culture medium (Thermo Fisher Scientific) was DMEM/F-12 for HCT116 cells and DMEM for HEK293T cells, each supplemented with 10% fetal bovine serum (Gemini Bio Products) and 100 units/ml penicillin-streptomycin (Thermo Fisher Scientific) at 37 °C in 5% CO2.
Measurement of Intracellular Levels of Pi, ATP, and Inositol Phosphates
For assays of intracellular ATP and Pi, 3 × 105 cells/well were seeded in a 12-well dish and cultured in DMEM/F-12 (containing 1 mm Pi) for 2 days at which point cultures were 70% confluent. For some experiments, an additional 5 mm Pi was added (as a NaH2PO4/Na2HPO4 mixture (pH 7.4)), for the times indicated (see “Results and Discussion”). In other experiments, cells were washed three times with Pi-free DMEM (ThermoFisher Scientific) and then incubated in either Pi-free DMEM or DMEM plus 2 mm NaH2PO4 for the times indicated (see “Results and Discussion”). To terminate these assays, cells were washed three times in ice-cold buffer containing 20 mm HEPES (pH 7.2), 150 mm NaCl and then lysed by agitation in 1 ml of wash buffer containing 1% Triton X-100 for 5 min at 4 °C. The ATP was assayed using a commercial kit (Molecular ProbesTM, A22066). For the Pi assays, the lysate was cleared of debris by a brief centrifugation, and then the resulting supernatant was centrifuged through an Amicon Ultra filter (10-kDa molecular mass cutoff; 5 min at 4 °C). The Pi in the eluate was generally assayed with a standard malachite green/molybdate method (44). Similar data were obtained (see figure legends) using an enzymatic assay (Cell Biolabs, Inc., catalogue number STA-685) that avoids potential contamination through acid-dependent hydrolysis of organic phosphates. The data that were obtained were normalized to protein concentration, which was measured using the PierceTM BCA protein assay kit (product number 23225).
To assay inositol phosphates, 1 × 106 cells were seeded in a 10-cm dish and cultured for 3 days in 7 ml of medium supplemented with 10 μCi/ml [3H]inositol (American Radiolabeled Chemicals) at which point cultures were 70% confluent. Cells were then incubated in medium with various Pi concentrations as described above. Cells were acid-quenched, and the inositol phosphates were extracted and analyzed by a 3 × 250-mm CarboPacTM PA200 HPLC column (ThermoFisher Scientific) all as described previously (27).
Preparation of Recombinant Proteins
Recombinant human DIPP1 and the PPIP5K2 kinase domain were prepared as described previously (17, 39). We also constructed pDEST515 plasmids (45) hosting wild-type hPPIP5K1 (BC057395.1) and hPPIP5K2 (NM_001345875), each with an N-terminal FLAG tag. Based on previous identifications of conserved catalytically essential residues (16, 46), we used the Q5 site-directed mutagenesis kit (New England Biolabs) to prepare constructs encoding single site mutant PPIP5Ks. The paired primers were as follows (mutagenic nucleotides are bold and underlined): hPPIP5K1: D332A: forward, TTTGTGTGTGCTGTCAATGGC; reverse, GGAATGACCATTGGCACG; R399A: forward, TGCAATTATTGCTCATGGGGATCGTACTC; reverse, ATGACACAACGAAGTTCC; hPPIP5K: D321A: forward, CTATGTCTGTGCTGTCAATGGCTTC; reverse, GACTGTCCATTGGCCCGT; R388A: forward, AGCTGTTATAGCTCATGGGGATCGAACAC; reverse, ATGACACATCTAAGTTCCATC. All constructs were confirmed by DNA sequencing. HEK293 cells were transiently transfected with each of these plasmids using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. Cells were harvested 16–20 h after transfection and lysed in ice-cold buffer containing 10 mm HEPES, 130 mm NaCl, 1% Triton X-100, 10 mm NaF, 10 mm Na2HPO4, 10 mm sodium pyrophosphate, and protease inhibitor mixture (Roche Applied Science). All subsequent steps were performed on ice in an anaerobic chamber (Bactron CAT180). The FLAG-tagged PPIP5K proteins were immunopurified using FLAG M2 affinity gel (Sigma). Purified FLAG-PPIP5K proteins were analyzed by SDS-PAGE and stained with Coomassie Blue. Densitometry analysis (ImageJ) was used to calculate the amounts of PPIP5K loaded onto the gel by reference to additional lanes loaded with known amounts of the human recombinant PPIP5K2 kinase domain.
Kinase and Phosphatase Assays
PPIP5K kinase activities were routinely measured at 37 °C in 1–2-h incubations comprising 100 μl of assay buffer containing 1 mm Na2EDTA, 50 mm KCl, 20 mm HEPES (pH 7.2), 7 mm MgCl2, 5 mm ATP, 0.5 mg/ml BSA, and either 1 μm InsP6 (Calbiochem) or 1 μm chemically synthesized 5-InsP7 (47). PPIP5K phosphatase activities were measured at 37 °C in 30-min incubations comprising 100 μl of assay buffer containing 1 mm Na2EDTA, 50 mm KCl, 20 mm HEPES (pH 7.2), 2 mm MgCl2, 0.5 mg/ml BSA, and either 1 or 0.05 μm chemically synthesized 1-InsP7 (48) or InsP8 (30). Each incubation also contained ∼2000 dpm of the corresponding 3H-labeled substrate. [3H]InsP6 was purchased from PerkinElmer Life Sciences; the [3H]inositol pyrophosphates were each prepared as described elsewhere (29).
DIPP1 phosphatase activity was measured at 37 °C in 30-min incubations comprising 100 μl of assay buffer containing 33 ng of enzyme, 1 μm [3H]InsP8 (approximately 4500 dpm), 1 mm Na2EDTA, 50 mm KCl, 20 mm HEPES (pH 7.2), 2 mm MgCl2, 0.5 mg/ml BSA, and, where indicated, 0.1–5 mm KH2PO4. All assays were quenched by addition of 0.2 volume of 2 m perchloric acid plus 1 mg/ml InsP6 and then neutralized (29). Reactions were then analyzed by HPLC chromatography using on a 4.6 × 125-mm Partisphere strong anion exchange HPLC column as previously described (29).
Author Contributions
C. G. performed most of the experiments and analyzed the results. H.-N. N. and H. W. also performed experiments and analyzed results. A. H. and H. J. J. synthesized essential reagents. C. G., H.-N. N., H. W., X. D., and S. B. S. contributed to project conception and the design of experiments. S. B. S. wrote most of the paper with contributions from all of the other coauthors.
Acknowledgment
We thank Dr. Jeremy Gunawardena for comments on a draft of the manuscript.
This work was supported in part by the Intramural Research Program of the NIEHS, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- InsP7
- diphosphoinositol pentakisphosphate
- 1-InsP7
- 1-diphosphoinositol 2,3,4,5,6-pentakisphosphate
- InsP6
- inositol hexakisphosphate
- 5-InsP7
- 5-diphosphoinositol 1,2,3,4,6-pentakisphosphate
- InsP8
- 1,5-bisdiphosphoinositol 2,3,4,6-tetrakisphosphate
- XPR1
- xenotropic and polytropic retrovirus receptor 1
- PPIP5K
- diphosphoinositol-pentakisphosphate kinase
- SPX
- SYG1/Pho81/XPR1 proteins
- IP6K
- inositol-hexakisphosphate kinase
- InsP5
- inositol pentakisphosphate
- DIPP
- diphosphoinositol-polyphosphate phosphohydrolase
- CRISPR
- clustered regularly interspaced short palindromic repeats.
References
- 1. Komaba H., and Fukagawa M. (2016) Phosphate-a poison for humans? Kidney Int. 90, 753–763 [DOI] [PubMed] [Google Scholar]
- 2. Bergwitz C., and Jüppner H. (2011) Phosphate sensing. Adv. Chronic. Kidney Dis. 18, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee Y. S., Huang K., Quiocho F. A., and O'Shea E. K. (2008) Molecular basis of cyclin-CDK-CKI regulation by reversible binding of an inositol pyrophosphate. Nat. Chem. Biol. 4, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lenburg M. E., and O'Shea E. K. (1996) Signaling phosphate starvation. Trends Biochem. Sci. 21, 383–387 [PubMed] [Google Scholar]
- 5. Wild R., Gerasimaite R., Jung J. Y., Truffault V., Pavlovic I., Schmidt A., Saiardi A., Jessen H. J., Poirier Y., Hothorn M., and Mayer A. (2016) Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 352, 986–990 [DOI] [PubMed] [Google Scholar]
- 6. Saiardi A. (2012) How inositol pyrophosphates control cellular phosphate homeostasis? Adv. Biol. Regul. 52, 351–359 [DOI] [PubMed] [Google Scholar]
- 7. Shears S. B. (2009) Diphosphoinositol polyphosphates: metabolic messengers? Mol. Pharmacol. 76, 236–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thota S. G., and Bhandari R. (2015) The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. J. Biosci. 40, 593–605 [DOI] [PubMed] [Google Scholar]
- 9. Voglmaier S. M., Bembenek M. E., Kaplin A. I., Dormán G., Olszewski J. D., Prestwich G. D., and Snyder S. H. (1996) Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc. Natl. Acad. Sci. U.S.A. 93, 4305–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saiardi A., Erdjument-Bromage H., Snowman A. M., Tempst P., and Snyder S. H. (1999) Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 9, 1323–1326 [DOI] [PubMed] [Google Scholar]
- 11. Lonetti A., Szijgyarto Z., Bosch D., Loss O., Azevedo C., and Saiardi A. (2011) Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. J. Biol. Chem. 286, 31966–31974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marks J., Lee G. J., Nadaraja S. P., Debnam E. S., and Unwin R. J. (2015) Experimental and regional variations in Na+-dependent and Na+-independent phosphate transport along the rat small intestine and colon. Physiol. Rep. 3, e12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shears S. B. (2015) Inositol pyrophosphates: why so many phosphates? Adv. Biol. Regul. 57, 203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saiardi A. (2012) Cell signalling by inositol pyrophosphates. Subcell. Biochem. 59, 413–443 [DOI] [PubMed] [Google Scholar]
- 15. Fridy P. C., Otto J. C., Dollins D. E., and York J. D. (2007) Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J. Biol. Chem. 282, 30754–30762 [DOI] [PubMed] [Google Scholar]
- 16. Mulugu S., Bai W., Fridy P. C., Bastidas R. J., Otto J. C., Dollins D. E., Haystead T. A., Ribeiro A. A., and York J. D. (2007) A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science 316, 106–109 [DOI] [PubMed] [Google Scholar]
- 17. Wang H., Nair V. S., Holland A. A., Capolicchio S., Jessen H. J., Johnson M. K., and Shears S. B. (2015) Asp1 from Schizosaccharomyces pombe binds a [2Fe-2S]2+ cluster which inhibits inositol pyrophosphate 1-phosphatase activity. Biochemistry 54, 6462–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Newsholme E. A., Arch J. R., Brooks B., and Surholt B. (1983) The role of substrate cycles in metabolic regulation. Biochem. Soc. Trans. 11, 52–56 [DOI] [PubMed] [Google Scholar]
- 19. Dasgupta T., Croll D. H., Owen J. A., Vander Heiden M. G., Locasale J. W., Alon U., Cantley L. C., and Gunawardena J. (2014) A fundamental trade-off in covalent switching and its circumvention by enzyme bifunctionality in glucose homeostasis. J. Biol. Chem. 289, 13010–13025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levine J., Kueh H. Y., and Mirny L. (2007) Intrinsic fluctuations, robustness, and tunability in signaling cycles. Biophys. J. 92, 4473–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Straube R. (2013) Sensitivity and robustness in covalent modification cycles with a bifunctional converter enzyme. Biophys. J. 105, 1925–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dexter J. P., Dasgupta T., and Gunawardena J. (2015) Invariants reveal multiple forms of robustness in bifunctional enzyme systems. Integr. Biol. 7, 883–894 [DOI] [PubMed] [Google Scholar]
- 23. Hedges S. B., Dudley J., and Kumar S. (2006) TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 22, 2971–2972 [DOI] [PubMed] [Google Scholar]
- 24. Azevedo C., and Saiardi A. (2017) Eukaryotic phosphate homeostasis: the inositol pyrophosphate perspective. Trends Biochem. Sci. 42, 219–231 [DOI] [PubMed] [Google Scholar]
- 25. Tobey S. L., and Anslyn E. V. (2003) Determination of inorganic phosphate in serum and saliva using a synthetic receptor. Org. Lett. 5, 2029–2031 [DOI] [PubMed] [Google Scholar]
- 26. Kestenbaum B., Glazer N. L., Köttgen A., Felix J. F., Hwang S. J., Liu Y., Lohman K., Kritchevsky S. B., Hausman D. B., Petersen A. K., Gieger C., Ried J. S., Meitinger T., Strom T. M., Wichmann H. E., et al. (2010) Common genetic variants associate with serum phosphorus concentration. J. Am. Soc. Nephrol. 21, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu C., Wilson M. S., Jessen H. J., Saiardi A., and Shears S. B. (2016) Inositol pyrophosphate profiling of two HCT116 cell lines uncovers variation in InsP8 levels. PLoS One 11, e0165286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Padmanabhan U., Dollins D. E., Fridy P. C., York J. D., and Downes C. P. (2009) Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J. Biol. Chem. 284, 10571–10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weaver J. D., Wang H., and Shears S. B. (2013) The kinetic properties of a human PPIP5K reveal that its kinase activities are protected against the consequences of a deteriorating cellular bioenergetic environment. Biosci. Rep. 33, e00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camalier C. E., Yi M., Yu L. R., Hood B. L., Conrads K. A., Lee Y. J., Lin Y., Garneys L. M., Bouloux G. F., Young M. R., Veenstra T. D., Stephens R. M., Colburn N. H., Conrads T. P., and Beck G. R. Jr. (2013) An integrated understanding of the physiological response to elevated extracellular phosphate. J. Cell. Physiol. 228, 1536–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 [DOI] [PubMed] [Google Scholar]
- 32. Soboll S., Scholz R., and Heldt H. W. (1978) Subcellular metabolite concentrations. Dependence of mitochondrial and cytosolic ATP systems on the metabolic state of perfused rat liver. Eur. J. Biochem 87, 377–390 [DOI] [PubMed] [Google Scholar]
- 33. Ando T., Imamura H., Suzuki R., Aizaki H., Watanabe T., Wakita T., and Suzuki T. (2012) Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 8, e1002561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dick C. F., Dos-Santos A. L., and Meyer-Fernandes J. R. (2011) Inorganic phosphate as an important regulator of phosphatases. Enzyme Res. 2011, 103980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Macdonald J. A., and Storey K. B. (2005) Temperature and phosphate effects on allosteric phenomena of phosphofructokinase from a hibernating ground squirrel (Spermophilus lateralis). FEBS J. 272, 120–128 [DOI] [PubMed] [Google Scholar]
- 36. Ingram S. W., Safrany S. T., and Barnes L. D. (2003) Disruption and overexpression of the Schizosaccharomyces pombe aps1 gene and the effects on growth rate, morphology, and intracellular diadenosine 5′,5‴-P1,P5-pentaphosphate and diphosphoinositol polyphosphate concentrations. Biochem. J. 369, 519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson M. S., Livermore T. M., and Saiardi A. (2013) Inositol pyrophosphates: between signalling and metabolism. Biochem. J. 452, 369–379 [DOI] [PubMed] [Google Scholar]
- 38. Wundenberg T., and Mayr G. W. (2012) Synthesis and biological actions of diphosphoinositol phosphates (inositol pyrophosphates), regulators of cell homeostasis. Biol. Chem. 393, 979–998 [DOI] [PubMed] [Google Scholar]
- 39. Wang H., Falck J. R., Hall T. M., and Shears S. B. (2011) Structural basis for an inositol pyrophosphate kinase surmounting phosphate crowding. Nat. Chem. Biol. 8, 111–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Safrany S. T., Caffrey J. J., Yang X., Bembenek M. E., Moyer M. B., Burkhart W. A., and Shears S. B. (1998) A novel context for the “MutT” module, a guardian of cell integrity, in a diphosphoinositol polyphosphate phosphohydrolase. EMBO J. 17, 6599–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gomes A. P., and Blenis J. (2015) A nexus for cellular homeostasis: the interplay between metabolic and signal transduction pathways. Curr. Opin. Biotechnol. 34, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu M., Chong L. S., Perlman D. H., Resnick A. C., and Fiedler D. (2016) Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 113, E6757–E6765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otto J. C., Kelly P., Chiou S. T., and York J. D. (2007) Alterations in an inositol phosphate code through synergistic activation of a G protein and inositol phosphate kinases. Proc. Natl. Acad. Sci. U.S.A. 104, 15653–15658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoenig M., Lee R. J., and Ferguson D. C. (1989) A microtiter plate assay for inorganic phosphate. J. Biochem. Biophys. Meth. 19, 249–251 [DOI] [PubMed] [Google Scholar]
- 45. Choi J. H., Williams J., Cho J., Falck J. R., and Shears S. B. (2007) Purification, sequencing, and molecular identification of a mammalian PP-InsP5 kinase that is activated when cells are exposed to hyperosmotic stress. J. Biol. Chem. 282, 30763–30775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pöhlmann J., Risse C., Seidel C., Pohlmann T., Jakopec V., Walla E., Ramrath P., Takeshita N., Baumann S., Feldbrügge M., Fischer R., and Fleig U. (2014) The vip1 inositol polyphosphate kinase family regulates polarized growth and modulates the microtubule cytoskeleton in fungi. PLoS Genet. 10, e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pavlovic I., Thakor D. T., Vargas J. R., McKinlay C. J., Hauke S., Anstaett P., Camuña R. C., Bigler L., Gasser G., Schultz C., Wender P. A., and Jessen H. J. (2016) Cellular delivery and photochemical release of a caged inositol-pyrophosphate induces PH-domain translocation in cellulo. Nat. Commun. 7, 10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Capolicchio S., Thakor D. T., Linden A., and Jessen H. J. (2013) Synthesis of unsymmetric diphospho-inositol polyphosphates. Angew. Chem. Int. Ed Engl. 52, 6912–6916 [DOI] [PubMed] [Google Scholar]
- 49. Machkalyan G., Trieu P., Pétrin D., Hébert T. E., and Miller G. J. (2016) PPIP5K1 interacts with the exocyst complex through a C-terminal intrinsically disordered domain and regulates cell motility. Cell. Signal. 28, 401–411 [DOI] [PubMed] [Google Scholar]