

Abstract
Estrogen receptor α (ER-α) is a nuclear hormone receptor that controls selected genes, thereby regulating proliferation and differentiation of target tissues, such as breast. Gene expression controlled by ER-α is modulated by Ca2+ via calmodulin (CaM). Here we present the NMR structure of Ca2+-CaM bound to two molecules of ER-α (residues 287–305). The two lobes of CaM bind to the same site on two separate ER-α molecules (residues 292, 296, 299, 302, and 303), which explains why CaM binds two molecules of ER-α in a 1:2 complex and stabilizes ER-α dimerization. Exposed glutamate residues in CaM (Glu-11, Glu-14, Glu-84, and Glu-87) form salt bridges with key lysine residues in ER-α (Lys-299, Lys-302, and Lys-303), which is likely to prevent ubiquitination at these sites and inhibit degradation of ER-α. Transfection of cells with full-length CaM slightly increased the ability of estrogen to enhance transcriptional activation by ER-α of endogenous estrogen-responsive genes. By contrast, expression of either the N- or C-lobe of CaM abrogated estrogen-stimulated transcription of the estrogen responsive genes pS2 and progesterone receptor. These data suggest that CaM-induced dimerization of ER-α is required for estrogen-stimulated transcriptional activation by the receptor. In light of the critical role of ER-α in breast carcinoma, our data suggest that small molecules that selectively disrupt the interaction of ER-α with CaM may be useful in the therapy of breast carcinoma.
Keywords: breast cancer, calmodulin (CaM), estrogen receptor, receptor structure-function, signaling, structure-function, transcription
Introduction
Estrogens exert pleiotropic functions in humans, ranging from neuroprotection and prevention of osteoporosis to cardioprotection and growth of breast tissue (1). These effects are produced through two members of the nuclear receptor superfamily, estrogen receptor-α (ER-α)4 and ER-β (2). The receptors share distinct structural and functional domains. These are the A/B regions, which contain a transactivation domain (AF1), the C region, which corresponds to the DNA-binding domain, the D region containing the hinge domain, and the E/F regions in which the ligand binding domain is located (2). Binding of 17β-estradiol (E2) to ER-α elicits effects in target tissues via the “classic” pathway or by non-genomic signaling through activation of protein kinase cascades (2, 3). In the classic signaling mechanism, E2 induces a conformational change in ER-α, which then dimerizes. The E2-bound receptor translocates to the nucleus where it binds to DNA at estrogen response elements (EREs) and recruits co-regulatory proteins. These co-activators and co-repressors modulate the transcriptional activation of genes by ER-α. A substantial body of evidence implicates perturbation of E2 signaling in cancer initiation, progression, and response to therapy (2). Although best characterized in breast cancer, ER also contributes to carcinoma of the endometrium and prostate. A clear understanding of the molecular mechanism of ER-α signaling is required to develop better therapeutic modalities for these neoplasms.
Recent data indicate that numerous proteins influence the structure and function of ER-α, stabilize the ER-α/DNA interaction, and modulate gene expression (4). These proteins are involved in cell cycle regulation, chromatin remodeling, protein turnover, and cell migration. Accumulating evidence shows that a group of ER-α modifying proteins participate in cell signaling. For example, the scaffold protein IQGAP1, which integrates diverse signaling pathways (5, 6), binds ER-α and modulates its transcriptional function (7).
Another signaling protein that influences ER-α is the Ca2+ modulator calmodulin (CaM). CaM is a highly conserved protein that contains 148 amino acids. Via direct interactions with numerous diverse proteins, CaM regulates several cellular functions including growth, proliferation, and cell cycle progression (8). CaM binds directly to ER-α, but not to ER-β (9, 10), in a Ca2+-regulated manner (11, 12). This interaction regulates the degradation of ER-α via the ubiquitin-proteasome pathway (13) and is also required for normal transcriptional function of ER-α (14). To gain insight into the molecular mechanism by which CaM produces these effects, we recently conducted structural analysis and solved the NMR structures of the individual lobes of CaM (N- and C-lobes) each bound to a functional fragment of ER-α (residues 287–305) (15). Shortly thereafter, Leclercq and colleagues (16) published a manuscript in which they analyzed the interaction of CaM with peptides corresponding to amino acid residues 287–311 and 295–311 of ER-α. These studies yielded two distinct models to describe the interaction between CaM and ER-α (15, 16). Our data revealed that one CaM molecule binds two molecules of ER-α in a 1:2 complex (15). We generated a model in which each lobe of CaM is attached to a separate hinge domain of ER-α, suggesting that CaM can facilitate dimerization of ER-α. By contrast, Carlier et al. (16) put forward a scheme in which the two lobes of CaM interact allosterically and CaM undergoes a concerted conformational change when binding ER-α. To reconcile these conflicting models and determine the correct mode of binding, we analyzed the structure of full-length CaM bound to two ER-α peptides. The data presented here reveal that this structure is identical to the structures of the separate CaM lobes each bound to ER-α. Thus, the two ER-α binding sites on CaM are completely independent. In addition, analysis of ER-α transcriptional activity in human cells transfected with separate lobes of CaM provides in vivo evidence that bolsters our model.
Results
NMR Structure of Full-length CaM Bound to Two Molecules of ER-α
Previously we solved NMR structures of individual CaM lobes (CaM-N and CaM-C) each bound to a peptide fragment of ER-α (residues 287–305, called ER(287–305)) (15). The N-terminal half (CaM-N, amino acids 1–74) and the C-terminal half (CaM-C, amino acids 75–148) of calmodulin each bound to one ER-α with nearly the same affinity as full-length CaM (CaM-F) binds to two ER-α (15). Thus, the two CaM lobes bind independently to two separate ER-α molecules, suggesting that CaM may stabilize dimerization of ER-α. In the current study, we present a NMR structural analysis of full-length CaM bound to two molecules of ER(287–305). The 1H-15N HSQC spectrum of 15N-labeled CaM bound to two molecules of ER(287–305) (Fig. 1A) looks similar to the NMR spectra of CaM-N (cyan in Fig. 1A) and CaM-C (magenta in Fig. 1A) each bound to one molecule of ER(287–305). This spectral similarity demonstrates that in the full-length protein both CaM lobes are independently folded and each lobe binds independently to a separate ER-α molecule. The constant time 1H-13C HSQC spectrum of 13C-labeled CaM bound to two molecules of unlabeled ER(287–305) also matches quite well to both spectra of the individual lobes (Fig. 1B). The agreement of the methyl chemical shifts in 1H-13C HSQC spectra (of CaM-F versus CaM-N and CaM-C) demonstrates that the structural environment of side chain methyl groups in the exposed hydrophobic core of both lobes that contact ER(287–305) must be unchanged in full-length CaM. Therefore, the structure of the individual lobes bound to ER-α determined previously (15) must have a similar structure in full-length CaM bound to two ER-α molecules. To further test this hypothesis, we solved the NMR structure of full-length CaM bound to two ER(287–305) molecules.
FIGURE 1.

NMR spectroscopy of CaM binding to ER(287–305). A, 15N-1H HSQC spectra of 15N-labeled full-length CaM (black), CaM-N (cyan), and CaM-C (magenta) in the presence of unlabeled ER(287–305). B, constant time 13C-1H HSQC spectra of 13C-labeled full-length CaM (black), CaM-N (cyan), and CaM-C (magenta) in the presence of unlabeled ER(287–305). The experimental conditions are defined under “Experimental Procedures” and were the same as described previously (15).
An NMR-derived structural model of full-length CaM bound to two molecules of ER(287–305) is shown in Fig. 2. Three-dimensional protein structures derived from the NMR assignments were calculated on the basis of NOE data, chemical shift analysis, 3JNHα spin-spin coupling constants, and residual dipolar coupling restraints as described previously (15) (see “Experimental Procedures”). Table 1 summarizes the structural statistics calculated for the 10 lowest energy conformers.
FIGURE 2.
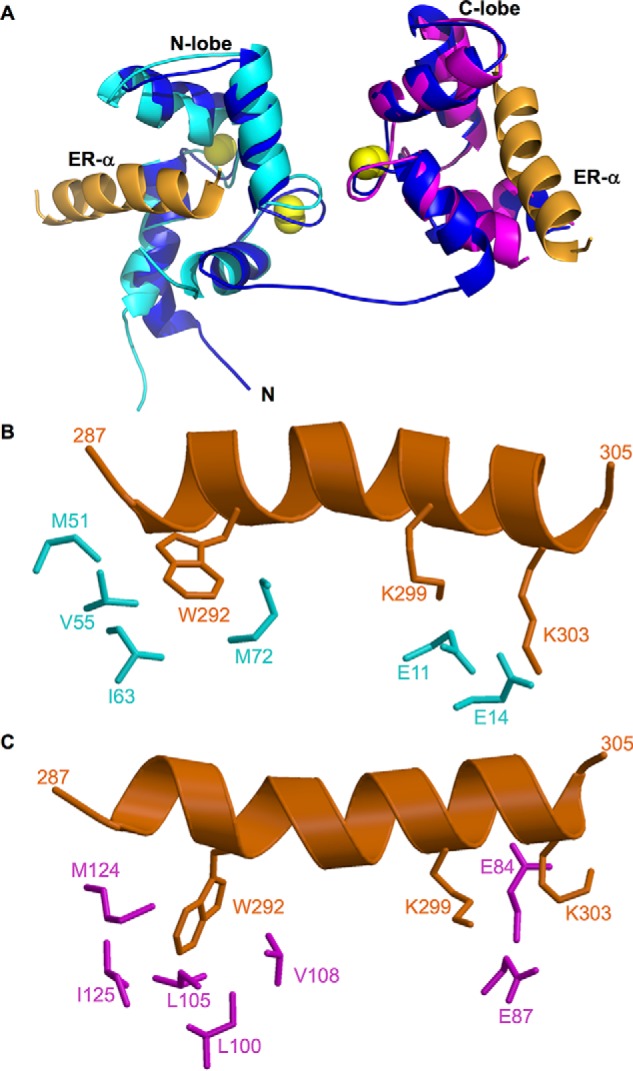
Structures of CaM·ER-α complexes. A, NMR structure of full-length CaM (dark blue) bound to two molecules of ER(287–305) (orange). The atomic coordinates were deposited into the Protein Data Bank (PDB accession no. 5T0X). Previous NMR structures of CaM-N (2LLO, cyan) and CaM-C (2LLQ, magenta) are overlaid on the full-length CaM structure for comparison. Bound Ca2+ are yellow spheres. The flexible interdomain linker (residues 74–82) forms an extended main chain conformation depicted by a blue line. B and C, exposed hydrophobic side chain atoms of CaM N-lobe (highlighted cyan in panel B) and C-lobe (highlighted magenta in panel C) interact with side chain atoms of key ER-α residues (orange) shown as sticks. Hydrophobic side chain atoms in ER-α (Trp-292) form detailed contacts with each lobe of CaM, and basic side chains in ER-α (Lys-299 and Lys-303) form salt bridges with Glu-14 (N-lobe) and Glu-84 (C-lobe) of CaM.
TABLE 1.
Structure statistics for NMR structures of CaM bound to ER(287–305)
| NMR restraints | |
| Short range NOEs for ER peptides | 30 |
| Dihedral angles | 79 |
| H-bonds | 66 |
| Total intermolecular NOEs | 96 |
| 1DHN RDC | 76 |
| Ramachandran plot | |
| Most favored region (%) | 80.5 |
| Allowed region (%) | 18.0 |
| Disallowed region (%) | 1.5 |
| Root mean squared deviation from average structure (Å) All backbone atoms | 0.59 |
The main chain structure of the N-lobe in full-length CaM bound to ER(287–305) looks quite similar to the structure of CaM-N bound to ER(287–305) (15). The root mean square deviation is <0.6 Å when comparing the main chain atoms of the full-length CaM N-lobe versus CaM-N. The EF-hand interhelical angles for the full-length CaM N-lobe (bound to ER(287–305) are 84° for EF1 and 90° for EF2, which are lower than those for free CaM (EF1, 103.8° and EF2, 101°). Exposed hydrophobic side chains in the full-length CaM N-lobe (Met-51, Val-55, Ile-63, and Met-72) interact with the aromatic side chain of Trp-292 from ER(287–305) (Fig. 2B). Also noteworthy are exposed glutamate side chains in full-length CaM N-lobe (Glu-11 and Glu-14) that form salt bridges with lysine residues in ER-α (Lys-299 and Lys-303).
The main chain structure of the C-lobe in full-length CaM bound to ER(287–305) (Fig. 2) looks similar to the structure of CaM-C bound to ER(287–305) (15). The root mean square deviation is <0.8 Å when comparing the main chain atoms of the full-length CaM C-lobe versus CaM-C. The EF-hand interhelical angles for the full-length CaM C-lobe bound to ER-α (103 °C for EF3 and 94 °C for EF4) are close to those for free CaM. Exposed hydrophobic residues in CaM (Ile-100, Leu-105, Val-108, Met-124, and Ile-125) form close contacts with the aromatic side chain of Trp-292 from ER(287–305) (Fig. 2C). Two lysine residues in ER-α (Lys-299 and Lys-303) form salt bridges with Glu-84 and Glu-87 in CaM.
The CaM Lobes Bind ER-α in Cell Lysates
Analysis was conducted to determine whether CaM-N and CaM-C could bind endogenous ER-α in a normal cell milieu. To evaluate this interaction, we wanted to transfect cells with the individual lobes of CaM and examine ER-α binding. Although expression of intact full-length CaM in cells was readily achieved, we encountered considerable difficulty expressing the isolated lobes of CaM in mammalian cells. After numerous attempts with several different plasmids that express the N- or C-terminal half of CaM, we were eventually successful with GFP-tagged constructs. We generated GFP-tagged constructs of full-length CaM and the two halves; the N-terminal half, amino acids 1–74, and the C-terminal half, amino acids 75–148. Transfection of HEK-293 or MCF-7 cells with each plasmid yielded clear, and reproducible, protein expression as detected with anti-GFP antibody (Figs. 3A, 4C, 5C, and 6C). Each protein migrated to its expected position on SDS-PAGE.
FIGURE 3.
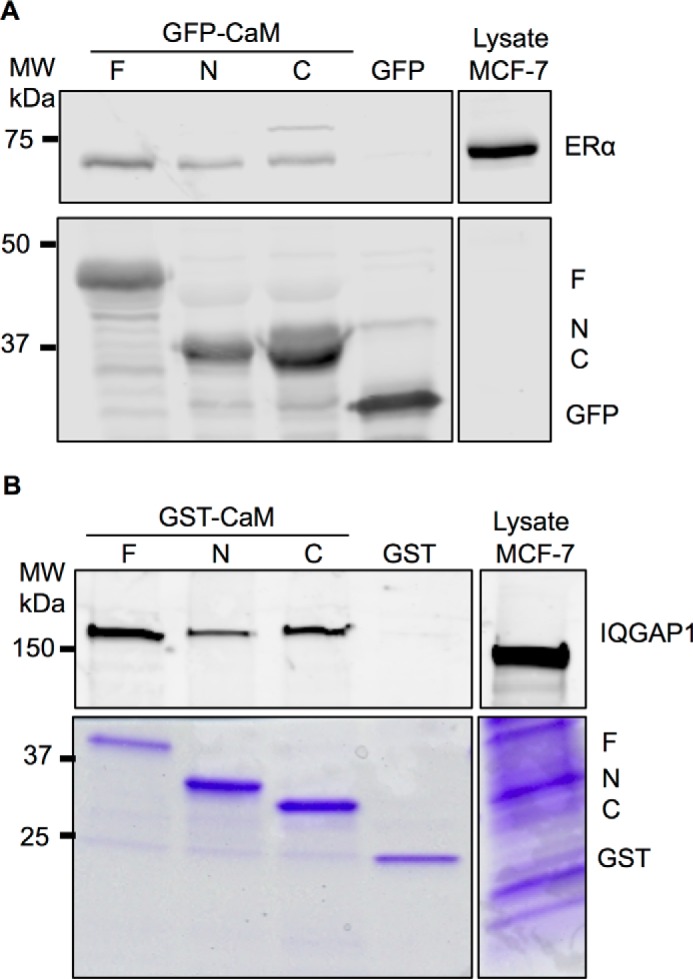
Binding of the CaM constructs to ER-α and IQGAP1. A, HEK-293 cells were transfected with GFP-tagged CaM-F (F), CaM-N (N), or CaM-C (C) or GFP alone (as control). GFP-tagged proteins were isolated with GFP-Trap_A agarose as described under “Experimental Procedures,” then incubated with equal amounts of protein lysate from MCF-7 cells. An aliquot of MCF-7 lysate that was not incubated with GFP-CaM or GFP was processed in parallel (Lysate). Proteins were resolved by SDS-PAGE and Western blots were probed with anti-ER-α (upper panel) and anti-GFP (lower panel) antibodies. Data are representative of 3 independent experiments. B, equal amounts of protein lysate from MCF-7 cells were incubated with GST-tagged CaM-F (F), CaM-N (N), or CaM-C (C) or GST alone (control). Complexes were isolated with glutathione-Sepharose. An aliquot of lysate was processed in parallel (Lysate). Proteins were resolved by SDS-PAGE and the gel was cut at ∼60 kDa. The top part of the gel was transferred to PVDF, whereas the lower part was stained with Coomassie Blue (lower panel). PVDF membranes were probed with anti-IQGAP1 antibodies (upper panel). Data are representative of 2 independent experiments.
FIGURE 4.
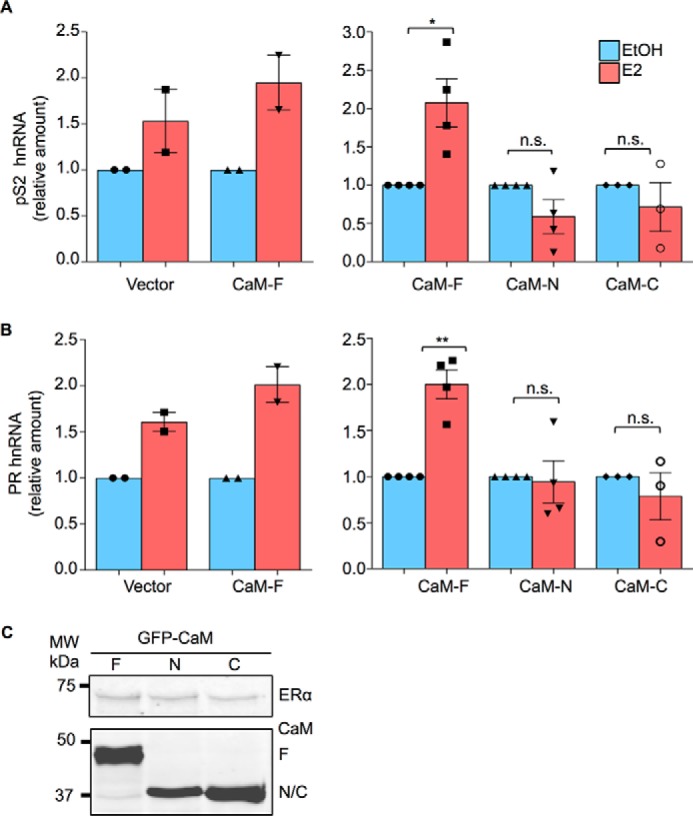
CaM alters ER-α function. A, HEK-293 cells were transiently transfected with both ER-α and GFP-tagged CaM-F, CaM-N, CaM-C, or pEGFP (Vector). Cells were cultured in phenol red-free medium for 24 h, then vehicle (EtOH, blue bars) or 100 nm E2 (red bars) was added to the medium. After incubation for 6 h, total RNA was isolated and quantitative RT-PCR analysis was performed to measure pS2 hnRNA. The amount of RNA in each sample was corrected for β-actin RNA in the same sample. Vehicle-treated cells were set as 1. The data represent the mean ± S.E. (error bars) of two independent experiments for vector and CaM-F (left panel) or three or four independent experiments for CaM-F, CaM-N, and CaM-C (right panel). Each condition was measured in triplicate. B, HEK-293 cells were transiently transfected with both ER-α and either pEGFP (Vector) or the GFP-tagged CaM plasmids. Following cell culture and E2 stimulation, performed as described for panel A, PR hnRNA was measured by quantitative RT-PCR. Samples were analyzed as described for pS2. The data represent the mean ± S.E. (error bars) of two independent experiments for vector and CaM-F (left panel) or three or four independent experiments for CaM-F, CaM-N, and CaM-C (right panel). Each condition was measured in triplicate. *, p < 0.05; **, p < 0.01. C, cells, transfected as outlined above, were lysed and equal amounts of protein lysate were resolved by Western blotting. Blots were probed with antibodies to ER-α (upper panel) and GFP (lower panel). A representative experiment of 2 is shown.
FIGURE 5.
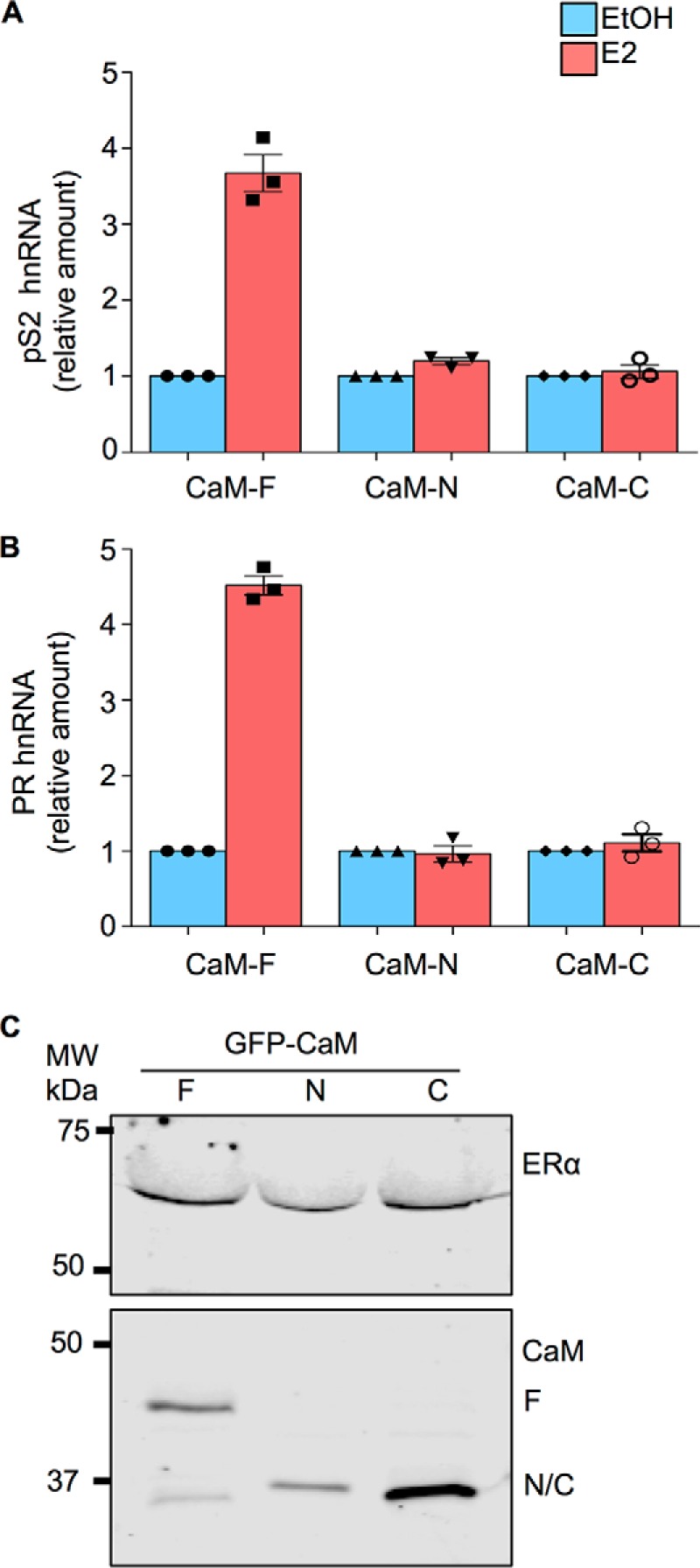
Individual lobes of CaM impair E2 stimulation of endogenous ER-α transcription. A and B, MCF-7 cells were transiently transfected with either GFP-tagged CaM-F, CaM-N, or CaM-C. Cells were cultured in phenol red-free medium for 24 h, then vehicle (EtOH, blue bars) or 100 nm E2 (red bars) was added to the medium. After incubation for 6 h, total RNA was isolated and quantitative RT-PCR analysis was performed to measure pS2 (panel A) or PR (panel B) hnRNA. The amount of RNA in each sample was corrected for β-actin RNA in the same sample. Vehicle-treated cells were set as 1. The data represent the mean ± S.E. (error bars) of an experiment performed in triplicate. C, MCF-7 cells, transfected as outlined above, were lysed and equal amounts of protein lysate were resolved by Western blotting. Blots were probed with antibodies to ER-α (upper panel) and GFP (lower panel). All data are representative of at least 3 independent experiments.
FIGURE 6.
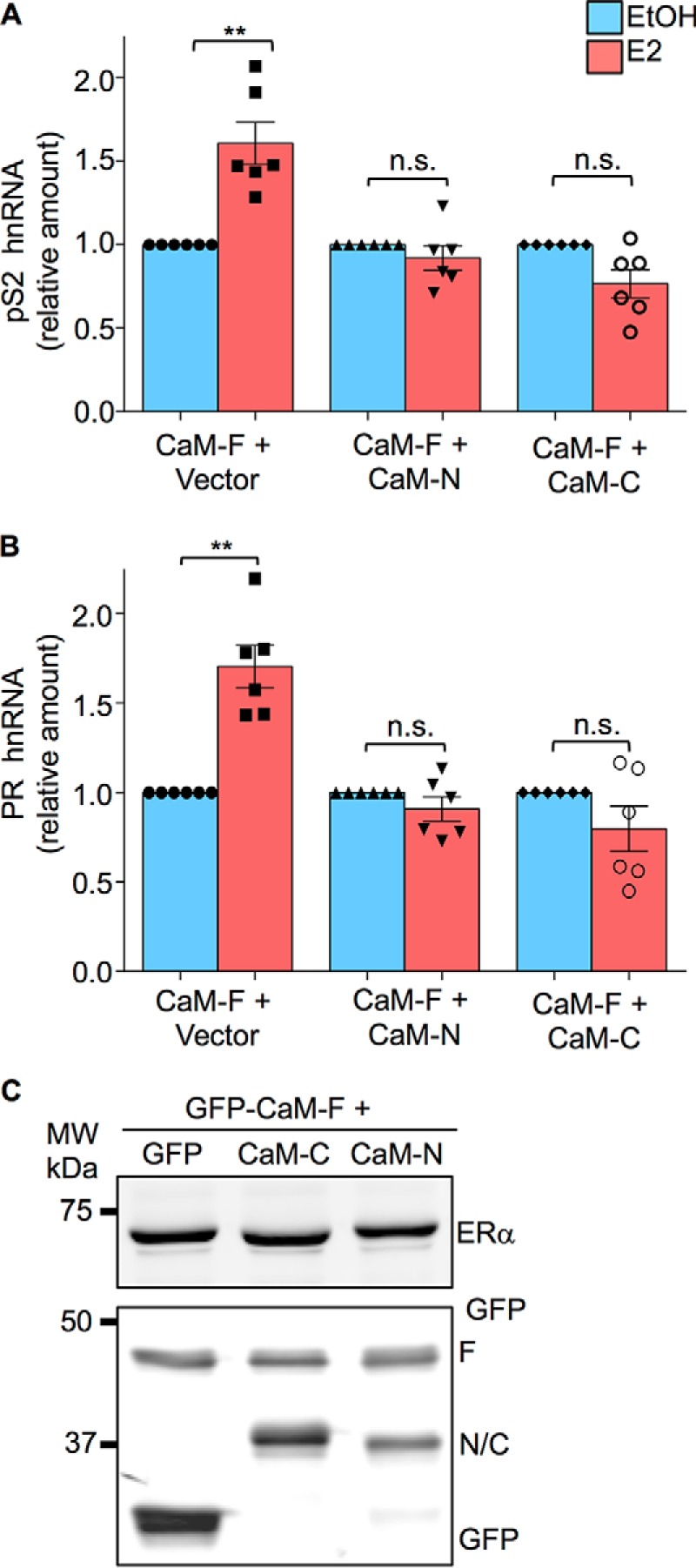
Individual lobes of CaM abrogate E2-stimulated transcriptional activation by ER-α. A, HEK-293 cells were transiently transfected with both ER-α and pEGFP-CaM-F as well as pEGFP (vector), pEGFP-CaM-N, or pEGFP-CaM-C. Cells were cultured in phenol red-free medium for 24 h, then vehicle (EtOH, blue bars) or 100 nm E2 (red bars) was added to the medium. After incubation for 6 h, total RNA was isolated and quantitative RT-PCR analysis was performed to measure pS2 hnRNA. The amount of RNA in each sample was corrected for β-actin RNA in the same sample. Vehicle-treated cells were set as 1. The data represent the mean ± S.E. (error bars) of six independent experiments, each performed in triplicate. B, HEK-293 cells were transfected and stimulated with E2 as described for panel A. PR hnRNA was measured by quantitative RT-PCR. Samples were analyzed as described for pS2. The data represent the mean ± S.E. (error bars) of six independent experiments, each performed in triplicate. C, cells, transfected as outlined above, were lysed and equal amounts of protein lysate were resolved by Western blotting. Blots were probed with antibodies to ER-α (upper panel) and GFP (lower panel). A representative experiment of 4 is shown. **, p < 0.005.
To examine binding, we transfected GFP-tagged CaM-F, CaM-N, and CaM-C into HEK-293 cells, isolated the proteins with GFP-Trap_A agarose, and then incubated them with MCF-7 cell lysates. GFP-tagged full-length CaM bound to endogenous ER-α in MCF-7 cell lysates (Fig. 3A). These findings are congruent with our prior documentation that endogenous ER-α co-immunoprecipitates with endogenous untagged CaM from MCF-7 human breast epithelial cells (11). Consistent with the in vitro data generated with pure CaM-N, CaM-C, and the fragment of ER-α (287–305) (15), we observed that the individual lobes of CaM could precipitate endogenous ER-α from cell lysates (Fig. 3A). Moreover, in keeping with the stoichiometry data, the amount of ER-α bound to CaM-F is considerably greater than that bound to CaM-N or CaM-C. No ER-α is present in samples containing GFP alone (Fig. 3A), validating the specificity of the interaction with the CaM constructs. Probing the blots for GFP showed similar amounts of each CaM construct (Fig. 3A, lower panel). These data reveal that the N- and C-halves of CaM independently bind ER-α in cell lysates.
To determine whether CaM-N and CaM-C are capable of binding other targets, we examined IQGAP1, which contains four IQ motifs that interact with CaM (17). We tagged CaM-F, CaM-N, and CaM-C with GST, expressed the proteins in Escherichia coli (Fig. 3B, lower panel) and incubated each with equal amounts of MCF-7 cell lysate. GST-tagged full-length CaM bound to IQGAP1 in cell lysates (Fig. 3B). Similarly, CaM-N and CaM-C each bind to endogenous IQGAP1. Thus, the individual lobes of CaM can bind to at least two targets that have different modes of association with CaM.
Individual Lobes of CaM Abrogate E2-stimulated Transcription by ER-α
If full-length CaM binds two molecules of ER-α, thus promoting receptor dimerization and facilitating ER-α transcriptional activity, one would anticipate that the individual lobes of CaM would elicit a different effect. In this situation, each lobe of CaM would bind to a separate molecule of ER-α, but the separate CaM lobes would not be able to induce receptor dimerization and ER-α transcriptional activity would therefore not be increased. The strategy we adopted to test this hypothesis was to transfect cells with the individual lobes of CaM and examine ER-α function.
The effect of the CaM constructs on ER-α function was evaluated by assessing the ability of ER-α to activate selected endogenous estrogen responsive genes. E2 promotes an increase of pS2 hnRNA in HEK-293 cells that were transfected with ER-α (Fig. 4A, left panel). (HEK-293 cells do not contain endogenous ER-α (data not shown).) Transfection of GFP-tagged full-length CaM slightly enhanced the ability of E2 to stimulate pS2 RNA. By contrast, expression of either CaM-N or CaM-C abrogated the effect of E2 on the transcriptional activation by ER-α (Fig. 4A, right panel). To extend these findings, we examined the effect of CaM on the ability of ER-α to activate progesterone receptor (PR), which is another endogenous estrogen-responsive gene. Analysis showed that either CaM-N or CaM-C blocked the increase in PR elicited by E2 (Fig. 4B). Western blotting of protein lysates confirmed that the cells expressed equivalent amounts of each CaM construct and had the same level of ER-α (Fig. 4C).
To determine whether the expression of GFP-tagged CaM-N or CaM-C can interfere with E2 stimulation of endogenous ER-α, MCF-7 were transfected with these constructs and transcription of pS2 and PR hnRNA was examined by quantitative RT-PCR. E2 stimulated transcription of pS2 by ER-α in MCF-7 cells transfected with GFP-tagged CaM-F (Fig. 5A). Analogous to the observations in HEK-293 cells, E2 was unable to increase pS2 transcription in MCF-7 cells transfected with CaM-N or CaM-C. Similarly, the ability of E2 to increase PR in MCF-7 cells was abrogated in the presence of CaM-N or CaM-C (Fig. 5B). The level of expression of each CaM construct was equivalent (Fig. 5C).
To ascertain whether the individual lobes of CaM could interfere with the effects of full-length CaM, we co-transfected full-length CaM with either CaM-N or CaM-C and examined E2-stimulated transcriptional activation. In HEK-293 cells transfected with full-length CaM and empty vector, E2 significantly stimulated the transcriptional activation of pS2 RNA (Fig. 6A). However, when cells were co-transfected with full-length CaM and CaM-N, E2 was unable to promote transcriptional activation of pS2 RNA. Similarly, transfecting CaM-C together with full-length CaM into cells completely prevented E2 from enhancing transcriptional activation of pS2 RNA (Fig. 6A). Essentially identical results were observed with PR. Expression of either CaM-N or CaM-C with full-length CaM abrogated the transcriptional activation of PR induced by E2 (Fig. 6B). These findings suggest that the individual lobes of CaM competitively inhibit full-length CaM. Western blotting of cell lysates verified the expression of equivalent levels of ER-α and the CaM constructs (Fig. 6C). Collectively these data indicate that the separate lobes of CaM are able to bind to ER-α, but they are unable to induce dimerization and promote activation of the receptor. It is noteworthy that binding of a single lobe of CaM, either the N- or C-lobe, to ER-α completely prevents E2 from stimulating transcriptional activation of the target genes we investigated.
Discussion
Previously, we solved the NMR structures of individual CaM lobes (CaM-N and CaM-C) each of which binds to the same site on ER-α (15). We showed that full-length CaM binds to two ER-α molecules, whereas CaM-N and CaM-C each binds to only one (15). On the basis of this earlier structural analysis, we proposed that each lobe of CaM binds to a separate ER-α molecule. In the current study, we now present the structure of full-length CaM bound to two ER-α peptides (Fig. 2). The structure reveals that each CaM lobe makes contact with the same residues of ER-α (residues 292, 299, and 303 in Fig. 2B). Furthermore, the structure of both lobes in full-length CaM (bound to two ER-α) looks identical to the previous structures of the individual lobes each bound to a single ER-α peptide (15). This structural independence of the two CaM lobes is consistent with our binding studies (15) that indicate each CaM lobe binds to ER-α with nearly the same binding affinity as full-length CaM. Finally, the structure of full-length CaM bound to two molecules of ER-α indicates a lack of structural contacts between the CaM lobes, which argues against allosteric conformational changes suggested by Carlier et al. (16). We conclude that one molecule of CaM connects two molecules of ER-α, which stabilizes its dimer structure and promotes transcriptional activation.
Our in vivo experiments provide biological data to support the model derived from the structural analysis. Prior work from our laboratory (11) and others (10, 12, 18), established that CaM interacts with ER-α in vitro and in cells. Binding of CaM modulates both ER-α degradation (10, 11, 13) and E2-stimulated transcriptional activity of ER-α (14). Interestingly, CaM is required for the formation of the ER-α·ERE complex and for activation of an estrogen responsive promotor (19). We previously generated an ER-α construct in which Ile-298 and Lys-299 are mutated, rendering the receptor unable to bind CaM (14). E2 failed to stimulate transcriptional activation of this mutant ER-α. Moreover, blocking CaM function in the nucleus of MCF-7 cells by transfecting a targeted specific CaM inhibitor peptide abrogated E2-induced transcriptional activation (9). Collectively, these data strongly suggest that an interaction between CaM and ER-α in the nucleus is required for E2 to stimulate transcriptional activation.
Consistent with these prior observations, we show here that overexpression of full-length CaM slightly, albeit not significantly, enhanced by 25% E2-induced transcriptional activation of two endogenous estrogen-responsive genes, namely pS2 and PR. The relatively small increase in E2-induced transcriptional activation is most likely due to the high concentrations of endogenous CaM in cells, which is 10−6 to 10−5 m, with even higher concentrations in rapidly growing cells (8). By contrast, expression of the separate lobes of CaM in cells abrogated the ability of E2 to stimulate ER-α transcriptional activity. A model illustrates how CaM regulates dimerization and activation of ER-α (Fig. 7). E2 binding induces dimerization of ER-α and translocation to the nucleus. Association of Ca2+/CaM with the E2-bound ER-α enables the receptor to activate transcription of target genes (Fig. 7). Note that CaM can stimulate ER-α dimerization in the absence of E2. When ER-α is bound to only one lobe of CaM instead of the full-length protein, CaM-induced dimerization of ER-α cannot occur and E2 is unable to promote transcription. Thus, binding of both E2 and CaM to ER-α is required for it to maximally activate transcription (Fig. 7).
FIGURE 7.
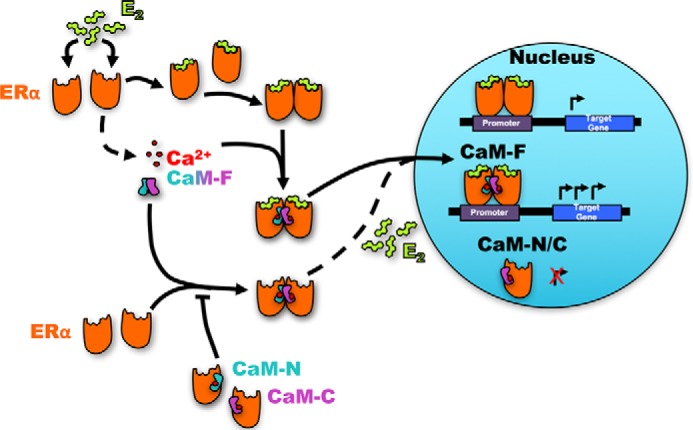
Model of the regulation of ER-α dimerization and transcription by CaM. The N- and C-lobes of CaM are shown in pale blue (CaM-N) and pink (CaM-C), respectively. Estrogen (E2) induces dimerization of ER-α. Full-length CaM (CaM-F) promotes ER-α dimerization independently of E2. In contrast, the individual lobes of CaM block ER-α dimerization, thereby preventing E2-stimulated transcriptional activation by ER-α. Both E2 and Ca2+/CaM are required for maximal activation of transcription.
The molecular mechanism by which the individual lobes of CaM abrogate E2-stimulated transcriptional activation by ER-α is unknown. There are several possible explanations. First, binding of CaM-N or CaM-C to ER-α may prevent receptor dimerization, which is necessary for transcriptional activation (2). The NMR structural data presented in this article strongly support this mechanism. Second, binding to one lobe of CaM may sterically block the interaction of ER-α with co-regulatory proteins, particularly co-activators, which modulate transcriptional activation (20). Third, the ER-α·CaM-N or ER-α·CaM-C complex may bind co-repressors rather than the co-activators, which bind to ER-α·CaM (full-length). It is known that the shape of the ligand·ER-α complex influences the specific co-regulators that are recruited (2). The components of the complex influence receptor activity, which activates or represses gene transcription. Fourth, ER-α may be titrated away from other co-regulators by the isolated CaM lobes. Fifth, the ER-α·CaM-N or ER-α·CaM-C complexes may be unable to translocate to the nucleus where transcription is activated. These mechanisms are not mutually exclusive and more than one may account for our data.
Regardless of the molecular mechanism, our observations elucidate the structure and biological function of the interaction between ER-α and CaM. It is estimated that 70% of breast malignancies are ER-α positive and therefore agents that suppress receptor function or E2 synthesis are used for therapy (21). Antagonism of ER-α with tamoxifen is the most widely used treatment for patients with breast carcinoma. Our structure and functional data suggest that altering the association of CaM with ER-α is a conceptually appealing therapeutic option. Consistent with this premise, published studies reveal that CaM antagonists attenuate the growth of breast cancer cell lines (22–24) and enhance antiestrogen therapy (25–27). It is possible that development of specifically targeted small molecules that selectively disrupt the CaM/ER-α interaction may be useful in the treatment of breast carcinoma and other ER-α-dependent malignancies.
Experimental Procedures
Materials
HEK-293 cells and MCF-7 breast epithelial cells were obtained from the American Type Culture Collection. All reagents for tissue culture were bought from Invitrogen. PVDF membranes were purchased from Millipore Corporation. Antibodies used in this study are listed in Table 2. Blocking buffer (927-50000) and infrared dye-conjugated (IRDye) antibodies, both goat anti-mouse IRDye 680LT (926–68020) and goat anti-rabbit IRDye 800CW (926–32211) were obtained from LI-COR Biosciences.
TABLE 2.
Antibodies used in this study
Construction of GFP-CaM and GST-CaM
The PCR products of full-length CaM (CaM-F), the N-terminal half (CaM-N, amino acids 1–74), and the C-terminal half (CaM-C, amino acids 75–148) of calmodulin were made using pDONR223-CaM1 as template. Primers were as follows: for CaM-F, forward primer, 5′-CGGGATCCGCTGATCAGCTGACCGAAGAACAG-3′ and reverse primer, 5′-GCTCTAGACTCGAGTCATTTTGCAGTCATCATCTGTACGAATTC-3′; for CaM-N, forward primer, 5′-CGGGATCCGCTGATCAGCTGACCGAAGAACAG-3′ and reverse primer, 5′-CCGGAATTCTCTAGATCAATCTTTCATTTTTCTAGCCATCATAG-3′; and for CaM-C, forward primer, 5′-CGGGATCCACAGATAGTGAAGAAGAAATCCG-3′ and reverse primer,5′-GCTCTAGACTCGAGTCATTTTGCAGTCATCATCTGTACGAATTC-3′. The PCR products of each CaM construct were digested with BamHI and XhoI. To generate GFP-CaM, PCR products were inserted into pEGFP-C1 at BglII and SalI sites. GST-CaM constructs were generated by inserting PCR products into pGEX-4T-1 at BamHI and XhoI sites. The sequences of all plasmids were verified by DNA sequencing. Plasmids were purified with QIAprep Spin Mini Prep Kit (Qiagen).
Cell Culture and Transfection
Cells were grown in DMEM supplemented with 10% FBS. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Preparation of GST Fusion Proteins
GST-CaM-F, GST-CaM-N, and GST-CaM-C were expressed in E. coli and purified by glutathione-Sepharose chromatography, essentially as described previously (28). All fusion proteins migrated to the expected position on SDS-PAGE. Proteins were at least 90% pure by Coomassie staining.
Binding Analysis
The binding of the CaM constructs to endogenous ER-α in MCF-7 lysates was evaluated. First, GFP-tagged CaM-F, CaM-N, or CaM-C (or GFP alone) were transfected into HEK-293 cells. Cells were cultured at 37 °C for 3 days, then lysed with 500 μl of Buffer A (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 1% Triton X-100) containing 1 mm EGTA, 1× Protease & Phosphatase Inhibitor Mixture (Thermo Scientific), and 1 mm PMSF. GFP was immunoprecipitated with GFP-Trap_A (anti-GFP VHH conjugated to agarose, ChromoTek) for 3 h at 4 °C, then washed 3 times with Buffer A containing 1 mm EGTA. MCF-7 cells were lysed with Buffer A containing 1 mm CaCl2, 1 mm PMSF, and 1× Protease & Phosphatase Inhibit Mixture. Equal amounts of MCF-7 protein lysate were incubated for 3 h at 4 °C with the GFP-tagged proteins on agarose beads. After 5 washes with Buffer A containing 1 mm CaCl2, samples were resolved by SDS-PAGE (29) and transferred to PVDF. The membrane was blocked with Blocking Buffer (LI-COR) for 1 h at 22 °C, probed overnight at 4 °C with anti-ER-α polyclonal antibodies, then incubated for 1 h with IRDye-conjugated anti-rabbit antibody. Antigen-antibody complexes were detected using the Odyssey Imaging System (LI-COR). GFP-tagged proteins were detected by probing the membrane with anti-GFP monoclonal antibody, followed by IRDye-conjugated anti-mouse antibody and detected with the Odyssey Imaging System.
Binding of CaM-F, CaM-N, and CaM-C to endogenous IQGAP1 was also performed. MCF-7 cells were lysed with 500 μl of Buffer A containing 2 mm EDTA, 1× Protease & Phosphatase Inhibitor Mixture, and 1 mm PMSF, then pre-cleared by rotating for 1 h at 4 °C with glutathione-Sepharose beads. Equal amounts of protein lysate were incubated with 40 μl of GST-CaM-F, GST-CaM-N, GST-CaM-C, or GST (control). All GST fusion proteins were bound to glutathione-Sepharose beads prior to adding lysate. After rotating at 4 °C for 3 h, samples were washed 5 times with Buffer A and resolved by SDS-PAGE (29). The gel was cut slightly above the 50-kDa molecular mass marker into two pieces. The lower portion of the gel was stained with Coomassie Blue. The upper portion of the gel was transferred to PVDF and processed essentially as described for ER-α, except the blots were probed with anti-IQGAP1 antibodies.
Quantitative RT-PCR
HEK-293 cells were cultured in phenol red-free medium with 10% charcoal-stripped fetal bovine serum (FBS), and transfected with pcDNA3-myc-ER-α and pEGFP-CaM-F, pEGFP-CaM-N, pEGFP-CaM-C, or pEGFP (vector). Where indicated, cells were transfected with both pcDNA3-myc-ER-α and pEGFP-CaM-F, as well as pEGFP-CaM-N, pEGFP-CaM-C, or pEGFP. The next day, the medium was replaced with fresh FBS-free medium. After 2 days, vehicle (EtOH) or E2 (to obtain a final concentration of 100 nm) were added to the medium. Following a 6-h incubation, total RNA was isolated from the cells using TRIzol (Invitrogen). 2 μg of RNA was reverse transcribed to cDNA using a High Capacity cDNA Reverse Transcriptase kit (Applied Biosystems) according to the manufacturer's instructions. RT-PCR was performed on a StepOnePlus Real Time PCR system (Applied Biosystems) using SYBR Green PCR master mix (Applied Biosystems) and 200 nm forward and reverse primers. The primers used were: for PR, forward primer, 5′-CCTCGGACACCTTGCCTGAA-3′, reverse primer, 5′-CGCCAACAGAGTGTCCAAGAC-3′; for pS2, forward primer, 5′-TTGGAGAAGGAAGCTGGATGG-3′, reverse primer, 5′-ACCACAATTCTGTCTTTCACGG-3′; and for β-actin, forward primer, 5′-TGCGTGACATTAAGGAGAAG-3′, reverse primer, 5′-GCTCGTAGCTCTTCTCCA-3′. RT-PCR enzyme activation was initiated for 10 min at 95 °C, then amplified by 40 cycles (15 s at 95 °C and 1 min at 60 °C). All samples were assayed in triplicate and β-actin was used as an internal control. Results were analyzed using the ΔΔCT method with StepOnePlus software (Applied Biosystems). RT-PCR with MCF-7 cells was performed essentially as described for HEK-293 cells, except the transfection with pcDNA3-myc-ER-α was omitted as MCF-7 cells have endogenous ER-α.
NMR Sample Preparation
NMR samples of isotopically labeled CaM bound to ER(287–305) were prepared as described previously (15). Purified CaM (3.5 mg) was initially dissolved in 10 ml of NMR buffer (20 mm Tris-d11, 5 mm CaCl2, 50 mm NaCl, 8% D2O, pH 7.0) and then added to 2 eq of ER(287–305). The CaM·ER complex was incubated at room temperature for 1 h, and concentrated to 0.4 ml using Amicon Ultra Centrifugal Filters Ultracel-3K (Millipore, UFC900324, 3 kDa cut-off).
NMR Spectroscopy
All NMR experiments were carried out at 310 K and performed using Bruker Avance III 800 MHz spectrometer equipped with a four-channel interface and triple-resonance cryoprobe (TCI). Two-dimensional NMR experiments, 15N-1H HSQC (and constant-time 13C-1H HSQC) spectra were recorded on a sample of 15N-labeled (13C-labeled) CaM in the presence of unlabeled ER(287–305). All three-dimensional NMR experiments for assigning backbone and side chain resonances were recorded on a double labeled sample (15N,13C-labeled CaM bound to unlabeled ER(287–305) as described previously (15). NMR distance restraints were obtained as described (15). NMR data were processed using NMRPipe (30) and analyzed with SPARKY (T. D. Goddard and D. G. Kneller, University of California at San Francisco).
Structure Calculation
The NMR structure of full-length CaM bound to ER(287–305) was calculated using NMR-derived distance restraints and residual dipolar couplings as described previously (15, 31–33). The structure of ER(287–305) bound to full-length CaM was verified to form an α-helix based on NOE distance restraints (HN-HN connectivity), chemical shift index (34), and circular dichroism analysis as described previously (15). The NMR-derived structures of CaM and ER(287–305) were then used as input for molecular docking within HADDOCK (35–38) using intermolecular NOE restraints and residual dipolar coupling data as described previously (15). The structure of ER(287–305) in the docking calculation was set as full flexible, and side chain atoms of CaM that exhibited intermolecular NOEs with ER(287–305) were allowed to move during simulated annealing. The final structures were further refined by including a final water refinement step.
Miscellaneous Methods
Statistical analysis was performed by Student's t test with Prism 6 (GraphPad). Protein concentrations were measured with the DC Protein Assay (Bio-Rad).
Author Contributions
J. B. A. and D. B. S. designed the study. Z. L., Y. Z., and A. C. H. conducted experiments. Z. L., Y. Z., A. C. H., J. B. A., and D. B. S. analyzed data. J. B. A. and D. B. S. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by the Intramural Research Program of the National Institutes of Health (to Z. L., A. C. H., and D. B. S.) and an extramural grant from the National Institutes of Health (EY012347) (to J. B. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5T0X) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ER-α
- estrogen receptor-α
- CaM
- calmodulin
- ERE
- estrogen-response element
- CaM-F
- full-length calmodulin
- CaM-C
- calmodulin C-terminal lobe
- CaM-N
- calmodulin N-terminal lobe
- E2
- 17-β-estradiol
- hnRNA
- heterogeneous nuclear RNA
- EGFP
- enhanced green fluorescent protein
- PR
- progesterone receptor
- HSQC
- heteronuclear single quantum coherence
- NOE
- nuclear Overhauser effect.
References
- 1. Gruber C. J., Tschugguel W., Schneeberger C., and Huber J. C. (2002) Production and actions of estrogens. N. Engl. J. Med. 346, 340–352 [DOI] [PubMed] [Google Scholar]
- 2. Thomas C., and Gustafsson J. Å. (2011) The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 11, 597–608 [DOI] [PubMed] [Google Scholar]
- 3. Levin E. R. (2009) Plasma membrane estrogen receptors. Trends Endocrinol Metab. 20, 477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schultz-Norton J. R., Ziegler Y. S., and Nardulli A. M. (2011) ERα-associated protein networks. Trends Endocrinol. Metab. 22, 124–129 [DOI] [PubMed] [Google Scholar]
- 5. Smith J. M., Hedman A. C., and Sacks D. B. (2015) IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 25, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hedman A. C., Smith J. M., and Sacks D. B. (2015) The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 16, 427–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erdemir H. H., Li Z., and Sacks D. B. (2014) IQGAP1 binds to estrogen receptor-α and modulates its function. J. Biol. Chem. 289, 9100–9112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chin D., and Means A. R. (2000) Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328 [DOI] [PubMed] [Google Scholar]
- 9. Li L., Li Z., and Sacks D. B. (2003) Calmodulin regulates the transcriptional activity of estrogen receptors: selective inhibition of calmodulin function in subcellular compartments. J. Biol. Chem. 278, 1195–1200 [DOI] [PubMed] [Google Scholar]
- 10. García Pedrero J. M., Del Rio B., Martínez-Campa C., Muramatsu M., Lazo P. S., and Ramos S. (2002) Calmodulin is a selective modulator of estrogen receptors. Mol. Endocrinol. 16, 947–960 [DOI] [PubMed] [Google Scholar]
- 11. Li Z., Joyal J. L., and Sacks D. B. (2001) Calmodulin enhances the stability of the estrogen receptor. J. Biol. Chem. 276, 17354–17360 [DOI] [PubMed] [Google Scholar]
- 12. Bouhoute A., and Leclercq G. (1992) Antagonistic effect of triphenylethylenic antiestrogens on the association of estrogen receptor to calmodulin. Biochem. Biophys. Res. Commun. 184, 1432–1440 [DOI] [PubMed] [Google Scholar]
- 13. Li L., Li Z., Howley P. M., and Sacks D. B. (2006) E6AP and calmodulin reciprocally regulate estrogen receptor stability. J. Biol. Chem. 281, 1978–1985 [DOI] [PubMed] [Google Scholar]
- 14. Li L., Li Z., and Sacks D. B. (2005) The transcriptional activity of estrogen receptor-α is dependent on Ca2+/calmodulin. J. Biol. Chem. 280, 13097–13104 [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y., Li Z., Sacks D. B., and Ames J. B. (2012) Structural basis for Ca2+-induced activation and dimerization of estrogen receptor α by calmodulin. J. Biol. Chem. 287, 9336–9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carlier L., Byrne C., Miclet E., Bourgoin-Voillard S., Nicaise M., Tabet J. C., Desmadril M., Leclercq G., Lequin O., and Jacquot Y. (2012) Biophysical studies of the interaction between calmodulin and the R(2)(8)(7)-T(3)(1)(1) region of human estrogen receptor α reveals an atypical binding process. Biochem. Biophys. Res. Commun. 419, 356–361 [DOI] [PubMed] [Google Scholar]
- 17. Li Z., and Sacks D. B. (2003) Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 278, 4347–4352 [DOI] [PubMed] [Google Scholar]
- 18. Castoria G., Migliaccio A., Nola E., and Auricchio F. (1988) In vitro interaction of estradiol receptor with Ca2+-calmodulin. Mol. Endocrinol. 2, 167–174 [DOI] [PubMed] [Google Scholar]
- 19. Biswas D. K., Reddy P. V., Pickard M., Makkad B., Pettit N., and Pardee A. B. (1998) Calmodulin is essential for estrogen receptor interaction with its motif and activation of responsive promoter. J. Biol. Chem. 273, 33817–33824 [DOI] [PubMed] [Google Scholar]
- 20. Lonard D. M., and O'Malley B. W. (2006) The expanding cosmos of nuclear receptor coactivators. Cell 125, 411–414 [DOI] [PubMed] [Google Scholar]
- 21. Le Romancer M., Poulard C., Cohen P., Sentis S., Renoir J. M., and Corbo L. (2011) Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr. Rev. 32, 597–622 [DOI] [PubMed] [Google Scholar]
- 22. Wei J. W., Hickie R. A., and Klaassen D. J. (1983) Inhibition of human breast cancer colony formation by anticalmodulin agents: trifluoperazine, W-7, and W-13. Cancer Chemother. Pharmacol. 11, 86–90 [DOI] [PubMed] [Google Scholar]
- 23. Strobl J. S., and Peterson V. A. (1992) Tamoxifen-resistant human breast cancer cell growth: inhibition by thioridazine, pimozide and the calmodulin antagonist, W-13. J. Pharmacol. Exp. Ther. 263, 186–193 [PubMed] [Google Scholar]
- 24. Gulino A., Barrera G., Vacca A., Farina A., Ferretti C., Screpanti I., Dianzani M. U., and Frati L. (1986) Calmodulin antagonism and growth-inhibiting activity of triphenylethylene antiestrogens in MCF-7 human breast cancer cells. Cancer Res. 46, 6274–6278 [PubMed] [Google Scholar]
- 25. Frankfurt O. S., Sugarbaker E. V., Robb J. A., and Villa L. (1995) Synergistic induction of apoptosis in breast cancer cells by tamoxifen and calmodulin inhibitors. Cancer Lett. 97, 149–154 [DOI] [PubMed] [Google Scholar]
- 26. Newton C. J., Eycott K., Green V., and Atkin S. L. (2000) Response of estrogen receptor containing tumour cells to pure antiestrogens and the calmodulin inhibitor, calmidzolium chloride. J. Steroid Biochem. Mol. Biol. 73, 29–38 [DOI] [PubMed] [Google Scholar]
- 27. Liu R., Zhang Y., Chen Y., Qi J., Ren S., Xushi M. Y., Yang C., Zhu H., and Xiong D. (2010) A novel calmodulin antagonist O-(4-ethoxyl-butyl)-berbamine overcomes multidrug resistance in drug-resistant MCF-7/ADR breast carcinoma cells. J. Pharm. Sci. 99, 3266–3275 [DOI] [PubMed] [Google Scholar]
- 28. Ho Y. D., Joyal J. L., Li Z., and Sacks D. B. (1999) IQGAP1 integrates Ca2+/calmodulin and Cdc42 signaling. J. Biol. Chem. 274, 464–470 [DOI] [PubMed] [Google Scholar]
- 29. Sacks D. B., Fujita-Yamaguchi Y., Gale R. D., and McDonald J. M. (1989) Tyrosine-specific phosphorylation of calmodulin by the insulin receptor kinase purified from human placenta. Biochem. J. 263, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 31. Park S., Li C., and Ames J. B. (2011) Nuclear magnetic resonance structure of calcium-binding protein 1 in a Ca2+-bound closed state: implications for target recognition. Protein Sci. 20, 1356–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y., Larsen C. A., Stadler H. S., and Ames J. B. (2011) Structural basis for sequence specific DNA binding and protein dimerization of HOXA13. PLoS ONE 6, e23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y., Matt L., Patriarchi T., Malik Z. A., Chowdhury D., Park D. K., Renieri A., Ames J. B., and Hell J. W. (2014) Capping of the N-terminus of PSD-95 by calmodulin triggers its postsynaptic release. EMBO J. 33, 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wishart D. S., Sykes B. D., and Richards F. M. (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31, 1647–1651 [DOI] [PubMed] [Google Scholar]
- 35. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., and Bonvin A. M. (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 [DOI] [PubMed] [Google Scholar]
- 36. de Vries S. J., van Dijk M., and Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 37. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 38. Schwieters C. D., Kuszewski J. J., Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 39. Sacks D. B., Porter S. E., Ladenson J. H., and McDonald J. M. (1991) Monoclonal antibody to calmodulin: development, characterization, and comparison with polyclonal anti-calmodulin antibodies. Anal. Biochem. 194, 369–377 [DOI] [PubMed] [Google Scholar]