

Abstract
The aggregation of amyloid-β (Aβ) on lipid bilayers has been implicated as a mechanism by which Aβ exerts its toxicity in Alzheimer's disease (AD). Lipid bilayer thinning has been observed during both oxidative stress and protein aggregation in AD, but whether these pathological modifications of the bilayer correlate with Aβ misfolding is unclear. Here, we studied peptide-lipid interactions in synthetic bilayers of the short-chain lipid dilauroyl phosphatidylcholine (DLPC) as a simplified model for diseased bilayers to determine their impact on Aβ aggregate, protofibril, and fibril formation. Aβ aggregation and fibril formation in membranes composed of dioleoyl phosphatidylcholine (DOPC) or 1- palmitoyl-2-oleoyl phosphatidylcholine mimicking normal bilayers served as controls. Differences in aggregate formation and stability were monitored by a combination of thioflavin-T fluorescence, circular dichroism, atomic force microscopy, transmission electron microscopy, and NMR. Despite the ability of all three lipid bilayers to catalyze aggregation, DLPC accelerates aggregation at much lower concentrations and prevents the fibrillation of Aβ at low micromolar concentrations. DLPC stabilized globular, membrane-associated oligomers, which could disrupt the bilayer integrity. DLPC bilayers also remodeled preformed amyloid fibrils into a pseudo-unfolded, molten globule state, which resembled on-pathway, protofibrillar aggregates. Whereas the stabilized, membrane-associated oligomers were found to be nontoxic, the remodeled species displayed toxicity similar to that of conventionally prepared aggregates. These results provide mechanistic insights into the roles that pathologically thin bilayers may play in Aβ aggregation on neuronal bilayers, and pathological lipid oxidation may contribute to Aβ misfolding.
Keywords: Alzheimer disease, amyloid-beta (AB), lipid bilayer, protein misfolding, toxicity
Introduction
The aggregation and fibrillation of amyloid-β (Aβ)2 in the brain has long been implicated in the neurotoxicity and pathogenesis of Alzheimer's disease (AD). Following its production via cleavage of the transmembrane amyloid precursor protein, Aβ undergoes aggregation along a nucleated growth pathway toward its end state amyloid fibril (1). It is believed that intermediate aggregates along this path are the primary culprit in AD-related cytotoxicity (2, 3). Early Aβ oligomers may perform their toxic function through interactions with the neuronal lipid bilayer (4, 5). Pore formation has been suggested to facilitate disruption of the ionic gradient across neuronal membranes (6, 7). Alternatively, elongation of the amyloid species on the bilayer may remove lipids from the membrane and create holes that allow for the uncontrolled transit of biomacromolecules across the membrane (8, 9). These two modes of membrane disruption are proposed to coexist; it is thought that porelike oligomers form first, whereas the fibril-induced perturbations occur later (8). Extensive work by Matsuzaki (5) has revealed that incorporating glycolipids, such as the ganglioside GM1, can promote the formation of toxic, membrane-associated Aβ aggregates linked to membrane disruption. Despite these insights, the overarching biological, chemical, and physical parameters that govern the interactions of Aβ with lipid bilayers and modulate the formation of toxic oligomers remain unclear.
Bilayer thickness, controlled by the acyl chain length, is one basic principle of membrane biophysics that has been shown to alter membrane protein folding and modulate energetic penalties caused by the hydrophobic mismatch (10, 11). It is therefore possible that alterations in the hydrophobic thickness can control amyloid aggregation and stabilize distinct intermediates. It remains unclear how on- and off-pathway amyloid oligomers may be stabilized by the lipid bilayer, and identifying the effects of the membrane's physical structure will provide greater understanding of how aggregation may occur on cellular bilayers. Additionally, it may identify conditions in which specific oligomeric intermediates may be stabilized for more thorough structural analysis than has been possible for Aβ oligomers to date. Herein we have used synthetic bilayers composed of dilauroyl phosphatidylcholine (DLPC; 20.9 nm thick), dioleoyl phosphatidylcholine (DOPC; 26.8 nm thick), or 1-palmitoyl-2-oleoyl phosphatidylcholine (POPC; 27.1 nm thick) to explore relationships between thin (DLPC) and thick (DOPC and POPC) bilayers, Aβ aggregation, and the stabilization of distinct Aβ oligomers (12, 13). We have previously shown that monomeric Aβ interacts with these three zwitterionic lipid bilayers through a conserved structure under non-aggregating conditions (14). This initial binding is driven by surface interactions with the self-recognition sequence and is minimally affected by the hydrophobic thickness of the membrane. It is more likely that the hydrophobic thickness of lipid bilayers influences the generation and/or stabilization of higher order membrane-associated species because of the ability of these Aβ aggregates to insert themselves within the bilayer (15). Both DOPC and POPC have been previously applied to examine the interactions between lipid bilayers and amyloidogenic proteins (8, 16–19). DLPC bilayers have been used in the study of a variety of membrane proteins and were observed to stabilize some membrane proteins in vitro that are less tractable with other lipid bilayer systems, but they have not been used previously in the study of Aβ (20, 21).
Through a suite of biophysical techniques, we have found that the thin, pathology-mimicking DLPC bilayers have a unique ability to stabilize prefibrillar oligomers within the bilayer while preventing the formation of mature fibrils. Surprisingly, it was observed that DLPC bilayers are also capable of remodeling mature Aβ fibrils into a semi-unfolded intermediate, a feat previously observed only after treatment with high concentrations of denaturants or exposure to high frequency sonication (22). These results show that thin bilayers are able to dramatically alter the aggregation of Aβ compared with conventional lipid bilayers, and this redirected misfolding can stabilize distinct, membrane-associated species not observed previously.
Results
Unlike DOPC and POPC, DLPC Liposomes Inhibit Aβ Fibrillation
Lipid bilayers can modulate the rate of amyloid formation by Aβ, although the extent of modulation varies depending on the bilayer composition, incubation conditions, and detection method (23–25). thioflavin-T (ThT), a dye that fluoresces in the presence of amyloid fibrils, can probe for the formation of fibrillar aggregates and is useful in delineating mechanistic deviations in amyloid aggregation (26–28). Thus, ThT was used to initially probe the aggregation propensity of Aβ in the presence of large unilamellar vesicles (LUVs) of DOPC, POPC, and DLPC and characterize kinetic differences that result from different interactions caused by variation in hydrophobic thickness.
Aβ aggregation was accelerated by increasing concentrations of both DOPC and POPC, as has been shown previously (Fig. 1, a–c) (23, 24). As the concentration of lipid increased from 0 to 200 μm, the lag phase became continually shorter, suggesting more rapid nucleation and earlier fibril formation. It is likely that the observed catalysis is the result of interactions between Aβ and the membrane, which serve to reduce the dimensionality of the search by individual Aβ monomers for binding partners and increase the local concentration of peptide (23). End state aggregates formed in the absence and presence of either DOPC or POPC were examined by CD spectroscopy and transmission electron microscopy (TEM) (Fig. 2). As expected, all three samples exhibited a canonical peak at 220 nm in CD spectra, representative of a β-strand fold and amyloid formation. TEM further confirmed the formation of amyloid fibers in the presence of both DOPC and POPC. These data support previous studies suggesting that both DOPC and POPC act as amyloid catalysts (23, 24).
FIGURE 1.

Effect of 100-nm phosphatidylcholine LUVs on Aβ aggregation kinetics. Both DOPC (a) and POPC (b) accelerate Aβ (10 μm) aggregation in a concentration-dependent manner as determined by ThT (20 μm) fluorescence. c, the lag phases suggest that kinetic acceleration is most potent at 500 μm lipid, although aggregation is rapidly accelerated at concentrations as low as 50 μm lipid. d, DLPC LUVs inhibit Aβ aggregation at stoichiometric and superstoichiometric concentrations. e, low concentrations of DLPC (nanomolar range) accelerate Aβ aggregation before reaching concentrations that eventually inhibit fibrillation. f, the lag phase of Aβ aggregation in the presence of DLPC LUVs reaches a maximum acceleration at ∼500–700 nm. Lag phases reported represent the average of three independent trials with error bars representing the S.D. of these measurements. All curves represent the average of three independent aggregation time courses.
FIGURE 2.

Secondary structure and morphology of Aβ aggregates. A, Aβ (30 μm) fibrils grown for 24 h in the absence and presence of DOPC, POPC, or DLPC LUVs (300 μm lipid) were compared with freshly prepared, monomeric Aβ by CD. B, TEM images were acquired of the end stage aggregates after a 24-h incubation for each sample used in CD analysis.
Incubation of DLPC LUVs with Aβ elicited a dramatically different result compared with DOPC and POPC (Fig. 1d). When Aβ aggregated in the presence of DLPC at stoichiometric (10 μm; 1:1 lipid/peptide ratio) or higher lipid concentrations, ThT fluorescence remained at baseline levels, which suggests no fibril formation. ThT will occasionally provide false positives of inhibition resulting from dye displacement or the inability of unique amyloid sequences to bind the dye, despite containing the canonical β-strand fold (29, 30). To confirm that the reduction in ThT fluorescence was the result of structural change, CD and TEM were performed on end stage aggregates (Fig. 2). CD results showed that the species formed in the presence of DLPC maintained a random coil structure after a 24-h incubation. The aggregates were also morphologically distinct from conventional amyloid fibrils. They were small and amorphous and had no discrete fibril structure, as shown by TEM. Combined, these data confirm that stoichiometric concentrations of DLPC LUVs slow amyloid fibril formation of Aβ.
To further characterize the potency of this inhibition, ThT was used to examine the kinetics of amyloid formation in the presence of substoichiometric concentrations of DLPC (Fig. 1, e and f). As the concentration of DLPC increased from 0 to 500 nm, the lag phase of aggregation shortened, suggesting accelerated Aβ fibrillation. Fiber formation catalyzed by 500 nm DLPC LUVs was further confirmed by both CD and TEM analysis (supplemental Fig. S1). Increasing the concentration beyond 700 nm lengthened the lag phase. Aggregation was halted by DLPC concentrations of >1 μm. Similar kinetic profiles were observed in the presence of DLPC LUVs of different diameters, suggesting that the effect of DLPC on aggregation is independent of bilayer curvature, unlike the effect of other model membranes (supplemental Fig. S2) (25). The profile by which DLPC initially accelerates and subsequently inhibits amyloid formation based on concentration is similar to the effect of detergents on amyloid formation (31). Amyloids are capable of aggregating in the presence of a variety of detergents, although this is concentration-dependent. As the concentration of detergent increases from 0 toward the critical micelle concentration (CMC) of the detergent, amyloid formation is accelerated, and the lag phase shortens. Catalysis is maximal around the CMC of the detergent. As detergent concentration is increased further, the lag phase increases, and aggregation slows as the interaction between micelles and the amyloidogenic peptide or protein is favored (31). This pattern is similar to what has been observed with DLPC LUVs. Additionally, DLPC is known to have detergent-like characteristics and has a CMC of ∼100 nm (32). This suggests that free DLPC, existing in equilibrium with DLPC encapsulated in LUVs, may play a role in modulating Aβ aggregation through a detergent-like mechanism. However, free DLPC only inhibited Aβ aggregation at much higher concentrations than DLPC in LUVs (15 μm; supplemental Fig. S3, A and B). Similar profiles have also been observed for various amyloids interacting with lipid bilayers (33, 34). As the LUV concentration increases in solution, the lag phase decreases as amyloidogenic peptides are able to bind to the same LUV, bind to each other more readily, and accelerate amyloid formation; this eventually reaches a point of critical acceleration. Beyond this concentration, aggregation is slowed as individual peptides or proteins now bind to individual LUVs and they become sequestered. It is likely that the observed acceleration and subsequent inhibition of Aβ fibrillation by DLPC may be due to interactions similar to those of both detergents and membranes. The ability of DLPC to act as both a detergent and a lipid suggests that it may be a combination of these two mechanisms.
Aβ Binding to DLPC Liposomes Induces Membrane Disruption and Unstructured Oligomers
To clarify the extent to which detergent-like activity of free DLPC is capable of modulating Aβ aggregation, it is essential to identify the source of free lipid. Although there is an equilibrium between bilayer-inserted and free DLPC, this equilibrium is not capable of generating high concentrations of free DLPC in the absence of peptide (35, 36). It has been shown, however, that interactions between amyloidogenic peptides and lipid bilayers can extract lipids and generate micelle-like particles (8, 37, 38). These species could exist as a result of Aβ-DLPC interactions and subsequently play a role in the observed modulation of Aβ aggregation. Differences between micelle-like lipid species and LUVs can be detected using 31P solid-state NMR (8, 38, 39). Due to their large size, intact vesicles of DLPC, DOPC, and POPC exhibit a powder pattern spectrum that spans from ∼−14 to ∼25 ppm. Only the perpendicular edge (∼−14 ppm) of the spectrum is highly sensitive (Fig. 3A). Following incubation with Aβ under aggregating conditions for 24 h, a new peak centered near 0 ppm was observed for LUVs composed of DLPC. This isotropic peak at 0 ppm suggests a new, small, lipid structure induced by Aβ (Fig. 3A). This provides a source for free DLPC; the interaction of Aβ with the surface of DLPC liposomes is capable of removing lipids from the bilayer, despite the lack of fibrillation. This is unique both in the lack of fiber formation, which has previously been suspected as the causative agent in amyloid-based membrane disruption, and in the absence of GM1 ganglioside in the bilayer, a lipid that is believed to be needed for the disruption of neuronal bilayers (5, 8, 37, 38).
FIGURE 3.

Membrane perturbations by Aβ. A, 31P solid-state NMR of 1000-nm LUVs (23 mm lipid) in the absence (black) and presence (red) of Aβ (230 μm). The anisotropic peak around −15 ppm represents the intact liposomes, whereas the small peak near 0 ppm in the DLPC + Aβ spectrum (noted by an arrow) indicates the formation of small, rapidly tumbling lipid species suggestive of membrane fragmentation. B, AFM (1 × 1-μm region) was performed on SLBs of DLPC, DOPC, or POPC incubated in the presence of Aβ as a function of time to observe both the deposition of Aβ aggregates on the membrane and defects induced in the bilayer surface as a result of aggregation. C, AFM measurements of the same 5 × 5-μm region of a DLPC SLB were taken at early time intervals following the addition of Aβ.
Although the disruption of DLPC LUVs by non-fibrillar Aβ species is novel, it is unlikely that the free DLPC generated is the sole cause of amyloid inhibition (supplemental Fig. S3, A and B). A second mechanism of inhibition is necessary to explain the observed potency of DLPC LUV inhibition. We initially hypothesized that changes in bilayer hydrophobic thickness may stabilize on- and/or off-pathway oligomers. To study intermediates formed on the surface of DLPC bilayers, we used atomic force microscopy (AFM), which is well suited for the interrogation and observation of bilayer-based aggregation of Aβ (7, 40–43). DLPC, DOPC, and POPC all formed smooth supported lipid bilayers (SLBs) that were stable for the duration of the experiments (supplemental Fig. S4) (44, 45). After incubating monomeric Aβ with SLBs of DOPC and POPC, early globular and prefibrillar oligomers were observed by AFM after 24 h in a liquid cell (Fig. 3B). Although ThT data suggested that mature fibrils exist by this point in aggregation, AFM was performed under quiescent conditions, which delays fibrillation (46). When DLPC SLBs were treated with Aβ, a large number of globular aggregates were present on the bilayer after 12 h. Aggregates persisted after a 24-h incubation, although the size of individual particles shrank. Defects in the membrane were also evident around the aggregates after both 12 and 24 h, suggesting that these aggregates perturb the bilayer integrity, in agreement with the 31P solid-state NMR results.
To quantify the membrane modification caused by Aβ, the root mean square roughness (Rq) and average roughness (Ra) of the samples with respect to time were calculated based on the AFM data (Table 1). Although all three bilayers initially formed with a similar smoothness and without defects, DLPC bilayers demonstrated a large increase in their roughness after 12 and 24 h of incubation with Aβ relative to both the initial bilayer smoothness and the observed smoothness for DOPC and POPC SLBs. The increase in both Rq and Ra is caused by both the formation of aggregates and the disruption of the bilayer integrity. Whereas Rq and Ra of DLPC bilayers decrease between 12 and 24 h of incubation, this is probably caused by aggregates equilibrating, becoming smaller and more evenly dispersed across the bilayer surface. To better understand the kinetics of the observed aggregate formation on the bilayer surface, AFM images were taken at early time points following the addition of Aβ to the DLPC SLBs (Fig. 3C). After just 5 min of incubation, holes were observed in the bilayer, indicative of dramatic disruption resulting from aggregation. Small, globular aggregates were also present. At later time points (15 and 20 min), the aggregates grew in size, whereas the membrane defects shrank slightly due to lipid diffusion within the membrane.
TABLE 1.
Quantifying membrane perturbation
The root mean square roughness (Rq) and the average roughness (Ra) of SLBs incubated with Aβ at different time points are calculated from the AFM images in Fig. 3.
| Sample | Rq ± S.D. | Ra ± S.D. |
|---|---|---|
| nm | nm | |
| DLPC | 0.36 ± 0.02 | 0.27 ± 0.01 |
| +Aβ (12 h) | 3 ± 1 | 1.7 ± 0.8 |
| +Aβ (24 h) | 1.5 ± 0.7 | 0.7 ± 0.2 |
| DOPC | 0.39 ± 0.04 | 0.29 ± 0.02 |
| +Aβ (12 h) | 0.40 ± 0.03 | 0.29 ± 0.01 |
| +Aβ (24 h) | 0.8 ± 0.1 | 0.51 ± 0.07 |
| POPC | 0.49 ± 0.03 | 0.39 ± 0.03 |
| +Aβ (12 h) | 0.5 ± 0.1 | 0.27 ± 0.06 |
| +Aβ (24 h) | 0.8 ± 0.5 | 0.4 ± 0.2 |
The AFM results point to a unique characteristic of DLPC bilayers. DLPC SLBs facilitate the rapid formation of globular aggregates on the bilayer surface of, and possibly within, the membrane. In contrast, most reports of membrane-mediated Aβ aggregation using AFM demonstrate the formation of conventional fibrils (41, 47, 48). This membrane-associated aggregation is probably responsible for the bilayer disruption, despite not occurring through the canonical amyloid formation pathway. Amorphous aggregation occurring selectively on DLPC SLBs suggests that it is at least partly the result of the uniquely small hydrophobic thickness of DLPC. These globular species are stabilized by the short hydrophobic distance, which prevents their progression toward mature fibrils. There are, therefore, two mechanisms by which DLPC LUVs appear to inhibit the formation of Aβ fibers. Aβ-DLPC interactions generate micelle-like lipid species that can prevent aggregation; driving inhibition is the stabilization of globular, membrane-associated aggregates, which are incapable of progressing toward fibrils.
DLPC Remodels Mature Aβ Fibrils
Although results to this point have illustrated how monomeric Aβ may interact with and aggregate on bilayers of varying thicknesses, it is equally important to investigate how preformed aggregates may interact with these bilayers. The addition of DLPC LUVs to premade fibrils caused a rapid reduction in ThT fluorescence in a dose-dependent manner (Fig. 4A). Similar fluorescence reduction has previously been observed in small molecule-induced disaggregation of various amyloid fibrils, suggesting that DLPC LUVs may alter the structure of the traditionally stable fiber (49–51). Although ThT displacement is a possibility, the stabilization of the final fluorescence at ∼40% of the original value, instead of returning to the baseline, suggests that ThT binding is maintained to some extent and that the changes are primarily structural (29). Additionally, the reduction in ThT fluorescence was independent of the concentration of ThT in solution, further supporting the change resulting from structural alterations (supplemental Fig. S5). Neither DOPC nor POPC LUVs caused a significant change in ThT fluorescence, and fiber structure was observed to persist based on both CD and TEM analysis following treatment with either DOPC or POPC (Fig. 4B and supplemental Fig. S6). This suggests that any structural changes to the fiber are specific to DLPC. The addition of free DLPC to preformed aggregates also minimally altered the ThT fluorescence of the aggregates, suggesting that the mechanism of remodeling is not through detergent-like effects (supplemental Fig. S3C).
FIGURE 4.

DLPC liposomes remodel preformed Aβ fibrils. A, when 100-nm LUVs of DLPC were added to preformed Aβ fibrils (10 μm monomer concentration; preaggregated for 24 h), ThT (20 μm) fluorescence was decreased in a dose-dependent manner. B, the addition of 100-nm LUVs of DOPC or POPC at much higher concentrations did not impact ThT fluorescence. All ThT curves represent the average of three independent aggregation time courses. C, the CD spectrum of fibrillar Aβ (30 μm) was monitored following the addition of 100-nm LUVs of DLPC (300 μm). D, TEM images of the aggregates induced by incubating preformed Aβ fibrils (30 μm monomer concentration) with 100-nm DLPC LUVs (300 μm lipid).
Assuming that the DLPC-induced fluorescence reduction is a function of structural changes, CD was used to examine the extent of these changes. Surprisingly, the β-strand signal at 220 nm persists over the course of 24 h despite the observed reduction in ThT fluorescence stabilizing after 4 h (Fig. 4C). Although seemingly contradictory, it has been demonstrated previously that amyloids can possess β-strand content and be unable to bind ThT (30). Moreover, the TEM images of the preformed fibers treated with DLPC displayed a morphology distinct from mature fibers formed in solution (Figs. 2B and 6d). The remodeled fibrils maintain some fiber-like characteristics, although they are much shorter and thinner than mature fibrils. These results confirm that DLPC LUVs are capable of remodeling mature Aβ aggregates, although not to the extent of some traditional studies of disaggregation (49–51).
FIGURE 6.
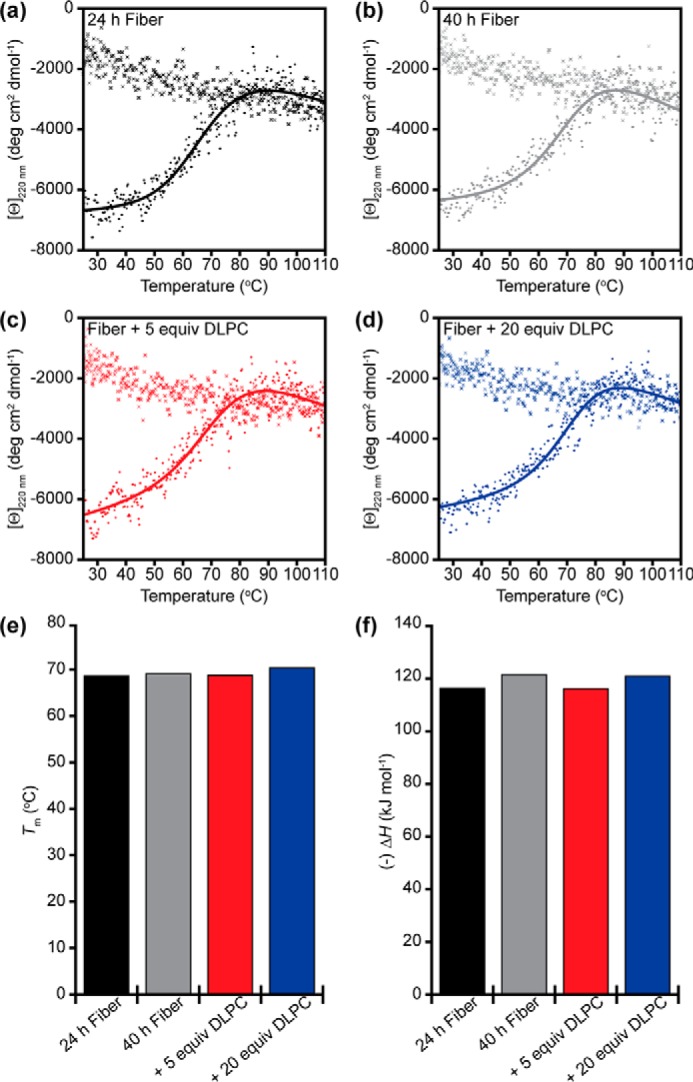
Thermodynamic stability of Aβ fibrils. Aβ fibrils (20 μm monomer concentration) formed after 24 h (a) and 40 h (b) were subjected to thermal denaturation, and the β-strand signal at 220 nm was observed as a function of time. Similar denaturation experiments were performed on Aβ fibrils formed by 24-h incubation and then treated with 5 eq (c) or 20 eq (d) of 100-nm DLPC LUVs. After the fiber was unfolded by heating, the sample was cooled from 110 °C down to 25 °C, and the structural signal was again observed for all four samples (marked by ×). These denaturation curves were fit to extrapolate both the melting temperature (Tm) (e) and the enthalpy of denaturation (ΔH) (f) of the fibril under different conditions.
There are, then, three likely mechanisms by which DLPC LUVs may remodel preformed Aβ fibrils: (i) DLPC changes the monomer-fiber equilibrium and causes additional monomers of Aβ to enter solution via dissociation from the ends of fibers, resulting in shorter aggregates (52); (ii) DLPC LUVs alter the hydrogen bonding network found within the fibril, generating distinct polymorphs of the aggregates that are less stable and can potentially become more similar to the immature protofibril structure (52); or (iii) DLPC LUVs rearrange the side chain packing of the mature fibrils and induce a partially unfolded state without significantly altering the hydrogen bonding of the fiber core (22).
We first explored the monomer-fiber equilibrium with 1H NMR. Whereas monomeric Aβ is highly dynamic and yields strong signals in solution NMR, the fibrillar aggregates, due to their large size and anisotropic tumbling, are essentially invisible (Fig. 5A). This allows for the selective observation of monomeric and small, soluble intermediates over large species. It is also possible to correlate changes in signal intensity with the equilibrium between monomeric and aggregated Aβ (52). DLPC LUVs were added to fibrillar Aβ, and spectra were continually recorded for 23 h (Fig. 5). Whereas previous disaggregation studies using similar methods observed an increase in relative signal intensity over time during disaggregation, DLPC induced a minimal change in signal (52). This suggests that the monomer-fiber equilibrium is minimally perturbed and thus insufficient to explain the remodeling of preformed fibrils.
FIGURE 5.
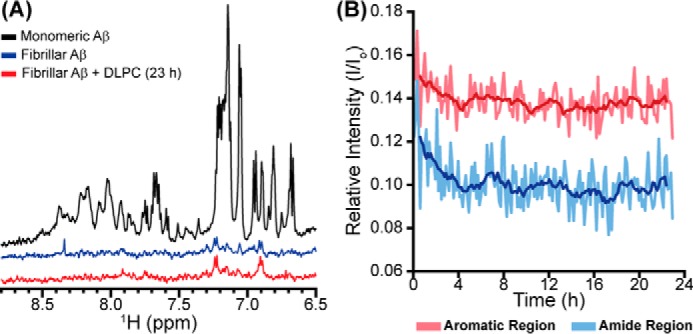
Monomer observation during fiber remodeling. A, the 1D 1H NMR spectrum of freshly prepared Aβ (50 μm) was observed before fibrillation. After 24 h, the fibrillar sample was again observed before the addition of DLPC LUVs (500 μm). B, the 1D 1H NMR spectrum was subsequently observed at 8-min intervals for 23 h. The relative intensity (compared with freshly prepared monomeric Aβ) of both the more flexible aromatic region (red) and the more rigid amide region (blue) were plotted versus time. The dark lines represent the moving average over a 40-min interval.
We next explored the possibility of DLPC altering the β-strand topology of the fiber backbone and the stability that it imparts. It has been shown through extensive solid-state NMR studies that Aβ fibers can adopt distinct backbone folds and polymorphs while maintaining the requisite β-strand prominence and fiber core based on aggregation conditions (53–56). It is possible that DLPC LUVs could exert a chaperone-like activity to convert one fiber polymorph into another. We explored the differences between the structures using thermal unfolding experiments. If fibers treated with DLPC LUVs result in altered β-strand folding and hydrogen bond networks, their unfolding temperatures and enthalpy of unfolding will probably be different as a result (57–59). To this end, we studied the stability of Aβ fibers formed for 24 and 40 h, as well as fibers formed for 24 h and subsequently treated with either 5 or 20 eq of DLPC in LUVs (Fig. 6). Samples were exposed to continuous shaking at 25 °C for the entirety of their aggregation and/or remodeling. All four samples exhibited similar melting temperatures (∼68 °C) and enthalpies of denaturation (115–120 kJ mol−1). This thermal unfolding was also found to be irreversible, in agreement with other studies (57). The melting temperature is slightly lower than has previously been published for the same isoform of Aβ, probably due to different aggregation conditions, highlighting this method's ability to distinguish distinct fiber polymorphs (57). The similarities in thermal denaturation suggest that, despite morphological differences based on TEM, the intermolecular forces responsible for aggregate stability are similar for all samples, and thus there is a low likelihood of distinct polymorphs being induced by the presence of DLPC.
It appears, then, that the DLPC-induced changes in ThT fluorescence and morphological differences observed by TEM are the result of more surface level disruptions. It has previously been shown that Aβ fibrils can adopt a pseudotransition state, termed the molten globule state, in which side chain packing at the fiber surface becomes perturbed (22). The molten globule state was defined by a reduction in ThT fluorescence and truncation of the fibrillar structure while the β-strand signal was maintained in CD spectroscopy, similar to what we have observed in the presence of DLPC (28). This molten globule state has previously only been observed following treatment of mature aggregates with ultrasonication. Treatment of fibrils with conventional chemical denaturants, such as detergents or guanidine hydrochloride, has previously resulted in the generation of the unfolded, monomeric form of Aβ (22). It is likely that DLPC is sufficiently mild in its perturbations so as to not fully denature and instead stabilize this unfolding transition state. Although we observed that DLPC inhibits Aβ aggregation through a combination of detergent-like interactions and stabilization of membrane-associated globulomers, it is likely that the fibrils are primarily altered through interactions with the surface of DLPC LUVs, which promote disorder in the traditionally ordered side chain packing of the fibril (22, 60). This increased disorder can facilitate a reduction in ThT binding and fluorescence. It may also increase the likelihood of Aβ fibrils to fragment to compensate for the increased side chain flexibility, explaining the truncated fibril structures observed by TEM (61, 62). These two modes of disruption would be relatively independent of the main chain interactions and result in the maintenance of the conventional β-strand structure, resulting in an aggregate species that resembles a protofibrillar structure (63, 64).
Effect of Lipid-Aβ Aggregates on Cell Viability
Having shown that DLPC LUVs are capable of generating two distinct, non-fibrillar Aβ aggregates when the lipids are present, the biological function of these two different species becomes of interest. It has been proposed that oligomers act as the toxic agent in amyloid-related diseases, and different oligomers can enact various toxic functions (2, 3). Stabilizing and characterizing the formation of many of these oligomers has been difficult, however. We therefore examined the toxic effects of stable species induced by preincubating monomeric Aβ with DLPC LUVs as well as those species generated by treatment of mature fibrils with DLPC LUVs (Fig. 7, A and B). Aβ aggregates formed in the absence of DLPC were observed to reduce cellular viability by ∼50%. Aggregates formed in the presence of DLPC, however, were found to be nontoxic (Fig. 7A). This further suggests that these Aβ oligomers formed in the presence of DLPC LUVs are off-pathway aggregates. Aβ fibrils remodeled by DLPC were found to induce cellular death to a similar extent as the untreated Aβ aggregate, suggesting that these molten globule aggregates resemble an on-pathway protofibrillar species, which has been suggested to be toxic (64).
FIGURE 7.
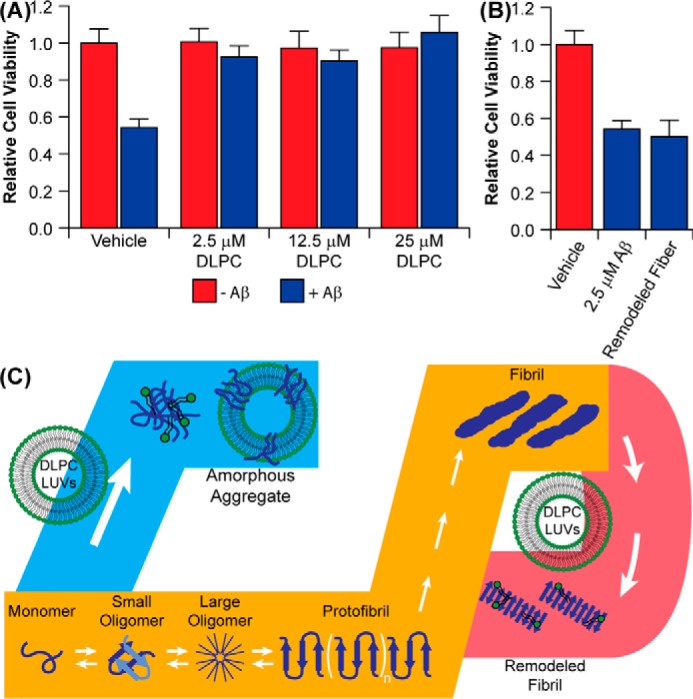
Function and identity of DLPC-induced Aβ aggregates. A, Aβ (2.5 μm) aggregates were generated in the presence and absence of 100-nm DLPC LUVs, and their toxicity against PC12 cells was compared by the MTT assay. B, the toxicity of preformed Aβ aggregates subsequently incubated with 10 eq (25 μm) of DLPC. C, the combined data from this study suggest that DLPC has two distinct modes of action against Aβ aggregation (orange pathway). When DLPC LUVs are added at the early stages of aggregation, they promote the formation of unstructured, off-pathway Aβ aggregates (blue pathway). When mature Aβ fibrils are treated with DLPC LUVs, the lipid induces a polymorphic change within the fibril without significantly altering the cross-β-strand structure of the fibril, resulting in a more protofibrillar structure in the final aggregates (red pathway).
Discussion
Although DLPC is not a lipid commonly found in eukaryotic membranes, it serves as a simplified model for membrane thinning associated with AD (65–68). Our findings suggest that bilayer thickness is able to play a key role in modulating the pathway of amyloid formation on the lipid bilayer. DLPC, as a model for a thin bilayer, inhibits the formation of Aβ amyloid fibers by stabilizing disordered aggregates on the bilayer surface, whereas DOPC and POPC, conventional bilayers, facilitate the formation of amyloid fibrils, as has been demonstrated previously (Fig. 7C) (23, 24). Aβ interaction with DLPC bilayers, in turn, induces membrane disruption, which causes lipids to shed from the bilayer and form micelle-like species above the CMC of DLPC (100 nm). Additionally, DLPC membranes are capable of reorganizing the surface and side chain orientation of mature fibrils. Interestingly, a lipid bilayer composed of dilauroyl phosphatidylserine, which shares the acyl chain length and hydrophobic thickness of DLPC, has been shown to promote the aggregation of α-synuclein, an amyloid implicated in Parkinson's disease (69). This finding suggests that the inhibition of Aβ aggregation by DLPC may be dependent on peptide, lipid headgroup, or possibly both.
Our results also suggest that the oligomers stabilized by their interaction with thin, DLPC bilayers are non-toxic (Fig. 7A). This, however, assumes that the act of forming these aggregates is also non-toxic. In these experiments, oligomers were preformed and subsequently added to live cells. However, it was also observed that the formation of these disordered oligomers is capable of disrupting the stability of the lipid bilayer, generating free lipid and large holes (Fig. 3). It has been previously suggested that conventional amyloid aggregation on cellular membranes is toxic, in part, because of its ability to destabilize the membrane and induce pores and holes (5, 8, 9). If a similar mechanism were to occur on locally thinned bilayers of cells during the formation of the disordered aggregates that we observe here, that mechanism would, itself, be detrimental to cellular health. Thus, although the exogenous addition of these aggregates is non-perturbing, their formation in the presence of cells would probably be detrimental and result in increased cellular death.
This potential mechanism has greater importance when we consider the wider biology of AD, which may promote the generation of locally thinned bilayers in vivo. The interaction of amyloidogenic sequences with lipid membranes and their aggregation on bilayers can reduce local bilayer thickness via mechanical strain (67, 68, 70–73). Additionally, short chain lipids may form within the AD-affected brain via lipid peroxidation promoted by the heightened oxidative stress found in the diseased state (65, 66, 74–77). Incorporation of these shortened lipids can be toxic and also may result in localized bilayer thinning (65, 66, 77). If aggregation were to occur around these regions of reduced bilayer hydrophobic thickness, caused either by initial aggregation and strain or the incorporation of peroxidized lipids, it is possible that a mechanism similar to that observed in the presence of DLPC could be replicated. Further clarification is needed to define whether this mechanism of Aβ modulation persists for all thin bilayers, independent of headgroup, and whether it holds true for thinned cellular membranes. It appears evident, however, that lipid bilayer hydrophobic thickness is capable of altering the aggregation pathway of Aβ and is able to stabilize distinct, membrane-associated aggregates.
Experimental Procedures
Materials and Reagents
All reagents were purchased from commercial suppliers and used as received unless noted otherwise. Aβ was purchased from BioBasic (Markham, Canada) (Aβ(1–40) = DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV) at >95% purity and used without further purification. DLPC, DOPC, and POPC were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). All other reagents were purchased from Sigma-Aldrich.
Vesicle Preparation
LUVs of DLPC, DOPC, and POPC were prepared from stock chloroform solutions. The resulting solution was dried under a nitrogen stream and placed under vacuum overnight to remove residual solvent. The resultant lipid film was rehydrated in buffer (20 mm PO4, pH 7.4, 50 mm NaCl) to yield a final lipid concentration of either 1, 10, or 24 mm (for solid-state NMR experiments). Dried lipid was resuspended by vortexing the solution and was subsequently extruded via 23 passes through a polycarbonate Nucleopore membrane filter (either 50, 100, 400, or 1000 nm, depending on experiment; Whatman) mounted to a mini-extruder (Avanti Polar Lipids) to obtain a homogeneous solution of LUVs. The formation of vesicles was subsequently confirmed by dynamic light scattering to ensure homogeneity.
Peptide Preparation
Aβ was received having been treated with hexafluoroisopropyl alcohol intended to monomerize the peptide. To disrupt any remaining aggregates, Aβ was dissolved in 5% (v/v) NH4OH at a concentration of 0.5 mg/ml. The peptide was aliquoted into 0.1-mg samples and lyopohilized overnight and stored at −20 °C until they were needed for an experiment. To prepare monomeric Aβ for experimentation, peptide was dissolved in buffer (20 mm PO4, pH 7.4, 50 mm NaCl) to a final concentration of 120 μm. The stock solution was sonicated for 10 s and then used immediately after preparation.
ThT Assay
The kinetics of amyloid formation were monitored by the fluorescent, amyloid-specific dye ThT. Samples were prepared by diluting the 120 μm Aβ stock to a final concentration of 10 μm in the presence of 20 μm ThT. To this was also added lipid from the prepared stock at a concentration ranging from 100 nm to 1 mm. Samples were plated in triplicate on uncoated Fisher 96-well polystyrene plates, maintained at 25 °C, and subjected to continuous, slow orbital shaking. Fluorescence readings were taken on a Biotek Synergy 2 microplate reader. Wells were read from the bottom with an excitation wavelength of 440 nm (30-nm bandwidth) and an emission wavelength of 485 nm (20-nm bandwidth) at 3-min intervals.
After data acquisition, raw fluorescence values were background-subtracted and then normalized. Normalized curves were then individually fit to Equations 1 and 2 to calculate the individual lag phase (tlag) for each curve (46). These values were then averaged across three separate trials.
| (Eq. 1) |
| (Eq. 2) |
For studies exploring fibril remodeling, fibrils were initially formed in the 96-well plate for 24 h. After mature fibrils were formed, lipid (either in LUVs or as free lipid) was added directly to wells at varying concentrations, diluting the volume by less than 1% to maintain signal intensity. The experiment was then restarted under conditions identical to those used to monitor fibril formation.
CD
Single point CD measurements were carried out at a concentration of 30 μm Aβ in the absence or presence of 300 μm (10 eq) 100-nm LUVs of either DLPC, DOPC, or POPC in buffer (20 mm PO4, pH 7.4, 50 mm NaF). CD measurements were performed on a JASCO J-1500 CD spectrometer using a 0.1-cm path length cell. Spectra were acquired at 25 °C using a bandwidth of 2 nm and a scan rate of 200 nm/min and averaging spectra over 20 scans. Measurements were taken immediately after mixing the sample (t = 0 h) and after 24 h of incubation at 25 °C under slow orbital shaking (conditions mimicking those used in ThT kinetic assays).
For time course analysis of fibril remodeling, fibrils were initially formed at 30 μm in buffer by incubating for 24 h at 25 °C under slow orbital shaking. After formation of the mature fibrils, LUVs were added to the solution, and the CD spectrum was monitored over time. Between reads, the sample was maintained at 25 °C under slow orbital shaking.
TEM
Samples used in TEM analysis were taken directly from the time course CD experiments after 24 h of incubation (both for the modulation of fiber formation and the remodeling of preformed fibrils). Glow-discharged grids (Formar/carbon 300 mesh, Electron Microscopy Sciences, Hatfield, PA) were treated with samples (5 μl) for 2 min at room temperature. Excess buffer was removed via blotting and then washed three times with double-distilled H2O. Each grid was then incubated with uranyl acetate staining solution (1% (w/v) in double-distilled H2O, 5 μl) for 1 min, and excess stain was blotted away. Images from each sample were taken on a JEOL 1400-plus TEM (80 kV) at ×50,000 magnification.
AFM
SLBs of POPC, DOPC, and DLPC were deposited on freshly cleaved mica by immersion of the substrates in vials containing the lipid vesicle dispersions in PBS (10 mm, pH 7.4, at 25 °C), kept in a sonication bath for 1 h. AFM images were acquired with a Cypher S (Asylum Research, Oxford Instruments) microscope in liquid AC mode using rectangular Si3N4 cantilever tips (BL-RC-150VB, Olympus, Tokyo, Japan) with a nominal spring constant of 0.03 newtons/m.
31P Solid-state NMR
Experiments were performed on a Varian 600-MHz solid-state NMR spectrometer. A Varian temperature control unit was used to maintain the sample temperature at 30 °C. All 31P spectra were collected using a single pulse under 42-kHz two-pulse phase modulation decoupling of protons. A typical 90° pulse length of 6.0 μs was used with a recycle delay of 3 s. The 31P chemical shift spectra are referenced with respect to 85% H3PO4 at 0 ppm. In each experiment, the 31P spectrum of 200 μl of 1000-nm LUVs composed of 23 mm DLPC, DOPC, or POPC was first acquired in the absence of peptide in buffer (20 mm Hepes, pH 7.4, 50 mm NaCl). After acquisition of the control spectrum, Aβ was added to the lipid solution at a concentration of 230 μm (100:1 lipid/peptide ratio), and the sample was incubated at 25 °C under slow orbital shaking for 24 h to induce aggregation. Following aggregation, a 31P spectrum was again acquired for each sample. Each spectrum is the result of 20,000 scans.
1H NMR
Experiments were performed on a Bruker 600-MHz NMR spectrometer equipped with a cryoprobe. A 1D 1H spectrum of monomeric Aβ (50 mm) was initially acquired in buffer (20 mm PO4, pH 7.4, 50 mm NaCl, 10% (v/v) D2O). Peptide aggregation was then induced by constant orbital shaking at 25 °C for 24 h. A spectrum was again taken of this aggregated sample. 100-nm DLPC LUVs were then added to the preformed aggregate sample (500 μm lipid), and acquisitions were taken continually for 23 h. Each spectrum was acquired at 25 °C and is the combination of 128 total scans. All spectra are normalized with respect to the initial monomer acquisition.
Thermal Denaturation
Four types of amyloid fibrils of 20 μm Aβ in 20 mm sodium phosphate buffer (pH 7.4) containing 50 mm NaCl prepared with constant shaking (50-s shaking, 10-s quiescence) at 37 °C were used for CD measurements. Aβ amyloid fibrils, which grew spontaneously from Aβ monomer solutions for 24 h without DLPC LUVs, were immediately used for CD measurements after confirming the stationary phase of fibrillation after the lag and exponential phase based on ThT fluorescence-based kinetics. The other three types of Aβ amyloid fibrils were obtained after a further 16-h incubation in the absence and presence of DLPC LUVs (100 and 400 μm DLPC) at 37 °C with the same shaking cycle above.
Heat scanning of all types of Aβ amyloid fibrils from 25 to 110 °C was performed by monitoring CD signals at 220 nm at a rate of 10 °C/min. CD measurements were performed with a J-820 spectropolarimeter (Jasco, Tokyo, Japan) equipped with a water-circulating cell holder for temperature control. Temperature was regulated using a PTC-423L Peltier unit (Jasco, Japan). A cell with a light path of 1 mm was sealed with a lid to prevent solvent evaporation during the heat treatment. CD signals were expressed as the mean residue ellipticity, [θ] (degrees cm2 dmol−1).
The melting temperature (Tm) and the enthalpy change (ΔH) of heat denaturation of Aβ amyloid fibrils were determined by a regression analysis using a nonlinear least squares fitting of data to the sigmoidal Equation 3 under the assumption of a two-state transition between unfolded monomers and amyloid fibrils. It should be noted that we performed thermodynamic analyses, although thermal denaturation of Aβ amyloid fibrils was irreversible,
| (Eq. 3) |
where θ is the signal intensity monitored by CD. The pre- and post-unfolding baselines are described by a + bT and c + dT. T and R indicate temperature and gas constant, respectively. The change in heat capacity is shown by ΔCp. Details of the derivation of the equation above are provided below. In our derivation, we assumed the two-state thermal denaturation model because the irreversible process in the Lumry-Eyring model can be neglected due to the high scan rate of 10 °C/min used (78, 79).
Derivation of the Fit Equation for Thermal Denaturation
A model of the two-state thermal denaturation of Aβ amyloid fibrils is defined as follows,
where F and U indicate the fibrillar and unfolded (i.e. denatured) states of Aβ, respectively.
The equilibrium constant of the unfolding reaction (KU) is given by the ratio of the fraction of each conformational state,
| (Eq. 4) |
where fF and fU represent the fraction of Aβ in the fibrillar and unfolded conformations, respectively.
The fraction is expressed as follows.
| (Eq. 5) |
Combining Reaction 1 and Equation 4 gives the following.
| (Eq. 6) |
| (Eq. 7) |
The observed CD intensity (θ) of Aβ at a given temperature is expressed as follows,
| (Eq. 8) |
where fF(T) and fU(T) are the fibrillar and unfolded fractions of Aβ at a given temperature (T), respectively. θF and θU indicate the CD signals of fibrillar and unfolded Aβ, respectively.
Using Equations 6 and 7, Equation 8 becomes the following.
| (Eq. 9) |
Rearranging Equation 6 gives the following.
| (Eq. 10) |
θF and θU are CD signals of fibrillar and unfolded Aβ, respectively, over a certain temperature range and are expressed as follows,
| (Eq. 11) |
| (Eq. 12) |
where a + bT and c + dT are the baselines of pre- and post-unfolding. a and c are intercepts, and b and d are slopes.
Substituting Equations 11 and 12 into Equation 10 gives the following equation.
| (Eq. 13) |
This is rearranged to the following equation.
| (Eq. 14) |
The change in enthalpy, ΔH(T), and entropy, ΔS(T), for the thermal denaturation of Aβ amyloid fibrils at a certain temperature is expressed as follows,
| (Eq. 15) |
| (Eq. 16) |
where Tm is the midpoint temperature of the thermal denaturation of Aβ amyloid fibrils, and ΔCp is the heat capacity change between fibrillar and unfolded A.
Equations 15 and 16 are solved as follows.
| (Eq. 17) |
| (Eq. 18) |
The temperature dependence of the change in Gibbs free energy, ΔG(T) at a certain temperature is defined by the following equations,
| (Eq. 19) |
| (Eq. 20) |
where R is the gas constant.
Equation 19 may be rearranged for KU,
| (Eq. 21) |
where “exp” represents an exponential function.
Substituting Equation 21 into Equation 14 gives the following.
| (Eq. 22) |
Substituting Equations 17 and 18 into Equation 20 gives the following.
| (Eq. 23) |
Equation 23 is rearranged to yield the following equation.
| (Eq. 24) |
Substituting Equation 24 into Equation 22 gives the following.
| (Eq. 25) |
Equation 24 is rearranged to yield the final equation, Equation 3.
Cellular Culture and Cytotoxicity Analysis
PC12 cells were grown in RPMI 1640 medium without l-glutamine supplemented with 5% fetal bovine serum, 10% horse serum, and 1% penicillin-streptomycin, in a humidified incubator at 37 °C with an environment of 5% CO2. Cells were kept between passage numbers 20 and 35. 10-cm cell culture dishes were used for culturing, and 96-flat bottom well trays were used for the MTT assays. Cells in a suspension of 90 μl (total of 104 cells) were dispensed in each well and allowed to adhere for 1 day before adding peptide aggregates.
Peptide aggregates were generated by incubating Aβ (25 μm) in the absence or presence of 100-nm DLPC LUVs (1–10 eq) for 18 h at 25 °C in buffer (20 mm PO4, pH 7.4, 50 mm NaCl). Aggregates were taken before complete fibrillation due to the purported toxicity of the intermediate aggregates (3, 80). 10 μl of aggregate solution was then added to the plated PC12 cells to a final volume of 100 μl (2.5 μm Aβ). After a 24-h incubation of cells with aggregates, the MTT cell proliferation assay (Promega, G4000) was used to determine the toxicity of samples following the manufacturer's protocol. 15 μl of the MTT dye solution was added to each well and set to incubate at 37 °C for 3–3.5 h. 100 μl of solubilization/stop buffer was added and set to incubate for 2–3 h. The absorbance of each well was then measured at both 570 and 700 nm (700 nm for background correction). All cellular viability values were normalized to cells treated with buffer. Values reported are the average of three independent trials, and the error is reported as the S.D. of these averages.
Author Contributions
K. J. K. and A. B. performed all kinetic and secondary structure analyses. K. J. K. and R. Z. performed all NMR experiments. C. S. and C. L. performed the AFM measurements. Y. L. and Y.-H. L. performed the thermodynamic stability analysis. K. J. K., M. D., and M. I. I. performed the toxicity studies. K. J. K. conceived the ideas for the project and wrote the paper under the guidance of A. R. and M. H. L. All authors reviewed the results and approved the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grant AG048934 (to A. R.), the Protein Folding Initiative at the University of Michigan (to M. H. L., M. I. I., and A. R.), National Research Foundation of Korea (NRF) Grant NRF-2014S1A2A2028270 funded by the Korean Government (to M. H. L. and A. R.), and University of Catania Grant FIR 2014 (to C. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S6.
- Aβ
- amyloid-β
- AD
- Alzheimer's disease
- DLPC
- dilauroyl phosphatidylcholine
- DOPC
- dioleoyl phosphatidylcholine
- POPC
- 1-palmitoyl-2-oleoyl phosphatidylcholine
- ThT
- thioflavin-T
- LUV
- large unilamellar vesicle
- TEM
- transmission electron microscopy
- CMC
- critical micelle concentration
- AFM
- atomic force microscopy
- SLB
- smooth supported lipid bilayer
- GM1
- Galβ1,3GalNAcβ1,4(NeuAcα2,3)-Galβ1,4Glc-ceramide.
References
- 1. Savelieff M. G., Lee S., Liu Y., and Lim M. H. (2013) Untangling amyloid-β, τ, and metals in Alzheimer's disease. ACS Chem. Biol. 8, 856–865 [DOI] [PubMed] [Google Scholar]
- 2. Benilova I., Karran E., and De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 3. Hardy J., and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 4. Kotler S. A., Walsh P., Brender J. R., and Ramamoorthy A. (2014) Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer's disease. Chem. Soc. Rev. 43, 6692–6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuzaki K. (2014) How do membranes initiate Alzheimer's disease? Formation of toxic amyloid fibrils by the amyloid β-protein on ganglioside clusters. Acc. Chem. Res. 47, 2397–2404 [DOI] [PubMed] [Google Scholar]
- 6. Jang H., Arce F. T., Ramachandran S., Capone R., Lal R., and Nussinov R. (2010) β-Barrel topology of Alzheimer's β-amyloid ion channels. J. Mol. Biol. 404, 917–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jang H., Arce F. T., Ramachandran S., Kagan B. L., Lal R., and Nussinov R. (2014) Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs. Chem. Soc. Rev. 43, 6750–6764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sciacca M. F., Kotler S. A., Brender J. R., Chen J., Lee D. K., and Ramamoorthy A. (2012) Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 103, 702–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yip C. M., and McLaurin J. (2001) Amyloid-β peptide assembly: a critical step in fibrillogenesis and membrane disruption. Biophys. J. 80, 1359–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fattal D. R., and Ben-Shaul A. (1993) A molecular model for lipid-protein interaction in membranes: the role of hydrophobic mismatch. Biophys. J. 65, 1795–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Killian J. A. (1998) Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta 1376, 401–415 [DOI] [PubMed] [Google Scholar]
- 12. Kucerka N., Liu Y., Chu N., Petrache H. I., Tristram-Nagle S., and Nagle J. F. (2005) Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using X-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys. J. 88, 2626–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kucerka N., Tristram-Nagle S., and Nagle J. F. (2005) Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 208, 193–202 [DOI] [PubMed] [Google Scholar]
- 14. Korshavn K. J., Bhunia A., Lim M. H., and Ramamoorthy A. (2016) Amyloid-β adopts a conserved, partially folded structure upon binding to zwitterionic lipid bilayers prior to amyloid formation. Chem. Commun. 52, 882–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arce F. T., Jang H., Ramachandran S., Landon P. B., Nussinov R., and Lal R. (2011) Polymorphism of amyloid β peptide in different environments: implications for membrane insertion and pore formation. Soft Matter 7, 5267–5273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar S., and Miranker A. D. (2013) A foldamer approach to targeting membrane bound helical states of islet amyloid polypeptide. Chem. Commun. 49, 4749–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jang H., Arce F. T., Ramachandran S., Kagan B. L., Lal R., and Nussinov R. (2013) Familial Alzheimer's disease Osaka mutant (DeltaE22) β-barrels suggest an explanation for the different Aβ1–40/42 preferred conformational states observed by experiment. J. Phys. Chem. B 117, 11518–11529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sciacca M. F., Lolicato F., Di Mauro G., Milardi D., D'Urso L., Satriano C., Ramamoorthy A., and La Rosa C. (2016) The role of cholesterol in driving IAPP-membrane interactions. Biophys. J. 111, 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stefanovic A. N., Lindhoud S., Semerdzhiev S. A., Claessens M. M., and Subramaniam V. (2015) Oligomers of Parkinson's disease-related α-synuclein mutants have similar structures but distinctive membrane permeabilization properties. Biochemistry 54, 3142–3150 [DOI] [PubMed] [Google Scholar]
- 20. Fleming P. J., Freites J. A., Moon C. P., Tobias D. J., and Fleming K. G. (2012) Outer membrane phospholipase A in phospholipid bilayers: a model system for concerted computational and experimental investigations of amino acid side chain partitioning into lipid bilayers. Biochim. Biophys. Acta 1818, 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rankenberg J. M., Vostrikov V. V., DuVall C. D., Greathouse D. V., Koeppe R. E. 2nd, Grant C. V., and Opella S. J. (2012) Proline kink angle distributions for GWALP23 in lipid bilayers of different thicknesses. Biochemistry 51, 3554–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Noda S., So M., Adachi M., Kardos J., Akazawa-Ogawa Y., Hagihara Y., and Goto Y. (2016) Thioflavin T-Silent denaturation intermediates support the main-chain-dominated architecture of amyloid fibrils. Biochemistry 55, 3937–3948 [DOI] [PubMed] [Google Scholar]
- 23. Aisenbrey C., Borowik T., Byström R., Bokvist M., Lindström F., Misiak H., Sani M. A., and Gröbner G. (2008) How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J. 37, 247–255 [DOI] [PubMed] [Google Scholar]
- 24. Gorbenko G. P., and Kinnunen P. K. (2006) The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 141, 72–82 [DOI] [PubMed] [Google Scholar]
- 25. Terakawa M. S., Yagi H., Adachi M., Lee Y. H., and Goto Y. (2015) Small liposomes accelerate the fibrillation of amyloid β(1–40). J. Biol. Chem. 290, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biancalana M., and Koide S. (2010) Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arosio P., Michaels T. C., Linse S., Månsson C., Emanuelsson C., Presto J., Johansson J., Vendruscolo M., Dobson C. M., and Knowles T. P. (2016) Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meisl G., Kirkegaard J. B., Arosio P., Michaels T. C., Vendruscolo M., Dobson C. M., Linse S., and Knowles T. P. (2016) Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 [DOI] [PubMed] [Google Scholar]
- 29. Suzuki Y., Brender J. R., Hartman K., Ramamoorthy A., and Marsh E. N. (2012) Alternative pathways of human islet amyloid polypeptide aggregation distinguished by (19)f nuclear magnetic resonance-detected kinetics of monomer consumption. Biochemistry 51, 8154–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong A. G., Wu C., Hannaberry E., Watson M. D., Shea J. E., and Raleigh D. P. (2016) Analysis of the amyloidogenic potential of pufferfish (Takifugu rubripes) islet amyloid polypeptide highlights the limitations of Thioflavin-T assays and the difficulties in defining amyloidogenicity. Biochemistry 55, 510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. So M., Ishii A., Hata Y., Yagi H., Naiki H., and Goto Y. (2015) Supersaturation-limited and unlimited phase spaces compete to produce maximal amyloid fibrillation near the critical micelle concentration of sodium dodecyl sulfate. Langmuir 31, 9973–9982 [DOI] [PubMed] [Google Scholar]
- 32. Marsh D. (1990) CRC Handbook of Lipid Bilayers, p. 816, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 33. Zhu M., and Fink A. L. (2003) Lipid binding inhibits α-synuclein fibril formation. J. Biol. Chem. 278, 16873–16877 [DOI] [PubMed] [Google Scholar]
- 34. Zhu M., Li J., and Fink A. L. (2003) The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 278, 40186–40197 [DOI] [PubMed] [Google Scholar]
- 35. La Rosa C., Scalisi S., Lolicato F., Pannuzzo M., and Raudino A. (2016) Lipid-assisted protein transport: a diffusion-reaction model supported by kinetic experiments and molecular dynamics simulations. J. Chem. Phys. 144, 184901. [DOI] [PubMed] [Google Scholar]
- 36. Buboltz J. T., and Feigenson G. W. (2005) Phospholipid solubility determined by equilibrium distribution between surface and bulk phases. Langmuir 21, 6296–6301 [DOI] [PubMed] [Google Scholar]
- 37. Sparr E., Engel M. F., Sakharov D. V., Sprong M., Jacobs J., de Kruijff B., Höppener J. W., and Killian J. A. (2004) Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 577, 117–120 [DOI] [PubMed] [Google Scholar]
- 38. Brender J. R., Dürr U. H., Heyl D., Budarapu M. B., and Ramamoorthy A. (2007) Membrane fragmentation by an amyloidogenic fragment of human islet amyloid polypeptide detected by solid-state NMR spectroscopy of membrane nanotubes. Biochim. Biophys. Acta 1768, 2026–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakazawa Y., Suzuki Y., Williamson M. P., Saitô H., and Asakura T. (2009) The interaction of amyloid Aβ(1–40) with lipid bilayers and ganglioside as studied by 31P solid-state NMR. Chem. Phys. Lipids. 158, 54–60 [DOI] [PubMed] [Google Scholar]
- 40. Connelly L., Jang H., Arce F. T., Ramachandran S., Kagan B. L., Nussinov R., and Lal R. (2012) Effects of point substitutions on the structure of toxic Alzheimer's β-amyloid channels: atomic force microscopy and molecular dynamics simulations. Biochemistry 51, 3031–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yates E. A., and Legleiter J. (2014) Preparation protocols of Aβ(1–40) promote the formation of polymorphic aggregates and altered interactions with lipid bilayers. Biochemistry 53, 7038–7050 [DOI] [PubMed] [Google Scholar]
- 42. Yates E. A., Owens S. L., Lynch M. F., Cucco E. M., Umbaugh C. S., and Legleiter J. (2013) Specific domains of Aβ facilitate aggregation on and association with lipid bilayers. J. Mol. Biol. 425, 1915–1933 [DOI] [PubMed] [Google Scholar]
- 43. Scalisi S., Sciacca M. F., Zhavnerko G., Grasso D. M., Marletta G., and La Rosa C. (2010) Self-assembling pathway of HiApp fibrils within lipid bilayers. Chembiochem 11, 1856–1859 [DOI] [PubMed] [Google Scholar]
- 44. Satriano C., Svedhem S., and Kasemo B. (2012) Well-defined lipid interfaces for protein adsorption studies. Phys. Chem. Chem. Phys. 14, 16695–16698 [DOI] [PubMed] [Google Scholar]
- 45. Satriano C., Lupo G., Motta C., Anfuso C. D., Di Pietro P., and Kasemo B. (2017) Ferritin-supported lipid bilayers for triggering the endothelial cell response. Colloids Surf. B Biointerfaces 149, 48–55 [DOI] [PubMed] [Google Scholar]
- 46. Batzli K. M., and Love B. J. (2015) Agitation of amyloid proteins to speed aggregation measured by ThT fluorescence: a call for standardization. Mater. Sci. Eng. C Mater. Biol. Appl. 48, 359–364 [DOI] [PubMed] [Google Scholar]
- 47. Yi X., Zhang Y., Gong M., Yu X., Darabedian N., Zheng J., and Zhou F. (2015) Ca2+ interacts with Glu-22 of Aβ(1–42) and phospholipid bilayers to accelerate the Aβ(1–42) aggregation below the critical micelle concentration. Biochemistry 54, 6323–6332 [DOI] [PubMed] [Google Scholar]
- 48. Canale C., Seghezza S., Vilasi S., Carrotta R., Bulone D., Diaspro A., San Biagio P. L., and Dante S. (2013) Different effects of Alzheimer's peptide Aβ(1–40) oligomers and fibrils on supported lipid membranes. Biophys. Chem. 182, 23–29 [DOI] [PubMed] [Google Scholar]
- 49. Pithadia A. S., Bhunia A., Sribalan R., Padmini V., Fierke C. A., and Ramamoorthy A. (2016) Influence of a curcumin derivative on hIAPP aggregation in the absence and presence of lipid membranes. Chem. Commun. 52, 942–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ahsan N., Mishra S., Jain M. K., Surolia A., and Gupta S. (2015) Curcumin pyrazole and its derivative (N-(3-nitrophenylpyrazole) curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of wild type and mutant α-synuclein. Sci. Rep. 5, 9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cao P., and Raleigh D. P. (2012) Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 51, 2670–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rezaei-Ghaleh N., Amininasab M., Kumar S., Walter J., and Zweckstetter M. (2016) Phosphorylation modifies the molecular stability of β-amyloid deposits. Nat. Commun. 7, 11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tycko R. (2014) Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 23, 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schütz A. K., Vagt T., Huber M., Ovchinnikova O. Y., Cadalbert R., Wall J., Güntert P., Böckmann A., Glockshuber R., and Meier B. H. (2015) Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew. Chem. Int. Ed. Engl. 54, 331–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Elkins M. R., Wang T., Nick M., Jo H., Lemmin T., Prusiner S. B., DeGrado W. F., Stöhr J., and Hong M. (2016) Structural polymorphism of Alzheimer's β-amyloid fibrils as controlled by an E22 switch: a solid-state NMR study. J. Am. Chem. Soc. 138, 9840–9852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ikenoue T., Lee Y. H., Kardos J., Saiki M., Yagi H., Kawata Y., and Goto Y. (2014) Cold denaturation of α-synuclein amyloid fibrils. Angew. Chem. Int. Ed. Engl. 53, 7799–7804 [DOI] [PubMed] [Google Scholar]
- 58. Cobb N. J., Apostol M. I., Chen S., Smirnovas V., and Surewicz W. K. (2014) Conformational stability of mammalian prion protein amyloid fibrils is dictated by a packing polymorphism within the core region. J. Biol. Chem. 289, 2643–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nyström S., Mishra R., Hornemann S., Aguzzi A., Nilsson K. P., and Hammarström P. (2012) Multiple substitutions of methionine 129 in human prion protein reveal its importance in the amyloid fibrillation pathway. J. Biol. Chem. 287, 25975–25984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Knowles T. P., Waudby C. A., Devlin G. L., Cohen S. I., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., and Dobson C. M. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 62. Tanaka M., Collins S. R., Toyama B. H., and Weissman J. S. (2006) The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 [DOI] [PubMed] [Google Scholar]
- 63. Nichols M. R., Colvin B. A., Hood E. A., Paranjape G. S., Osborn D. C., and Terrill-Usery S. E. (2015) Biophysical comparison of soluble amyloid-β(1–42) protofibrils, oligomers, and protofilaments. Biochemistry 54, 2193–2204 [DOI] [PubMed] [Google Scholar]
- 64. O'Nuallain B., Freir D. B., Nicoll A. J., Risse E., Ferguson N., Herron C. E., Collinge J., and Walsh D. M. (2010) Amyloid β-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. 30, 14411–14419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beranova L., Cwiklik L., Jurkiewicz P., Hof M., and Jungwirth P. (2010) Oxidation changes physical properties of phospholipid bilayers: fluorescence spectroscopy and molecular simulations. Langmuir 26, 6140–6144 [DOI] [PubMed] [Google Scholar]
- 66. Negre-Salvayre A., Auge N., Ayala V., Basaga H., Boada J., Brenke R., Chapple S., Cohen G., Feher J., Grune T., Lengyel G., Mann G. E., Pamplona R., Poli G., Portero-Otin M., et al. (2010) Pathological aspects of lipid peroxidation. Free Radic. Res. 44, 1125–1171 [DOI] [PubMed] [Google Scholar]
- 67. Last N. B., and Miranker A. D. (2013) Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc. Natl. Acad. Sci. U.S.A. 110, 6382–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee C. C., Sun Y., and Huang H. W. (2012) How type II diabetes-related islet amyloid polypeptide damages lipid bilayers. Biophys. J. 102, 1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Galvagnion C., Brown J. W., Ouberai M. M., Flagmeier P., Vendruscolo M., Buell A. K., Sparr E., and Dobson C. M. (2016) Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 113, 7065–7070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pannuzzo M., Milardi D., Raudino A., Karttunen M., and La Rosa C. (2013) Analytical model and multiscale simulations of Aβ peptide aggregation in lipid membranes: towards a unifying description of conformational transitions, oligomerization and membrane damage. Phys. Chem. Chem. Phys. 15, 8940–8951 [DOI] [PubMed] [Google Scholar]
- 71. Tofoleanu F., and Buchete N. V. (2012) Molecular interactions of Alzheimer's Aβ protofilaments with lipid membranes. J. Mol. Biol. 421, 572–586 [DOI] [PubMed] [Google Scholar]
- 72. Sokolov Y., Kozak J. A., Kayed R., Chanturiya A., Glabe C., and Hall J. E. (2006) Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J. Gen. Physiol. 128, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lockhart C., and Klimov D. K. (2014) Binding of Aβ peptide creates lipid density depression in DMPC bilayer. Biochim. Biophys. Acta 1838, 2678–2688 [DOI] [PubMed] [Google Scholar]
- 74. Praticò D. (2008) Oxidative stress hypothesis in Alzheimer's disease: a reappraisal. Trends Pharmacol. Sci. 29, 609–615 [DOI] [PubMed] [Google Scholar]
- 75. Bradley-Whitman M. A., and Lovell M. A. (2015) Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch. Toxicol. 89, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chiurchiù V., Orlacchio A., and Maccarrone M. (2016) Is modulation of oxidative stress an answer? The state of the art of redox therapeutic actions in neurodegenerative diseases. Oxid. Med. Cell Longev. 2016, 7909380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Axelsen P. H., Komatsu H., and Murray I. V. (2011) Oxidative stress and cell membranes in the pathogenesis of Alzheimer's disease. Physiology 26, 54–69 [DOI] [PubMed] [Google Scholar]
- 78. La Rosa C., Milardi D., Grasso D., Guzzi R., and Sportelli L. (1995) Thermodynamics of the thermal unfolding of azurin. J. Phys. Chem. 99, 14864–14870 [Google Scholar]
- 79. Manetto G. D., La Rosa C., Grasso D. M., and Milardi D. (2005) Evaluation of thermodynamic properties of irreversible protein thermal unfolding measured by DSC. J. Therm. Anal. Calorim. 80, 263–270 [Google Scholar]
- 80. Ono K., Condron M. M., and Teplow D. B. (2009) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.