

Abstract
Many diseases, especially cancers, result from aberrant or overexpression of pathogenic proteins. Specific inhibitors against these proteins have shown remarkable therapeutic effects, but these are limited mainly to enzymes. An alternative approach that may have utility in drug development relies on selective degradation of pathogenic proteins via small chimeric molecules linking an E3 ubiquitin ligase to the targeted protein for proteasomal degradation. To this end, we recently developed a protein knockdown system based on hybrid small molecule SNIPERs (Specific and Nongenetic IAP-dependent Protein Erasers) that recruit inhibitor of the apoptosis protein (IAP) ubiquitin ligases to specifically degrade targeted proteins. Here, we extend our previous study to show a proof of concept of the SNIPER technology in vivo. By incorporating a high affinity IAP ligand, we developed a novel SNIPER against estrogen receptor α (ERα), SNIPER(ER)-87, that has a potent protein knockdown activity. The SNIPER(ER) reduced ERα levels in tumor xenografts and suppressed the growth of ERα-positive breast tumors in mice. Mechanistically, it preferentially recruits X-linked IAP (XIAP) rather than cellular IAP1, to degrade ERα via the ubiquitin-proteasome pathway. With this IAP ligand, potent SNIPERs against other pathogenic proteins, BCR-ABL, bromodomain-containing protein 4 (BRD4), and phosphodiesterase-4 (PDE4) could also be developed. These results indicate that forced ubiquitylation by SNIPERs is a useful method to achieve efficient protein knockdown with potential therapeutic activities and could also be applied to study the role of ubiquitylation in many cellular processes.
Keywords: estrogen receptor, proteasome, tumor therapy, ubiquitin, X-linked inhibitor of apoptosis protein (XIAP), LCL161, SNIPER
Introduction
Pharmacological inhibitors of oncogenic kinases, such as imatinib and crizotinib, have demonstrated remarkable therapeutic activities against malignant cells expressing the respective target proteins (1). However, there are many pathogenic proteins without enzymatic activity to which pharmacological inhibitors can hardly be developed (2, 3). An alternative approach is to down-regulate the expression of the pathogenic proteins, which is usually achieved in vitro by genetic methods using oligonucleotides, such as antisense DNA and double-stranded RNA. Oligonucleotides, however, are scarcely incorporated into cells without transfection reagents. When systemically administered in vivo, they are mostly taken up by hepatocytes, and the delivery to the desired target tissues cannot be easily achieved, which complicates the clinical application of this technology (4, 5).
As a new method to down-regulate pathogenic proteins in a nongenetic manner, we and others have devised a protein knockdown system to induce selective degradation of target proteins by using small molecules, called Proteolysis Targeting Chimeras (PROTACs)3 and Specific and Nongenetic IAP-dependent Protein Erasers (SNIPERs), which possess sufficient membrane permeability (6, 7). These compounds are chimeric molecules containing two different ligands connected by a linker; one is a ligand for an E3 ubiquitin ligase and the other is for the target protein, which are designed to cross-link these proteins to induce polyubiquitylation and proteasomal degradation of the target proteins in the cells. To recruit von Hippel-Lindau (VHL) E3 ligase complex and cereblon (CRBN) E3 ligase complex, a VHL inhibitor based on the HIF-1α peptide and a phthalimide moiety were respectively conjugated in PROTACs (8–10), whereas bestatin was incorporated into SNIPERs to recruit cellular inhibitor of apoptosis protein 1 (cIAP1) E3 ligase (11).
To date, several SNIPER compounds have been developed targeting cellular retinoic acid-binding proteins (CRABPs), nuclear receptors such as estrogen receptor α (ERα), and spindle regulatory protein-transforming acidic coiled-coil-3 (TACC3), which specifically down-regulate the respective target proteins (12–15). These SNIPERs contain bestatin as a ligand for cIAP1, which possesses modest binding affinity but induces autoubiquitylation and proteasomal degradation of cIAP1 E3 ligase (16), which may limit the protein knockdown efficacy. To demonstrate a proof of concept of this technology in vivo, SNIPERs with more potent activity are required.
IAPs are a family of antiapoptotic proteins containing one to three baculoviral IAP repeat (BIR) domains (17–19). Some family members, such as cIAP1, cIAP2, and X chromosome-linked IAP (XIAP), directly interact with and regulate caspases, executioners of apoptosis, via the BIR domain (20–23). These IAPs are attractive targets for tumor therapy because IAPs are overexpressed in multiple human malignancies and implicated in promoting tumor progression, treatment failure, and poor prognosis (24–26). SMAC/DIABLO is an endogenous inhibitory protein of the IAPs, which binds to the proteins via its N-terminal IAP-binding motif. Based on the IAP-binding tetrapeptides of SMAC, many potent and cell-permeable peptidomimetic IAP antagonists (also known as SMAC mimetics) have been developed, some of which are under evaluation in clinical phase studies as antitumor drugs (17, 27). These IAP antagonists interact with the BIR domains of IAP proteins to induce autoubiquitylation and proteasomal degradation of IAPs (28–30). Because these IAP antagonists show higher affinity to IAPs than bestatin, we reasoned that novel SNIPERs with potent protein knockdown activity could be developed by incorporating IAP antagonists into SNIPERs.
Here, we demonstrated that the incorporation of an LCL161 derivative as an IAP ligand into SNIPERs allowed us to develop potent SNIPERs against several target proteins. We also showed in vivo protein knockdown of ERα and growth inhibition of ERα-positive human breast tumors in a xenograft model by SNIPER(ER).
Results
Development of Potent SNIPER(ER)s by Incorporation of IAP Antagonists
We previously developed a SNIPER(ER) by conjugating bestatin to 4-hydroxytamoxifen (4-OHT) (SNIPER(ER)-14 in this paper), which induced proteasomal degradation of ERα mediated by cIAP1 in MCF-7 breast tumor cells at 10 μm (14). To improve the protein knockdown activity, we replaced the bestatin moiety with an IAP antagonist, MV1, which shows higher affinity to IAPs than bestatin; the resulting SNIPER(ER)-19 reduced the ERα protein at 30 nm (Fig. 1, A and B). Then we synthesized a series of SNIPER(ER)s containing different ER ligands or different linker lengths, and SNIPER(ER)-20 showed the most potent activity among them (Fig. 1, A–C).
FIGURE 1.

SNIPER(ER)s composed of various ERα and IAP ligands. A, structural schema of SNIPER(ER)s containing various ERα and IAP ligands. The detailed chemical structures of SNIPER compounds are provided in the supplemental material. B, protein knockdown activities of SNIPER(ER)s. C, effect of linker length on the protein knockdown activity of the MV1-based SNIPER(ER)s. MCF-7 cells were treated with the indicated concentrations of SNIPER(ER)s or a mixture of the ligands for 6 and 24 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent the ERα/actin ratio normalized by the vehicle control as 100.
To further improve the activity, we incorporated several IAP antagonists and found that SNIPER(ER)-87 with an LCL161 derivative reduces the ERα level more potently than SNIPER(ER)-20 (Fig. 2, A and B). The effective knockdown (50% reduction) of ERα by SNIPER(ER)-87 was observed at a concentration as low as 3 nm, and maximum activity was observed at around 100 nm. SNIPER(ER)-87 and -88, the latter of which has a longer linker, rapidly reduced the ERα protein within 1 h after treatment, and the reduction was sustained for 48 h (Fig. 2C). SNIPER(ER)-89 with a shorter linker than SNIPER(ER)-87 showed attenuated knockdown activity (Fig. 2D). Combination treatment with the ER ligand (4-OHT) and the IAP antagonist (LCL161 derivative) did not decrease the ERα protein, indicating that linking the two ligands into a single molecule is critical for protein knockdown (Fig. 2, C and D). SNIPER(ER)-87 also showed protein knockdown activity in other human breast tumor T47D and ZR75-1 cells (Fig. 2E). In addition to ERα protein, SNIPER(ER)-87 effectively reduced the level of cIAP1, but only slightly reduced that of XIAP (Fig. 2B), indicating that SNIPER(ER)-87 simultaneously activates autoubiquitylation and proteasomal degradation of cIAP1 as observed with other SNIPERs and IAP antagonists (11–17, 28–30).
FIGURE 2.

Novel SNIPER(ER) with potent protein knockdown activity. A, chemical structure of novel SNIPER(ER). B, protein knockdown activity of SNIPER(ER)-87. MCF-7 cells were treated with the indicated concentrations of SNIPER(ER)-87 for 6 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent ERα/actin ratio normalized by vehicle control as 100. Data in the bar graph are the mean ± S.D. of three independent experiments; asterisks indicate p < 0.05 compared with vehicle control. C, SNIPER(ER)-87 or -88 rapidly down-regulates ERα protein levels. MCF-7 cells were treated with the indicated concentrations of SNIPER(ER)s for the indicated periods. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent the ERα/actin ratio normalized by the vehicle control as 100. D, optimization of linker length in the SNIPER(ER). MCF-7 cells were treated with the indicated concentrations of SNIPER(ER)s or a mixture of the ligands for 24 h and then analyzed by Western blotting. E, SNIPER(ER)-87 degrades ERα in human breast tumor T47D and ZR75-1 cells.
SNIPER(ER)-87 Specifically Induces Degradation of the ERα Protein by the Ubiquitin-Proteasome System (UPS)
To explore the mechanism of SNIPER(ER)-87-induced reduction of the ERα protein, we first examined the effect of UPS inhibitors. The decrease in the ERα protein by SNIPER(ER)-87 detected by Western blotting and immunocytochemical analysis was abrogated by the proteasome inhibitor MG132 (Fig. 3, A and B). Similar results were obtained by treatment with another proteasome inhibitor, bortezomib, and a ubiquitin-activating enzyme inhibitor, MLN7243, indicating that SNIPER(ER)-87 induces UPS-dependent degradation of the ERα protein (Fig. 3C). MLN4924, an inhibitor of NEDD8-activating enzyme, did not affect the activity, suggesting that cullin-based ubiquitin ligases are not involved in the ERα degradation (Fig. 3C). Next, we performed the ubiquitylation assay. MCF-7 cells were transfected with an expression vector of HA-tagged ubiquitin and then treated with SNIPER(ER)-87 or control compounds in the presence of MG132. The cell lysates were immunoprecipitated with anti-HA (ubiquitin) antibody, and the precipitates were analyzed by Western blotting with an anti-ERα antibody to detect ubiquitylated ERα protein. SNIPER(ER)-87, but not the LCL161 derivative with 4-OHT, greatly induced the polyubiquitylation of ERα protein, as did fulvestrant, a clinically approved ERα degrader (Fig. 3D). Thus, SNIPER(ER)-87 induces polyubiquitylation of the ERα protein in cells.
FIGURE 3.

SNIPER(ER)-87 selectively degrades ERα protein via UPS. A and B, proteasomal degradation of ERα by SNIPER(ER)-87. MCF-7 cells were treated with 100 nm SNIPER(ER)-87 or a mixture of the LCL161 derivative and 4-OHT in the presence or absence of 10 μm MG132 for 6 h. Cell lysates were analyzed by Western blotting (A). The treated cells were stained with the indicated antibodies and Hoechst 33342 (B). Scale bars, 20 μm. C, effect of UPS inhibitors on the SNIPER(ER)-87-induced degradation of ERα protein. MCF-7 cells were treated with the indicated concentrations of UPS inhibitors in the presence or absence of 30 nm SNIPER(ER)-87 for 6 h and then analyzed by Western blotting. D, DMSO; SN, SNIPER(ER)-87. D, ubiquitylation of ERα by SNIPER(ER)-87. MCF-7 cells that had been transfected with HA-ubiquitin were treated with the indicated compounds in the presence of 10 μm MG132 for 3 h. Whole-cell lysates (lower panels) and lysates immunoprecipitated with anti-HA antibody (upper panel) were analyzed by Western blotting with the indicated antibodies. IP, immunoprecipitation; IB, immunoblot. E, SNIPER(ER)-87 selectively induces ERα degradation. MCF-7 cells were treated with 10 nm SNIPER(ER)-87 or 10 μg/ml cycloheximide (CHX) for 3 and 6 h, and cell lysates were analyzed by Western blotting.
To understand the selectivity to the target protein, the effect of SNIPER(ER)-87 on the levels of various proteins was examined. SNIPER(ER)-87 induced degradation of the ERα protein, but not short-lived proteins, and the proteins degraded in a cell cycle-dependent manner, whereas a protein synthesis inhibitor, cycloheximide, rapidly reduced these proteins (Fig. 3E). In addition, SNIPER(ER)-87 did not degrade other nuclear receptors and the proteins targeted by different SNIPERs. This indicates that SNIPER(ER)-87 selectively degrades ERα and cIAP1.
XIAP Is Required for ERα Degradation by SNIPER(ER)-87
SNIPER(ER)-87 shows binding affinity to cIAP1, cIAP2, and XIAP, which reflects the ability of LCL161 to bind these IAPs. To determine which IAP is recruited to ERα by SNIPER(ER)-87, we carried out a coprecipitation assay. We focused on XIAP and cIAP1, because cIAP2 is not expressed in MCF7 cells. Cells were treated with or without SNIPER(ER)-87 in the presence of MG132; the cell lysates were immunoprecipitated with anti-ERα antibody, and the precipitates were analyzed by Western blotting to detect the IAPs (Fig. 4A). XIAP and cIAP1 were coprecipitated only when cells had been treated with SNIPER(ER)-87, indicating that these IAPs interact with ERα in a manner dependent on SNIPER(ER)-87. Compared with the IAP protein levels in total lysates, XIAP was more efficiently recruited to ERα than was cIAP1. Depletion of XIAP by siRNA greatly increased the SNIPER(ER)-mediated interaction between ERα and cIAP1, whereas that of cIAP1 did not affect the interaction of ERα with XIAP (Fig. 4B). These results indicate that SNIPER(ER)-87 preferentially recruits XIAP to ERα. The SNIPER(ER)-mediated recruitment of XIAP to ERα was confirmed by reciprocal precipitation with anti-XIAP antibody (Fig. 4C). In line with this, depletion of XIAP significantly suppressed the SNIPER(ER)-87-induced degradation of the ERα protein, whereas depletion of cIAP1 minimally suppressed it (Fig. 5A). As a control experiment, ERα degradation induced by fulvestrant and β-estradiol was similarly analyzed, but depletion of XIAP did not abrogate the ERα degradation induced by these agents (Fig. 5B). These results indicate that SNIPER(ER)-87 preferentially cross-links ERα with XIAP, and XIAP is the primary E3 ligase responsible for the ERα degradation, which is mechanistically distinct from ERα degradation induced by fulvestrant and β-estradiol.
FIGURE 4.
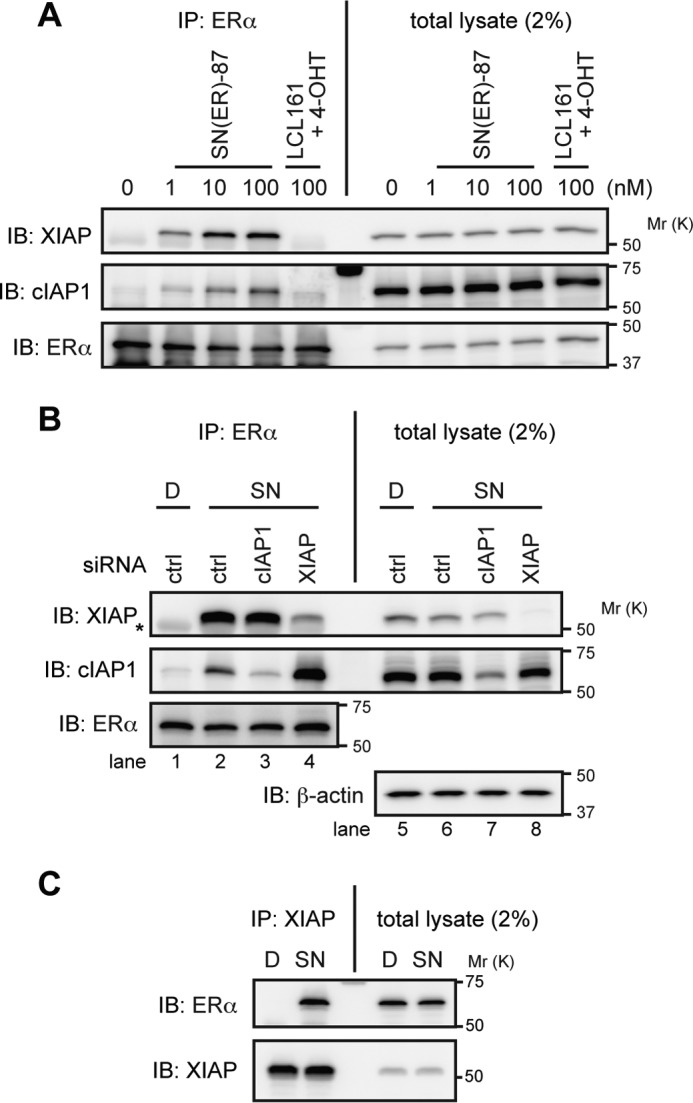
SNIPER(ER)-87 preferentially recruits XIAP to ERα. A, MCF-7 cells were treated with the indicated compounds in the presence of 10 μm MG132 for 3 h. Immunoprecipitates of anti-ERα (IP) and whole-cell lysates (total lysate) were analyzed by Western blotting. B, MCF-7 cells were transfected with the indicated siRNA for 42 h and were then treated with 10 nm SNIPER(ER)-87 in the presence of 10 μm MG132 for 3 h. Immunoprecipitates (IP) of anti-ERα and whole-cell lysates (total lysate) were analyzed by Western blotting. Asterisk in the XIAP panel indicates an IgG heavy chain band. C, MCF-7 cells were treated with 10 nm SNIPER(ER)-87 in the presence of 10 μm MG132 for 3 h. Cell lysates were immunoprecipitated with anti-XIAP antibody, and the precipitates were analyzed by Western blotting. D, DMSO; SN, SNIPER(ER)-87; IB, immunoblot.
FIGURE 5.

XIAP is required for the degradation of ERα by SNIPER(ER)-87. A, depletion of XIAP suppresses the SNIPER(ER)-87-induced degradation of ERα. MCF-7 and T47D cells were transfected with the indicated siRNA for 42 h and treated with 10 nm SNIPER(ER)-87 for 3 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent ERα/actin ratio normalized by vehicle control as 100. Three different siRNAs against XIAP and cIAP1 were used. B, depletion of XIAP does not inhibit the ERα degradation by fulvestrant and β-estradiol. MCF-7 cells were transfected with the indicated siRNA for 42 h and treated with 10 nm of the indicated compounds for 3 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent the ERα/actin ratio normalized by the vehicle control as 100. C, essential role of XIAP RING domain in the SNIPER(ER)-induced ERα degradation. MCF-7 cells were transfected with the indicated siRNA for 24 h. Then cells were infected with the indicated lentiviral vectors for 45 h and treated with 10 nm SNIPER(ER)-87 for 3 h. Whole-cell lysates were analyzed by Western blotting. D, XIAP ΔRing suppresses the SNIPER(ER)-87-induced degradation of ERα. Cells were infected with indicated lentiviral vectors for 45 h and then treated with 10 nm SNIPER(ER)-87 for 3 h. Whole-cell lysates were analyzed by Western blotting. D, DMSO; SN, SNIPER(ER)-87.
We next studied the role of the RING domain of XIAP. The ERα degradation by SNIPER(ER)-87 was suppressed by siRNA-mediated depletion of XIAP as above. When siRNA-resistant wild-type XIAP (WT) was added back to the cells by infecting them with a lentiviral expression vector, the SNIPER(ER)-87-induced ERα degradation was restored (Fig. 5C). However, adding back XIAP RING mutants (ΔRing and H467A) did not restore the degradation activity. In addition, overexpression of XIAP ΔRing inhibited ERα degradation by SNIPER(ER)-87 (Fig. 5D). These results suggest that the XIAP E3 ligase activity associated with the RING domain is required for the SNIPER(ER)-87-induced ERα degradation.
The involvement of IAPs in the SNIPER(ER)-87-induced ERα degradation was further studied by an approach based on chemical biology. SNIPER(ER)-143, in which an N-methylated analog of the LCL161 derivative was conjugated, showed binding affinity to ERα comparable with that of SNIPER(ER)-87 but lost the ability to bind IAP proteins (Figs. 2A and 6A). Consequently, SNIPER(ER)-143 did not cross-link ERα with XIAP and cIAP1 in MCF-7 cells (Fig. 6B) nor induce the degradation of ERα (Fig. 6C). These findings indicate that the IAP binding ability is critical for the ERα knockdown activity of SNIPER(ER)-87.
FIGURE 6.
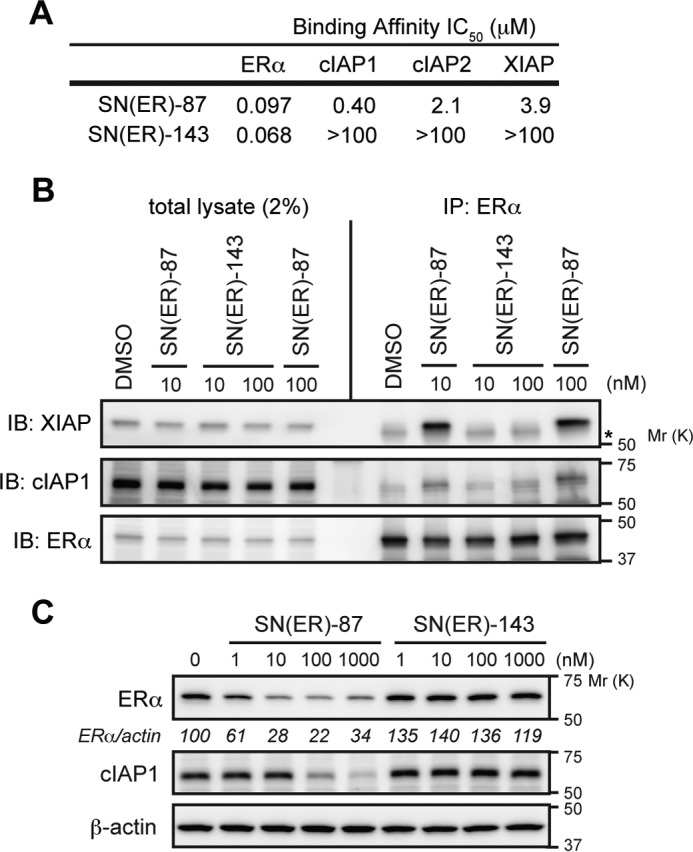
SNIPER(ER) without IAP binding ability loses its protein knockdown activity. A, binding affinities of SNIPER(ER) to ERα and IAPs. IC50 values (concentrations of SNIPER(ER)s required to inhibit the probe binding to each protein by 50%) are presented. B, inactive SNIPER(ER)-143 does not cross-link ERα with XIAP. MCF-7 cells were treated with the indicated compounds in the presence of 10 μm MG132 for 3 h. Immunoprecipitates (IP) of anti-ERα and whole-cell lysates (total lysate) were analyzed by Western blotting. Asterisk in the XIAP panel indicates an IgG heavy chain band. C, inactive SNIPER(ER)-143 does not degrade ERα protein. MCF-7 cells were treated with the indicated compounds for 6 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent ERα/actin ratio normalized by vehicle control as 100. IB, immunoblot.
In Vivo Protein Knockdown by SNIPER(ER)-87
To evaluate the knockdown activity of SNIPER(ER)-87 in vivo, we first measured the level of ERα in ovary. When female BALB/c mice were intraperitoneally injected with SNIPER(ER)-87 (10 or 30 mg/kg body weight), the ERα protein levels in ovary were significantly reduced (Fig. 7A). Then, to explore the in vivo protein knockdown in a tumor model, we next developed MCF-7 breast tumor xenografts in nude mice. Again, SNIPER(ER)-87 reduced the ERα protein level in ovary (Fig. 7B). In addition, the ERα protein levels in the orthotopic tumors were significantly reduced to ∼50% in SNIPER(ER)-87-treated mice compared with those in vehicle-treated mice (Fig. 7C). Meanwhile, administration of inactive SNIPER(ER)-143 did not affect the ERα levels in tumors (Fig. 7D). Thus, SNIPER(ER)-87 shows in vivo protein knockdown activity in tumors.
FIGURE 7.

In vivo protein knockdown by SNIPER(ER)-87 in mice. A, in vivo protein knockdown in ovary. Female BALB/c mice were injected with vehicle or 10 or 30 mg/kg SNIPER(ER)-87. After 6 or 24 h, the mice were sacrificed, and their ovaries were collected and analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent the ERα/actin ratio normalized by the vehicle control as 100 (average of each group). B–D, MCF-7 human breast tumor cells were inoculated into mammary fat pads of 6-week-old female BALB/c nude mice. The tumor-bearing mice were intraperitoneally injected with SNIPER(ER)-87 and -143. After 24 h, mice were sacrificed and ERα protein levels in ovary (B) and tumor xenografts (C and D) were analyzed by Western blotting; representative data are shown. Bar graphs represent the mean ± S.D. of each group. B, n = 5; C, n = 12; D, n = 8. *, p < 0.001 in two-sided Student's t test. IP, immunoprecipitates; IB, immunoblot.
SNIPER(ER)-87 Inhibits Estrogen Signaling and Estrogen-dependent Tumor Growth
Because ERα plays an essential role in estrogen signaling and the growth of certain ERα-positive breast tumor cells (31, 32), we next examined the effect of SNIPER(ER)-87 on estrogen-dependent gene expression and tumor growth. In luciferase assays with an estrogen-response element reporter, SNIPER(ER)-87 effectively inhibited the ERα-dependent transcriptional activation by β-estradiol (Fig. 8A), which is in good agreement with the ERα knockdown activity (Fig. 2B). In addition, SNIPER(ER)-87 efficiently suppressed the growth of ERα-positive breast tumor cells (IC50 values were 15.6 nm in MCF-7 and 9.6 nm in T47D), but not that of ERα-negative breast tumor cells (MDA-MB-231) (Fig. 8B), which is consistent with the results of cell cycle distribution analyzed by flow cytometry (Fig. 8C). Thus, SNIPER(ER)-87 shows activities to inhibit estrogen signaling and growth of ERα-positive tumor cells. To demonstrate the therapeutic significance of these findings, we also evaluated the in vivo antitumor activity of SNIPER(ER)-87 in an MCF-7 tumor xenograft mouse model. As pharmacokinetic studies indicated that intraperitoneally administered SNIPER(ER)-87 was eliminated in 24 h (Fig. 8, D and E), tumor-bearing mice were treated daily with SNIPER(ER)-87 by intraperitoneal injection (30 mg/kg body weight) for 14 days. Treatment with SNIPER(ER)-87 attenuated tumor progression as assessed by measuring tumor volume (Fig. 8F). The inhibition of tumor cell proliferation by SNIPER(ER)-87 was confirmed by immunohistochemistry stained with S phase-related proliferating cell nuclear antigen (PCNA) (Fig. 8G). Notably, no obvious toxicities, including body weight changes, were observed throughout the 2 weeks of administration of SNIPER(ER)-87 (Fig. 8H). These results imply the potential utility of SNIPER(ER)-87 in the treatment of ERα-positive breast tumors.
FIGURE 8.

Antitumor activity of SNIPER(ER)-87. A, inhibition of estrogen-dependent gene expression by SNIPER(ER)-87. MCF-7 cells were treated with the indicated concentrations of SNIPER(ER)-87 in the presence or absence of 0.1 nm β-estradiol for 24 h. The ERα-dependent transcriptional activity was evaluated by luciferase assay using a luciferase reporter containing three tandem repeats of consensus estrogen-responsive element motif. The data represent mean ± S.D. (n = 3). B, growth inhibition of ERα-positive human breast tumor cells by SNIPER(ER)-87. Cells were treated with the indicated concentrations of SNIPER(ER)-87 for 72 h, and the cell growth was evaluated by cell viability assay. The data represent mean ± S.D. (n = 3). C, cell cycle arrest and apoptosis observed in ERα-positive human breast tumor cells. Cells were treated with 100 nm SNIPER(ER)-87 for 48 h and analyzed in a flow cytometer. Cell cycle distribution was quantified by MultiCycle software. D and E, pharmacokinetic data of SNIPER(ER)-87 administered to mice. F, SNIPER(ER)-87 inhibits the growth of MCF-7 orthotopic breast tumor xenografts in nude mice. The tumor volume represents the mean ± S.D. of each group (mice n = 9 each; tumor n = 18 each; *, p < 0.0001 in two-sided Student's t test). Mice were administered vehicle or SNIPER(ER)-087 (30 mg/kg, intraperitoneally, every 24 h). Representative tumors are shown in the top panel. Scale bar, 10 mm. G, immunohistochemical staining of PCNA on a representative tumor from a vehicle- or a SNIPER(ER)-87-treated mouse. Scale bar, 150 μm. H, treatment with SNIPER(ER)-87 (30 mg/kg, intraperitoneally, every 24 h) did not induce significant body weight loss in mice after 14 days.
Development of LCL161-based SNIPERs against Different Target Proteins
To investigate the utility of the LCL161 derivative in the development of SNIPERs, we conjugated the LCL161 derivative to dasatinib, JQ-1, and a PDE4 inhibitor, ligands for BCR-ABL, BRD4, and PDE4 proteins, respectively (Fig. 9A). These LCL161-based SNIPERs showed efficient protein knockdown activities against target proteins at nanomolar concentrations (Fig. 9, B and C), which were suppressed by MG132 and MLN7243 (Fig. 9D). These results suggest that the LCL161 derivative is a useful IAP ligand in SNIPER to degrade a variety of target proteins.
FIGURE 9.

Development of potent SNIPERs against BCR-ABL, BRD4, and PDE4 by using the LCL161 derivative as an IAP ligand. A, chemical structure of SNIPERs against BCR-ABL, BRD4, and PDE4. B and C, reduction of target proteins by SNIPERs. K562, LNCaP, and HT1080 cells were treated for 6 h (B) or 24 h (C) with SNIPER(ABL)-38, SNIPER(BRD4)-1, and SNIPER(PDE4)-9, respectively, and the cell lysates were analyzed by Western blotting. Numbers below the target protein panels represent target/actin ratio normalized by vehicle control as 100. Data in the bar graphs are the mean ± S.D. of three independent experiments; asterisks indicate p < 0.05 compared with the vehicle control. D, degradation of target proteins by SNIPERs via the UPS. Cells were treated with SNIPERs in the presence or absence of 10 μm MG132 or 10 μm MLN7243 for 6 h. Cell lysates were analyzed by Western blotting.
Discussion
In this study, we incorporated an LCL161 derivative with the capacity to bind with high affinity to cIAP1, cIAP2, and XIAP into SNIPERs, and we successfully developed potent SNIPERs against ERα, PDE4, BRD4, and BCR-ABL, which induce the degradation of respective target proteins via UPS in a highly specific manner. SNIPER(ER)-87 showed activity of reducing the ERα protein at nanomolar concentrations in an in vitro cell culture system and also in tumor xenografts in vivo. Analysis of the mechanism involved in this revealed that SNIPER(ER)-87 preferentially recruits XIAP to ERα in the cells, and XIAP, but not cIAP1, is the primary E3 ubiquitin ligase responsible for the SNIPER(ER)-87-induced ERα degradation. It is not clear why SNIPER(ER)-87 preferentially recruits XIAP as compared with cIAP1, despite its binding affinity to cIAP1 being 10-fold higher than that to XIAP. It is likely that the relative amounts and subcellular localization of ERα and IAPs or the latency of the IAP-BIR3 domains to which the LCL161 moiety binds could influence the SNIPER(ER)-87-induced recruitment of IAPs to ERα in the cells.
SNIPER(ER)-87 shows fairly good metabolic stability in serum. When SNIPER(ER)-87 was administered to mice intraperitoneally, a significant concentration was retained in the blood (Fig. 8, D and E), and ERα proteins in tumor xenografts and ovary were effectively reduced. In addition, daily administration of SNIPER(ER)-87 inhibited the growth of ERα-positive human breast tumor xenografts in mice. Similar in vivo protein knockdown and potential therapeutic activities were recently reported with PROTACs and dBET1 targeting BRD4 protein by recruiting VHL and CRBN E3 ligases, respectively (10, 33). These observations imply the potential utility of the protein knockdown technology to clinical applications. Because these E3 ligases, including cIAP1 and XIAP, are ubiquitously expressed in various types of cell, targeted degradation of proteins with this class of molecule could be applicable in a variety of cells. It should be noted that IAPs are frequently overexpressed in tumor cells, which is involved in resistance to tumor therapy (17, 34). Therefore, the ability of SNIPERs to degrade cIAP1, and XIAP to some extent, simultaneously with the target proteins suggests that they could be particularly advantageous in killing tumor cells.
In addition to SNIPER(ER)s against ERα protein, we also developed potent SNIPERs targeting BRD4, PDE4, and BCR-ABL proteins by incorporating the LCL161 derivative as an E3 ligand. These SNIPERs effectively degrade respective target proteins, indicating the utility of the LCL161 derivative for the development of various SNIPERs. The protein knockdown efficacy by most SNIPER molecules, as well as that by PROTACs, was suppressed at higher concentrations (Figs. 1B, 2B, and 9B) (8, 10), which is known as a hook effect. This effect is explained by the inhibition of ternary complex formation (E3-SNIPER-target) by an excess amount of bivalent compounds such as SNIPERs and PROTACs.
With respect to the PROTACs against BCR-ABL protein, CRBN-based but not VHL-based PROTACs can degrade BCR-ABL protein (35), suggesting that an appropriate E3 ligase should be recruited to the target proteins. It is likely that correct exposure of the lysine residues on the surface of target proteins to an appropriate E3 ligase is critically important for the efficient ubiquitylation and subsequent degradation of the target proteins. Therefore, for each target, finding the most appropriate E3 ligase recruited to the target proteins by PROTACs or SNIPERs might be important for the maximum protein knockdown activity.
Although we focused on the proteasomal degradation of target proteins by SNIPER-mediated forced ubiquitylation in this study, ubiquitylation plays a role in a variety of cellular processes, such as autophagy (36), DNA repair (37), generation of peroxisome (38), and sorting membrane proteins in recycling endosomes (39). Because of its modular structure, SNIPER-mediated forced ubiquitylation can be applied to other target proteins to study the role of ubiquitylation in such cellular processes. In addition, endogenous proteins can be ubiquitylated by SNIPERs without involving genetic engineering, which implies a potential utility of SNIPERs in drug development that induces a particular cellular response, not limited to proteasomal degradation, by forced ubiquitylation.
Experimental Procedures
Chemistry
The chemical synthesis and physicochemical data on SNIPER compounds are provided in the supplemental material.
Biology
Cell Culture
Human breast carcinoma MCF-7, T47D, and ZR-75-1 cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and 100 μg/ml kanamycin with or without 10 μg/ml insulin (Sigma). Human myeloid leukemia K562 and human prostate carcinoma LNCaP cells were maintained in RPMI 1640 medium containing 10% FBS and 100 μg/ml kanamycin. Human fibrosarcoma HT1080 cells were maintained in Dulbecco's modified Eagle's medium containing 10% FBS and 100 μg/ml kanamycin. Cells were treated with various concentrations of the compounds for the indicated times.
Western Blotting
Cells were lysed with SDS lysis buffer (0.1 m Tris-HCl, pH 8.0, 10% glycerol, 1% SDS) and immediately boiled for 10 min to obtain clear lysates. The protein concentration was measured by the BCA method (Pierce), and the lysates containing equal amounts of proteins were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Darmstadt, Germany) for Western blotting analysis using the appropriate antibodies. The immunoreactive proteins were visualized using the Immobilon Western chemiluminescent HRP substrate (Millipore) or Clarity Western ECL substrate (Bio-Rad), and light emission intensity was quantified with an LAS-3000 lumino-image analyzer equipped with ImageGauge version 2.3 software (Fuji, Tokyo, Japan). The antibodies used in this study were as follows: anti-ERα rabbit monoclonal antibody (mAb) (Cell Signaling Technology, Danvers, MA; catalog no. 8644); anti-ERα rabbit polyclonal antibody (pAb) (Santa Cruz Biotechnology, Dallas, TX; catalog nos. sc-542 and sc-543); anti-cIAP1 goat pAb (R&D Systems, Minneapolis, MN; catalog no. AF8181); anti-cIAP1 rat mAb (Enzo Life Sciences, Farmingdale, NY; catalog no. 1E1-1-10); anti-β-actin mouse mAb (Sigma, catalog no. A5316); anti-XIAP rabbit pAb (Cell Signaling Technology, catalog no. 2042); anti-Myc tag mouse mAb (Cell Signaling Technology, catalog no. 2276); anti-GFP mouse mAb (BD Biosciences; catalog no. 632375); anti-MCL-1 mouse mAb (BD Biosciences, catalog no. 559027); anti-p53 mouse mAb (Santa Cruz Biotechnology, catalog no. sc-126); anti-FLIP rat mAb (AdipoGen Life Sciences, San Diego; catalog no. AG-20B-0005-C100); anti-cyclin B mouse mAb (Santa Cruz Biotechnology, catalog no. sc-245); anti-cyclin A rabbit pAb (Santa Cruz Biotechnology, catalog no. sc-751); anti-TACC3 rabbit pAb (Santa Cruz Biotechnology, catalog no. sc-22773); anti-p27 mouse mAb (BD Biosciences, catalog no. 610242); anti-p21 mouse mAb (Santa Cruz Biotechnology, catalog no. sc-6246); anti-AR rabbit mAb (Cell Signaling Technology, catalog no. 5153); anti-AhR rabbit mAb (Cell Signaling Technology, catalog no. 13790); anti-VDR rabbit mAb (Cell Signaling Technology, 12550); anti-CRABP2 rabbit pAb (Bethyl Laboratories, Montgomery, TX; catalog no. A300-809A); anti-c-Abl rabbit mAb (Cell Signaling Technology, catalog no. 2862); anti-BRD4 rabbit mAb (Cell Signaling Technology, catalog no. 13440); and anti-PDE4 rabbit pAb (Santa Cruz Biotechnology, catalog no. sc-25810).
Immunostaining
MCF-7 cells were treated with the indicated compounds in combination with 10 μm MG132 for 3 h. They were then fixed in 100% methanol on ice for 10 min, washed four times with PBS, and blocked in PBS containing 3% BSA and 0.1% Triton X-100 (PBS-TB) for 1 h at room temperature. Next, the cells were incubated for 2 h with anti-ERα rabbit mAb (Cell Signaling, 8644) or anti-α-tubulin mouse mAb (Sigma, T-5168) as the primary antibodies, and for 1 h with Alexa Fluor 488-conjugated anti-rabbit IgG or Alexa Fluor 568-conjugated anti-mouse IgG (Life Technologies, Inc.) as the secondary antibodies with Hoechst 33342 (Life Technologies, Inc.). Fluorescent images were obtained using a BZ-9000 (Keyence, Osaka, Japan).
Immunoprecipitation
MCF-7 cells were treated with the indicated concentrations of the indicated compounds in combination with 10 μm MG132 for 3 h. Cells were lysed using IP lysis buffer (10 mm HEPES, pH 7.4, 142.5 mm KCl, 5 mm MgCl2, 1 mm EGTA, and 0.1% Triton X-100), containing protease inhibitor mixtures, rotated for 15 min at 4 °C, and centrifuged at 15,000 rpm for 10 min at 4 °C to obtain the supernatants. The lysates that had been precleared with naked protein G-Sepharose were immunoprecipitated with protein G-Sepharose beads preincubated anti-ERα rabbit pAb (Santa Cruz Biotechnology, sc-543) or anti-XIAP mouse mAb (MBL, Nagoya, Japan; catalog no. M044-3) for 2 h at 4 °C. The precipitates were washed with IP lysis buffer four times and analyzed by Western blotting.
Ubiquitylation Assay
MCF-7 cells were transfected with pcDNA3-HA-ubiquitin for 24 h. The cells were then incubated with the indicated compounds in the presence of MG132 (10 μm) for 3 h before being harvested and lysed in SDS lysis buffer. The cell lysates were boiled for 10 min, diluted 10 times with 0.1 m Tris-HCl, pH 7.5, and immunoprecipitated with anti-HA agarose-conjugated beads (Sigma, catalog no. E6779). The precipitates were extensively washed and analyzed by Western blotting using anti-ERα antibody (Cell Signaling Technology, catalog no. 8644).
siRNA Transfection
MCF-7 or T47D cells were transiently transfected with a gene-specific short interfering RNA (siRNA) or a negative control siRNA (Qiagen, Valencia, CA) using Lipofectamine RNAi MAX reagent (Life Technologies, Inc.). The siRNA sequences used in this study were as follows: human cIAP1-1 (5′-UCUAGAGCAGUUGAAGACAUCUCUU-3′); cIAP1-2 (5′-GCUGUAGCUUUAUUCAGAAUCUGGU-3′); cIAP1-3 (5′-GGAAAUGCUGCGGCCAACAUCUUCA-3′); XIAP-1 (5′-ACACUGGCACGAGCAGGGUUUCUUU-3′); XIAP-2 (5′-GAAGGAGAUACCGUGCGGUGCUUUA-3′); and XIAP-3 (5′-CCAGAAUGGUCAGUACAAAGUUGAA-3′).
Lentivirus Infection
Lentivirus expression plasmids for WT, H467A, and ΔRing XIAP were constructed by inserting a fragment coding for XIAP-3 siRNA-resistant Myc-XIAP (WT, H467A, and ΔRing, respectively) into CSII-EF-MCS-IRES-Venus (RIKEN, Japan). The fragments were generated using PCR products. All constructs were verified by sequencing. To prepare the lentiviruses, 293T cells were transfected with a lentiviral expression plasmid together with a packaging (pCAG-HIVgp) and a VSV-G-/Rev-expressing (pCMV-VSV-G-RSV-Rev) plasmid by calcium phosphate transfection. After 48 h of transfection, the medium containing lentiviruses was collected and filtered, and the lentiviruses were then concentrated by centrifugation with PEG-it. MCF-7 cells were infected with the lentiviruses with 10 μg/ml Polybrene by a centrifugation method (2,500 rpm, 90 min). The cells were then incubated with a fresh culture medium.
Luciferase Assay
MCF-7 cells were transfected with firefly luciferase reporter plasmid containing three tandem copies of estrogen-response element and control Renilla luciferase plasmid-SV40 using Lipofectamine LTX (Life Technologies, Inc.) in phenol red-free medium containing 4% charcoal/dextran-treated FBS. After 24 h, cells were treated with the indicated concentrations of SNIPER(ER)-87 in the presence or absence of 0.1 nm β-estradiol in phenol red-free medium containing 0.2% charcoal/dextran-treated FBS for 24 h. The firefly luciferase activity in cell lysates was measured and normalized with Renilla luciferase activity. The data represent means ± S.D. (n = 3).
Cell Viability Assay
Cell viability was evaluated by crystal violet staining. Cells were treated with graded concentrations of the compounds for 72 h, and then stained with 0.1% crystal violet (Wako, Osaka, Japan) in 1% ethanol for 15 min at room temperature. The cells were rinsed thoroughly with distilled water and then lysed in 1% SDS. The absorbance of cell lysate at 600 nm was measured using EnVision Multilabel Plate Reader (PerkinElmer Life Sciences).
In Vivo Protein Knockdown
Mice were housed in pathogen-free animal facilities with 12-h light/dark cycles and were fed rodent chow and water ad libitum at the National Institute of Health Sciences. All experiments were conducted in accordance with the guidelines approved by the National Institute of Health Sciences. For Fig. 7A, female 6-week-old BALB/c mice (Clea Japan, Tokyo, Japan) were randomized and divided into six treatment groups as follows: 1) vehicle treatment for 6 h (n = 3); 2) 10 mg/kg SNIPER(ER)-087 treatment for 6 h (n = 2); 3) 30 mg/kg SNIPER(ER)-087 treatment for 6 h (n = 2); 4) vehicle treatment for 24 h (n = 4); 5) 10 mg/kg SNIPER(ER)-087 treatment for 24 h (n = 3); and 6) 30 mg/kg SNIPER(ER)-087 treatment for 24 h (n = 3). For Fig. 7, B–D, each suspension of 1 × 107 MCF-7 cells was mixed with an equal volume of Matrigel (Corning Life Sciences) and injected (100 μl total) into the left and right mammary fat pads of 6-week-old female BALB/c nude mice (Clea Japan). After cell inoculation, β-estradiol solution was subcutaneously injected into the neck twice at intervals of 6 days. Fourteen days after the last β-estradiol injection, tumor-bearing mice were randomized and divided into two or three treatment groups as follows: for Fig. 7, B and C: 1) vehicle treatment for 24 h (n = 5); and 2) 30 mg/kg SNIPER(ER)-087 treatment for 24 h (n = 5); for Fig. 7D: 1) vehicle treatment for 24 h (n = 4); 2) 30 mg/kg SNIPER(ER)-087 treatment for 24 h (n = 4); and 3) 30 mg/kg SNIPER(ER)-143 treatment for 24 h (n = 4). Compounds were administered via intraperitoneal injection. After the indicated times, the mice were sacrificed, and tissues were excised. Total lysates from the ovaries and tumors were analyzed by Western blotting.
In Vivo Tumor Growth Inhibition
Each suspension of 1 × 107 MCF-7 cells was mixed with an equal volume of Matrigel (BD Biosciences) and inoculated (100 μl total) into the left and right mammary fat pads of 6-week-old female BALB/c nude mice (Clea Japan) that had received an β-estradiol pellet (6 μg per day) (Innovative Research of America, Sarasota, FL) under the neck skin. After 4 days, mice bearing ∼100-mm3 tumors were randomized and divided into two groups (n = 9). One group served as a control for dosing vehicle, whereas the other group was administered SNIPER(ER)-087 (30 mg/kg, intraperitoneally, every 24 h). Tumor volumes were measured every 2 days using a caliper and calculated according to the standard formula: (length × width2)/2. At 2 weeks, mice were sacrificed, and the tumors were excised.
Immunohistochemistry
Tumor tissues were fixed in 10% buffered formalin (Wako, Osaka, Japan) and embedded in paraffin, and 6-μm sections were prepared. Sections were deparaffinized and stained with hematoxylin and eosin (40). To detect PCNA, antigens were retrieved, and colorimetric detection was performed with anti-PCNA rabbit mAb (Cell Signaling Technology, catalog no. 13110).
Cell Cycle Analysis
After treatment, cells were gently trypsinized and washed with serum-containing medium. Cells were collected by centrifugation, additionally washed with PBS, and fixed in 70% ice-cold ethanol for 1 h on ice. The cells were then washed, treated with 1 mg/ml RNase A for 1 h at 37 °C, and stained in propidium iodide solution (50 μg/ml in 0.1% sodium citrate, 0.1% Nonidet P-40). The stained cells were analyzed in a FACScan flow cytometer (BD Biosciences).
Statistical Analysis
Student's t test was used to determine the significance of differences among the experimental groups. Values of p < 0.05 were considered significant.
Measurement of Binding Affinity
Binding experiments were performed in white 384-well plates (PerkinElmer Life Sciences, catalog no. 6007290). A total of 5 μl of His-tagged IAP proteins (40 nm XIAP_BIR3, 15 nm cIAP1_BIR3, and 35 nm cIAP2_BIR3) and 5 μl of increasing concentrations of compounds were added to wells in the assay buffer (20 mm HEPES, 150 mm NaCl, 0.1% BSA, 0.01% Tween 20, 0.1 mm DTT, pH 7.5). After shaking at room temperature, 5 μl of biotinyl-SMAC (20 nm XIAP_BIR3, 40 nm cIAP1_BIR3, and 120 nm cIAP2_BIR3 dissolved in assay buffer) was added to the well, followed by the addition of 5 μl of a mixture of anti-His6 cryptate and SA-XLent, and 160× dilution with HTRF detection buffer. After overnight incubation at room temperature in the dark, HTRF measurement was carried out on a multilabel reader (EnVision, PerkinElmer Life Sciences) with the following settings: measurement mode, time-resolved fluorescence; excitation, 320 nm; emission donor, 615 nm; and emission acceptor, 665 nm.
Fluorescence at 615 nm (F615 nm) represents the total europium cryptate signal, and fluorescence at 665 nm (F665 nm) represents the FRET signal. The ratio = (F665 nm/F615 nm) × 10,000 was calculated, and IC50 values were determined using this ratio by nonlinear regression curve fitting with the program XLfit.
The binding between test compounds and the human ERα protein was determined using the PolarScreenTM Estrogen Receptor-α Competitor Assay Green (Thermo Fisher Scientific, catalog no. A15882), containing recombinant ERα full-length protein, Fluormone ES2 Green2, and ES2 screening buffer. Purified ERα and Fluormone ES2 were diluted with assay buffer to final concentrations of 25 and 4.5 nm, respectively, and 4 μl of the dilution was added to each well of a 384-well black low-volume assay plate (Greiner catalog no. 784076). Then, 2 μl of ES2 screening buffer containing test compounds or DMSO was added to the well. The plate was subjected to centrifugation and incubated at room temperature for 1 h, and the intensity of the fluorescence polarization signal was measured by a plate reader (Envision, PerkinElmer Life Sciences). The wells containing ERα and Fluormone ES2 were used as a positive control, and the wells containing only Fluormone ES2 were used as a negative control. IC50 values were calculated by XLfit (ID Business Solutions, fit model 204) from the data expressed as % control inhibition.
Pharmacokinetics
Female BALB/c mice were purchased from Charles River Japan, maintained with 12-h light/dark cycles, and allowed free access to a chow diet (CE-2; Clea Japan) and drinking water. Animal study was conducted with a 7-day acclimation period after arrival. SNIPER(ER)-87 was dissolved in 10% DMSO, 10% Cremophor EL, 20% PEG 400, 60% distilled water, and mice were injected with 10 mg/kg SNIPER(ER)-87 intraperitoneally. After 0.5, 1, 2, 4, 8, and 24 h, blood was obtained in a tube with heparin under isoflurane-induced anesthesia and centrifuged at 12,000 rpm for 5 min to collect the plasma for measurement of compound concentrations. The care and use of the animals and the experimental protocols used in this research were approved by the Experimental Animal Care and Use Committee of Takeda Pharmaceutical Co., Ltd.
Author Contributions
N. O., K. O., N. S., and M. N. designed the experiments; N. O., K. O., N. S., T. H., O. S., R. K., H. F., M. T., and H. M. performed the experiments and analyzed the data; M. I., K. N., O. U., K. S., Y. I., H. N., and N. C. designed and synthesized the compounds; N. O., M. I., K. N., O. U., K. S., and M. N. wrote the manuscript; and M. N. supervised all research. All authors discussed and checked the manuscript.
Supplementary Material
Acknowledgments
We thank Mariko Seki for measurement of the protein knockdown activities; Yasumi Kumagai and Motoo Iida for NMR measurement and analysis; Mitsuyoshi Nishitani for small molecule X-ray analysis; Chie Kushibe and Katsuhiko Miwa for HPLC purification and analysis; Mihoko Kunitomo and Toshiyuki Nomura for advice on the in vivo experiments; and Genostaff for histochemical analysis.
This work was supported in part by Japan Society for the Promotion of Science KAKENHI Grants 26860049 (to N. O.) and 16H05090 and 16K15121 (to M. N.), Japan Agency for Medical Research and Development Grants 15ak0101029h1402 and 16ak0101029j1403 (to M. N.) and 16cm0106124j0001 (to N. O.), the Ministry of Health and Labor Welfare, Japan (to M. N.), and Takeda Science Foundation (to N. O.). M. I., K. N., O. U., K. S., O. S., R. K., H. F., M. T., H. M., Y. I., H. N., and N. C. are employees of Takeda Pharmaceutical Co., Ltd. The authors filed a patent for the novel compounds described in this paper.

This article contains supplemental Schemes S1–S13, Chemistry, and Fig. S1.
- PROTAC
- Proteolysis Targeting Chimera
- SNIPER
- Specific and Nongenetic Inhibitor of apoptosis Protein (IAP)-dependent protein Eraser
- ERα
- estrogen receptor α
- XIAP
- X-linked inhibitor of apoptosis protein
- VHL
- von Hippel-Lindau
- CRBN
- cereblon
- CRABP
- cellular retinoic acid-binding protein
- UPS
- ubiquitin-proteasome system
- PCNA
- proliferating cell nuclear antigen
- 4-OHT
- 4-hydroxytamoxifen
- HTRF
- homogeneous time-resolved fluorescence.
References
- 1. Zhang J., Yang P. L., and Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wells J. A., and McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 3. Arkin M. R., and Wells J. A. (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev. Drug Discov. 3, 301–317 [DOI] [PubMed] [Google Scholar]
- 4. Mansoori B., Sandoghchian Shotorbani S., and Baradaran B. (2014) RNA interference and its role in cancer therapy. Adv. Pharm. Bull. 4, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ozcan G., Ozpolat B., Coleman R. L., Sood A. K., and Lopez-Berestein G. (2015) Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 87, 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ohoka N., Shibata N., Hattori T., and Naito M. (2016) Protein knockdown technology: application of ubiquitin ligase to cancer therapy. Curr. Cancer Drug Targets 16, 136–146 [DOI] [PubMed] [Google Scholar]
- 7. Toure M., and Crews C. M. (2016) Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 55, 1966–1973 [DOI] [PubMed] [Google Scholar]
- 8. Bondeson D. P., Mares A., Smith I. E., Ko E., Campos S., Miah A. H., Mulholland K. E., Routly N., Buckley D. L., Gustafson J. L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., et al. (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J. D., Crew A. P., Coleman K., and Crews C. M. (2015) Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., and Bradner J. E. (2015) Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Itoh Y., Ishikawa M., Naito M., and Hashimoto Y. (2010) Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 132, 5820–5826 [DOI] [PubMed] [Google Scholar]
- 12. Okuhira K., Ohoka N., Sai K., Nishimaki-Mogami T., Itoh Y., Ishikawa M., Hashimoto Y., and Naito M. (2011) Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that cross-link cIAP1 and the target protein. FEBS Lett. 585, 1147–1152 [DOI] [PubMed] [Google Scholar]
- 13. Demizu Y., Okuhira K., Motoi H., Ohno A., Shoda T., Fukuhara K., Okuda H., Naito M., and Kurihara M. (2012) Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett. 22, 1793–1796 [DOI] [PubMed] [Google Scholar]
- 14. Okuhira K., Demizu Y., Hattori T., Ohoka N., Shibata N., Nishimaki-Mogami T., Okuda H., Kurihara M., and Naito M. (2013) Development of hybrid small molecules that induce degradation of estrogen receptor-α and necrotic cell death in breast cancer cells. Cancer Sci. 104, 1492–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ohoka N., Nagai K., Hattori T., Okuhira K., Shibata N., Cho N., and Naito M. (2014) Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis. 5, e1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sekine K., Takubo K., Kikuchi R., Nishimoto M., Kitagawa M., Abe F., Nishikawa K., Tsuruo T., and Naito M. (2008) Small molecules destabilize cIAP1 by activating autoubiquitylation. J. Biol. Chem. 283, 8961–8968 [DOI] [PubMed] [Google Scholar]
- 17. Fulda S., and Vucic D. (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 11, 109–124 [DOI] [PubMed] [Google Scholar]
- 18. Deveraux Q. L., and Reed J. C. (1999) IAP family proteins–suppressors of apoptosis. Genes Dev. 13, 239–252 [DOI] [PubMed] [Google Scholar]
- 19. Salvesen G. S., and Duckett C. S. (2002) IAP proteins: blocking the road to death's door. Nat. Rev. Mol. Cell Biol. 3, 401–410 [DOI] [PubMed] [Google Scholar]
- 20. Deveraux Q. L., Takahashi R., Salvesen G. S., and Reed J. C. (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304 [DOI] [PubMed] [Google Scholar]
- 21. Hao Y., Sekine K., Kawabata A., Nakamura H., Ishioka T., Ohata H., Katayama R., Hashimoto C., Zhang X., Noda T., Tsuruo T., and Naito M. (2004) Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 6, 849–860 [DOI] [PubMed] [Google Scholar]
- 22. Suzuki Y., Nakabayashi Y., and Takahashi R. (2001) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 98, 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kikuchi R., Ohata H., Ohoka N., Kawabata A., and Naito M. (2014) APOLLON protein promotes early mitotic CYCLIN A degradation independent of the spindle assembly checkpoint. J. Biol. Chem. 289, 3457–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Imoto I., Tsuda H., Hirasawa A., Miura M., Sakamoto M., Hirohashi S., and Inazawa J. (2002) Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 62, 4860–4866 [PubMed] [Google Scholar]
- 25. Imoto I., Yang Z. Q., Pimkhaokham A., Tsuda H., Shimada Y., Imamura M., Ohki M., and Inazawa J. (2001) Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 61, 6629–6634 [PubMed] [Google Scholar]
- 26. Tamm I., Kornblau S. M., Segall H., Krajewski S., Welsh K., Kitada S., Scudiero D. A., Tudor G., Qui Y. H., Monks A., Andreeff M., and Reed J. C. (2000) Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res. 6, 1796–1803 [PubMed] [Google Scholar]
- 27. Cohen P., and Tcherpakov M. (2010) Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 143, 686–693 [DOI] [PubMed] [Google Scholar]
- 28. Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., and Vucic D. (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 [DOI] [PubMed] [Google Scholar]
- 29. Vince J. E., Wong W. W., Khan N., Feltham R., Chau D., Ahmed A. U., Benetatos C. A., Chunduru S. K., Condon S. M., McKinlay M., Brink R., Leverkus M., Tergaonkar V., Schneider P., Callus B. A., et al. (2007) IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 [DOI] [PubMed] [Google Scholar]
- 30. Bertrand M. J., Milutinovic S., Dickson K. M., Ho W. C., Boudreault A., Durkin J., Gillard J. W., Jaquith J. B., Morris S. J., and Barker P. A. (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 [DOI] [PubMed] [Google Scholar]
- 31. Hall J. M., and McDonnell D. P. (2005) Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol. Interv. 5, 343–357 [DOI] [PubMed] [Google Scholar]
- 32. Nadji M., Gomez-Fernandez C., Ganjei-Azar P., and Morales A. R. (2005) Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am. J. Clin. Pathol. 123, 21–27 [DOI] [PubMed] [Google Scholar]
- 33. Raina K., Lu J., Qian Y., Altieri M., Gordon D., Rossi A. M., Wang J., Chen X., Dong H., Siu K., Winkler J. D., Crew A. P., Crews C. M., and Coleman K. G. (2016) PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 7124–7129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pedersen J., LaCasse E. C., Seidelin J. B., Coskun M., and Nielsen O. H. (2014) Inhibitors of apoptosis (IAPs) regulate intestinal immunity and inflammatory bowel disease (IBD) inflammation. Trends Mol. Med. 20, 652–665 [DOI] [PubMed] [Google Scholar]
- 35. Lai A. C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E., Hines J., and Crews C. M. (2016) Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 55, 807–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Katsuragi Y., Ichimura Y., and Komatsu M. (2015) p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672–4678 [DOI] [PubMed] [Google Scholar]
- 37. Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T. K., Choudhary C., Bekker-Jensen S., and Mailand N. (2015) Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527, 389–393 [DOI] [PubMed] [Google Scholar]
- 38. Honsho M., Yamashita S., and Fujiki Y. (2016) Peroxisome homeostasis: mechanisms of division and selective degradation of peroxisomes in mammals. Biochim. Biophys. Acta 1863, 984–991 [DOI] [PubMed] [Google Scholar]
- 39. Mohapatra B., Ahmad G., Nadeau S., Zutshi N., An W., Scheffe S., Dong L., Feng D., Goetz B., Arya P., Bailey T. A., Palermo N., Borgstahl G. E., Natarajan A., Raja S. M., et al. (2013) Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 1833, 122–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shibata N., Ohoka N., Sugaki Y., Onodera C., Inoue M., Sakuraba Y., Takakura D., Hashii N., Kawasaki N., Gondo Y., and Naito M. (2015) Degradation of stop codon read-through mutant proteins via the ubiquitin-proteasome system causes hereditary disorders. J. Biol. Chem. 290, 28428–28437 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.