

Abstract
A hallmark of cancer cells is the ability to survive and proliferate when challenged with stressors such as growth factor insufficiency. In this study, we report a novel glycosylation-dependent mechanism that protects tumor cells from serum growth factor withdrawal. Our results suggest that the β-galactoside α-2,6-sialyltransferase 1 (ST6Gal-I) sialyltransferase, which is up-regulated in numerous cancers, promotes the survival of serum-starved cells. Using ovarian and pancreatic cancer cell models with ST6Gal-I overexpression or knockdown, we find that serum-starved cells with high ST6Gal-I levels exhibit increased activation of prosurvival signaling molecules, including pAkt, p-p70S6K, and pNFκB. Correspondingly, ST6Gal-I activity augments the expression of tumor-promoting pNFκB transcriptional targets such as IL-6, IL-8, and the apoptosis inhibitor cIAP2. ST6Gal-I also potentiates expression of the cell cycle regulator cyclin D2, leading to increased phosphorylation and inactivation of the cell cycle inhibitor pRb. Consistent with these results, serum-starved cells with high ST6Gal-I expression maintain a greater number of S phase cells compared with low ST6Gal-I expressors, reflecting enhanced proliferation. Finally, selective enrichment in clonal variants with high ST6Gal-I expression is observed upon prolonged serum deprivation, supporting the concept that ST6Gal-I confers a survival advantage. Collectively, these results implicate a functional role for ST6Gal-I in fostering tumor cell survival within the serum-depleted tumor microenvironment.
Keywords: β-galactoside α-2,6-sialyltransferase 1 (ST6Gal-I); cell cycle; glycosylation; growth factor; sialyltransferase
Introduction
Cell surface glycans lie at the interface between the extracellular milieu and intracellular signaling networks that regulate cellular response to the microenvironment. Glycosylation of surface receptors can have profound effects on cell signaling through glycan-dependent modulation of receptor conformation, clustering, surface retention, and/or interaction with other membrane proteins. The profile of glycans on membrane receptors changes dramatically in correspondence with numerous pathophysiologic conditions, including malignant transformation (1–4). However, the functional role of aberrant glycosylation as a transducer of tumor microenvironmental cues remains poorly understood.
One important glycosyltransferase dysregulated in cancer cells is the ST6Gal-I2 sialyltransferase (5–8). ST6Gal-I adds α2-6-linked sialic acids to N-glycans on select receptors, including the β1 integrin receptor (9–11), the Fas and TNFR1 death receptors (12, 13), the growth factor receptor EGF receptor (EGFR) (14), and multiple other membrane proteins. ST6Gal-I expression is markedly up-regulated in numerous malignancies, including colon, pancreatic, and ovarian adenocarcinoma (15–17). In ovarian cancer, high levels of ST6Gal-I correlate with reduced progression-free and overall patient survival, and ST6Gal-I expression is enriched in metastatic versus primary ovarian tumors (16). Contrarily, ST6Gal-I expression is negligible in the differentiated epithelium of normal colon, pancreas, and ovary (15, 16), whereas, notably, a subset of cells with high ST6Gal-I expression is found within the base of colon crypts, a stem cell niche (15). ST6Gal-I is also highly expressed in other epithelium-related stem cell compartments (15, 16) as well as in embryonic and induced pluripotent stem cells (15, 18–20).
Consistent with a potential function in conferring stem cell-like properties, we recently reported that ST6Gal-I promotes a cancer stem cell (CSC) phenotype (16). ST6Gal-I expression correlates with other CSC markers, including ALDH1 and CD133 (15), and ST6Gal-I activity is critical for CSC behaviors such as tumor spheroid growth, chemoresistance and tumor-initiating potential (16). Additionally, ST6Gal-I up-regulation in tumor cells induces the expression of important CSC-associated transcription factors, including Sox9 and Slug (16). We hypothesize that one of the main functions of ST6Gal-I in cancer cells, including CSCs, is to protect against diverse cytotoxic stimuli through sialylation-dependent modulation of cell surface receptors. For example, α2-6 sialylation of the Fas receptor inhibits apoptosis by preventing Fas internalization, a requisite step in apoptotic signaling (12). Furthermore, α2-6 sialylation of TNFR1 blocks TNFα-stimulated cell death (13), whereas α2-6 sialylation of certain integrins impedes apoptosis induced by galectins (21, 22), a family of galactose-binding lectins. In immune cells, ST6Gal-I-mediated sialylation of CD45 prevents galectin-dependent CD45 clustering and subsequent apoptosis (23). Finally, α2-6 sialylation of the platelet endothelial cell adhesion molecule receptor prevents endothelial cell death (24).
In this study, we describe a new survival-associated function for ST6Gal-I: protection against serum growth factor insufficiency. Using ovarian and pancreatic cancer cell models with ST6Gal-I overexpression or knockdown, we show that ST6Gal-I facilitates the survival of cells grown under serum-depleted culture conditions. Serum-starved cells with high ST6Gal-I expression maintain activation of prosurvival signaling nodes, including Akt and NFκB, and retain proliferative capacity via ST6Gal-I-dependent up-regulation of cyclin D2. These findings highlight a novel role for the tumor glycome in sustaining the viability of tumor cells exposed to serum-depleted environments, such as those found within hypovascularized regions of large, solid tumors.
Results
Cells with High ST6Gal-I Expression Are Resistant to Cytotoxic Stress Induced by Serum Deprivation
To interrogate the role of ST6Gal-I in protection against serum withdrawal, ST6Gal-I was stably overexpressed in the OV4 ovarian cancer cell line, which is one of the few cancer lines that lack detectable ST6Gal-I protein (Fig. 1A). As expected, overexpression (OE) of ST6Gal-I led to a pronounced increase in surface α2-6 sialylation relative to empty vector (EV) control cells, as measured by the binding of SNA (Fig. 1B). The SNA lectin specifically detects α2-6 sialic acids. Contrarily, ST6Gal-I overexpression had no effect on α2-3 sialylation, evidenced by equivalent binding of the MAA lectin to EV and OE cells (Fig. 1C).
FIGURE 1.
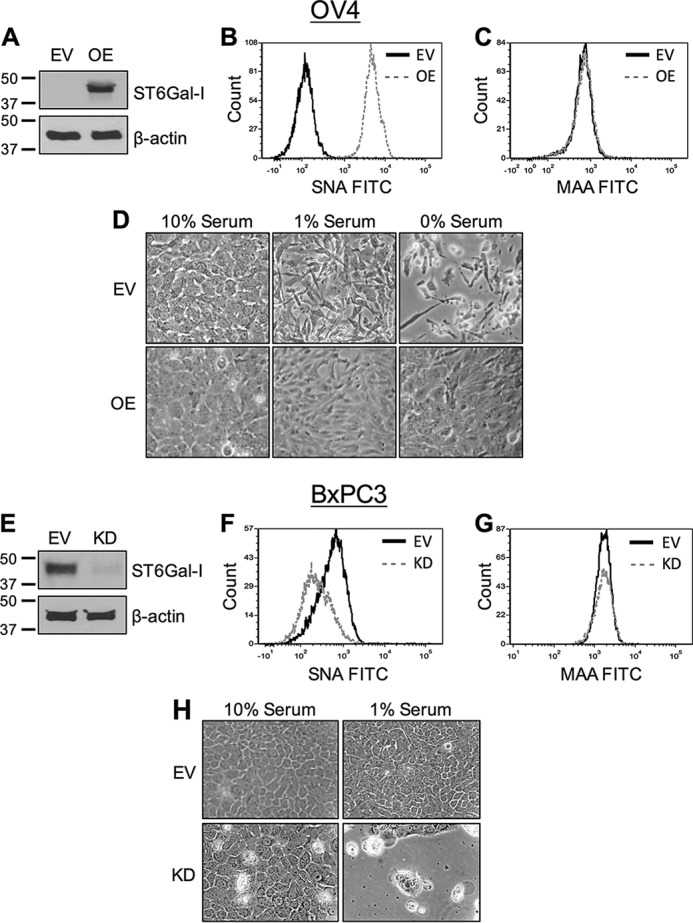
Cells with high ST6Gal-I expression are resistant to stress induced by serum withdrawal. A, OV4 cells were stably transduced with a lentivirus encoding ST6Gal-I, and ST6Gal-I OE was confirmed by immunoblotting. Control cells were generated by stable transduction of an EV lentiviral construct. B, OV4 EV and OE cells were stained with SNA-FITC and evaluated by flow cytometry. C, OV4 EV and OE cells were stained with MAA-FITC and evaluated by flow cytometry. D, OV4 EV and OE cells were cultured in 10%, 1%, or 0% FBS-containing medium for 1 week and imaged to observe differences in cell morphology. E, BxPC3 cells were stably transduced with shRNA for ST6Gal-I using a lentivirus, and ST6Gal-I KD was verified by immunoblotting. F, BxPC3 cells were stained with SNA-FITC and evaluated by flow cytometry. G, BxPC3 cells were stained with MAA-FITC and evaluated by flow cytometry. H, EV and KD BxPC3 cells were cultured in 10% or 1% FBS-containing medium for 1 week and then imaged.
OV4 EV or OE cells were grown in medium containing 10%, 1%, or 0% FBS for 1 week. As shown in Fig. 1D, EV cells grown in 1% FBS displayed a spindle-shaped morphology, reflecting cell stress, whereas EV cells grown in 0% FBS exhibited rounding and detachment from the plate, indicative of cell death. In contrast to EV cells, OE cells maintained a spread morphology and formed a confluent monolayer, even in 0% FBS, suggesting that ST6Gal-I promotes cell survival.
In complementary studies, the effect of ST6Gal-I knockdown was examined. BxPC3 pancreatic cancer cells, which have moderate levels of endogenous ST6Gal-I, were stably transduced with a lentivirus encoding shRNA for ST6Gal-I (Fig. 1E). ST6Gal-I knockdown (KD) cells had reduced surface expression of α2-6 sialic acids (indicated by decreased SNA staining, Fig. 1F), whereas α2–3 sialylation was unaffected (Fig. 1G).
BxPC3 EV and KD cells were grown for 1 week in 10% or 1% FBS. EV cell morphology was indistinguishable in the 10% versus 1% FBS cultures; however, serum-starved KD cells showed evidence of cytotoxicity and cell detachment (Fig. 1H). We also assessed BXPC3 growth in 0% FBS; however, there were few surviving cells in the KD population (data not shown). Therefore, all subsequent studies were conducted with 1% FBS for the BxPC3 line.
To further probe the role of ST6Gal-I in protection against serum withdrawal, OV4 and BxPC3 cells were grown for 1 week in 10% or 1% FBS, lysed, and immunoblotted for survival-associated signaling molecules. Under conditions of serum-depletion, OV4 OE cells expressed higher levels of activated (phosphorylated) Akt as well as the apoptosis inhibitor cIAP2 relative to EV cells (representative blots in Fig. 2A and densitometric analyses of three independent blots in Fig. 2B). Interestingly, pAkt and cIAP2 expression was also higher in the OE versus EV cultures grown in 10% FBS, suggesting that ST6Gal-I enhances basal activation of these molecules. Consistent with these results, serum-starved BxPC3 cells with ST6Gal-I knockdown had reduced levels of pAkt and cIAP2 compared with EV cells (representative blots in Fig 2C and densitometry in Fig. 2D). We also immunoblotted for apoptosis-inducing factor (AIF), which is a caspase-independent cell death marker associated with the intrinsic apoptotic pathway. OV4 OE cells had lower expression of AIF than EV cells under conditions of both 10% and 1% FBS. However, unlike OV4 cells, AIF expression in BxPC3 cells was equivalent in the serum-starved EV and KD lines. Additionally, we evaluated Beclin expression, a marker for autophagy. No significant correlations were noted between Beclin levels and ST6Gal-I activity in the two cell models, suggesting that autophagy may not be a major factor in the protective effect of ST6Gal-I, although further studies are clearly needed.
FIGURE 2.

ST6Gal-I enhances survival signaling in serum-starved cells. A, OV4 cells were serum deprived for 1 week and then immunoblotted for survival (pAkt and cIAP2) or cell death (Beclin and AIF) markers. Representative blots are shown. B, densitometric analyses of p-Akt, total Akt, cIAP2, AIF, and Beclin. Three distinct blots (from three independently generated cell lysates) were evaluated using ImageJ, and data were plotted as mean ± S.D. *, p < 0.05. For Akt, pAkt and total Akt were each normalized to β-tubulin, and then data were plotted as pAkt/total Akt (p/t Akt). For cIAP2, AIF, and Beclin, values were normalized to β-tubulin. C, BxPC3 EV and KD cells were serum-deprived for 1 week and immunoblotted for survival or cell death markers as in A. D, densitometric analyses of three independent blots for pAkt, total Akt, cIAP2, AIF, and Beclin. Data were normalized to β-tubulin as described in B. *, p < 0.05.
Other studies from our group have suggested that cytotoxic stimuli can exert selective pressure, leading to the expansion of clonal variants with high ST6Gal-I expression. For example, cell lines developed to grow continuously in the presence of the chemotherapeutics cisplatin or irinotecan have up-regulated ST6Gal-I (15, 25). Accordingly, we investigated whether prolonged exposure to serum-depleted culture conditions would select for cells with high ST6Gal-I expression. OV4 EV and OE cells were grown in 10% or 1% FBS for 1 week, and then cells were lysed and immunoblotted for ST6Gal-I (Fig. 3, A and B). Although the levels of ST6Gal-I remained undetectable in EV cells, OE cells showed a distinct enrichment in ST6Gal-I expression in 1% compared with 10% FBS culture conditions. The ST6Gal-I OE line is a polyclonal population generated by lentiviral transduction, and the ST6Gal-I construct is driven by a constitutive promoter. Thus, increased ST6Gal-I levels in the 1% FBS OE cultures are more likely a consequence of clonal selection rather than induction in ST6Gal-I gene expression. Similar results were noted for BxPC3 cells (Fig. 3, C and D); robust ST6Gal-I up-regulation was apparent in EV cells grown in 1% versus 10% serum.
FIGURE 3.
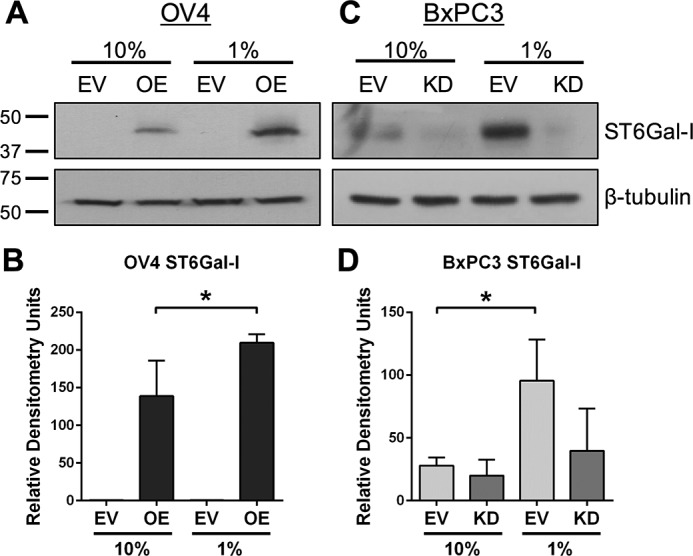
ST6Gal-I levels are enriched in serum-starved cells. A, representative immunoblot showing an increased level of ST6Gal-I in OV4 OE cells upon serum starvation. B, densitometry of three independent blots for ST6Gal-I in OV4 cells. C, representative immunoblot showing an increased level of ST6Gal-I in BxPC3 EV cells upon serum starvation. D, densitometry of three blots for ST6Gal-I in BxPC3 cells. All densitometric values were normalized to β-tubulin. *, p < 0.05.
ST6Gal-I Promotes Survival Signaling in Cells Exposed to Acute Serum Deprivation
To monitor acute responses to serum deprivation, signaling was examined in cells incubated for 4, 18, or 24 h in medium containing either 10% or 1% FBS. As shown in Fig. 4, A and B, OV4 OE cells displayed higher levels of pAkt than EV cells at all time points under 1% FBS culture conditions. Of note, there was a substantial decrease in pAkt in EV cells exposed to 1% FBS relative to 10% FBS, consistent with repression of survival signaling networks. We also evaluated phosphorylation of p70S6K, a kinase downstream of Akt that plays an important role in protein synthesis (26). The translation-promoting activity of p70S6K is necessary for growth factor-induced cell cycle progression (27). Although there were no differences in p-p70S6K levels at 4 and 18 h between OV4 OE and EV cells, at 24 h, p-p70S6K levels were higher in the serum-starved OE population (Fig. 4, A and C). Studies using BxPC3 cells corroborated a role for ST6Gal-I in survival signaling. Compared with BxPC3 EV cells, KD cells exposed to 1% FBS exhibited a dramatic loss of pAkt at all time points (Fig. 5, A and B). Levels of p-p70S6K were also reduced in serum-starved KD versus EV cells at the 24-h time point (Fig. 5, A and C).
FIGURE 4.

ST6Gal-I activity promotes the activation of prosurvival molecules upon acute serum deprivation. A, OV4 cells were serum-deprived for 4, 18, or 24 h and then immunoblotted for pAkt, total Akt, p-p70S6K, and total p70S6K. B, densitometric analyses for pAkt and total Akt for three blots. Values for pAkt and total Akt were each normalized to β-tubulin, and then data were plotted as pAkt/total Akt. *, p < 0.05. C, densitometric analyses for p-p70S6K and total p70S6K for three blots. Values for p-p70S6K and total p70S6K were each normalized to β-tubulin, and then data were plotted as p-p70S6K/total p70S6K. *, p < 0.05.
FIGURE 5.

ST6Gal-I knockdown inhibits activation of prosurvival molecules upon acute serum deprivation. A, BxPC3 cells were serum-deprived for 4, 18, or 24 h and then immunoblotted for pAkt, total Akt, p-p70S6K, and total p70S6K. B, densitometric analyses for pAkt and total Akt from three blots. Values for pAkt and total Akt were each normalized to β-tubulin, and then data were plotted as pAkt/total Akt. *, p < 0.05. C, densitometric analyses for p-p70S6K and total p70S6K from three blots. Values for p-p70S6K and total p70S6K were each normalized to β-tubulin, and then data were plotted as p-p70S6K/total p70S6K. *, p < 0.05.
ST6Gal-I Potentiates NFκB Signaling
The NFκB signaling axis is another essential prosurvival pathway, and enhanced NFκB activation is common in cancer cells (28). We therefore evaluated the effects of serum starvation on NFκB (p65) phosphorylation. Compared with the EV control, OV4 OE cells displayed increased pNFκB and total NFκB when grown for 24 h in 1% FBS (Fig. 6, A and C). Contrarily, serum-starved BxPC3 KD cells had reduced levels of pNFκB and total NFκB compared with EV cells (Fig. 6, B and D).
FIGURE 6.

ST6Gal-I activity up-regulates the expression of NFκB pathway proteins. A and B, OV4 (A) and BxPC3 (B) cells were grown in 10% or 1% FBS for 24 h and then immunoblotted for pNFκB and total NFκB (p65). C, densitometry for three OV4 immunoblots. Values for pNFκB and total NFκB were each normalized to β-tubulin. pNFκB and total NFκB were graphed separately (rather than phospho/total) because differences were noted in total NFκB levels. *, p < 0.05. D, densitometry for three BxPC3 immunoblots. Values for pNFκB and total NFκB were each normalized to β-tubulin. *, p < 0.05. E and F, OV4 (E) and BxPC3 (F) cells were serum-starved for 6 or 24 h and then analyzed by qRT-PCR for various prosurvival molecules known to be transcriptionally up-regulated by NFκB. Values represent the averages of at least four independent experiments ± S.D., with each independent experiment performed in triplicate. *, p < 0.05.
We next assessed the expression of known NFκB transcriptional targets. As shown in Fig. 6E, OV4 OE cells expressed significantly higher levels of cIAP2, IL-6, and IL-8 mRNA than EV cells at 24 h regardless of the FBS concentration. IL-6 and IL-8, but not cIAP2, were also elevated in OE cells at the 6-h time point. Knockdown of ST6Gal-I in BxPC3 cells inhibited expression of these same targets (Fig. 6F). At 24 h, cIAP2 and IL-8 expression was reduced in KD cells relative to EV cells in 1% FBS medium, whereas IL-6 was decreased in KD cells in both 10% and 1% FBS medium. These data suggest that NFκB signaling pathways are more active in cells with high ST6Gal-I expression.
ST6Gal-I Regulates Cyclin D2 Expression and pRb Phosphorylation
One of the key downstream targets of NFκB is cyclin D2, a crucial cell cycle regulatory protein. Typically, when cells lack access to growth factors, they exit the cell cycle and enter G1 arrest (29). Because cyclin D2 controls the G1/S transition (29), we investigated the role of ST6Gal-I in regulating cyclin D2 expression. As shown in Fig. 7A, OV4 OE cells expressed higher levels of cyclin D2 mRNA than EV cells when grown in either 10% or 1% FBS. Intriguingly, at 24 h, cyclin D2 expression was higher in OE cells grown in 1% versus 10% FBS, which may reflect a compensatory survival response. ST6Gal-I also regulated cyclin D2 expression in BxPC3 cells (Fig. 7B). KD cells had reduced levels of cyclin D2 mRNA relative to EV cells regardless of FBS concentration. These results were validated by immunoblotting, which showed striking differences in cyclin D2 protein expression in cells with differential ST6Gal-I activity. OV4 OE cells had substantially greater levels of cyclin D2 than EV cells, which were further enriched in OE cells grown in 1% FBS (Fig. 7, C and E). Similarly, BxPC3 EV cells had greater levels of cyclin D2 than KD cells (Fig. 7, D and F).
FIGURE 7.

ST6Gal-I regulates cyclin D2 expression and pRb phosphorylation. A and B, OV4 (A) and BxPC3 (B) cells were serum-starved for 6 or 24 h, and cyclin D2 mRNA was measured by qRT-PCR. Data are from at least four independent experiments, with each independent experiment performed in triplicate. *, p < 0.05. C and D, OV4 (C) and BxPC3 (D) cells were serum-starved for 24 h, and cell lysates were immunoblotted for cyclin D2. E and F, OV4 (E) and BxPC3 (F) immunoblots of cyclin D2 were evaluated by densitometry (3 blots/cell line). *, p < 0.05. G and H, OV4 (G) and BxPC3 (H) cells were immunoblotted for the downstream cyclin D2 target pRb. I and J, OV4 (I) and BxPC3 (J) immunoblots for p-Rb and total pRb were evaluated by densitometric analyses of three independent blots. Values for p-pRb and total pRb were normalized to β-tubulin and plotted separately because differences were noted in both phosphorylated and total pRb. *, p < 0.05.
Cyclin D2 partners with select cyclin-dependent kinases to mediate phosphorylation of the retinoblastoma protein (pRb), a molecule that represses transition into S phase (29). Phosphorylation of pRB leads to pRb degradation, thus allowing the cell cycle to proceed. Serum-starved OV4 OE cells exhibited increased levels of both phosphorylated and total pRB compared with EV cells (Fig. 7, G and I), whereas serum-starved BxPC3 KD cells had diminished p-pRb and total pRb relative to EV cells (Fig. 7, H and J). These results suggest that ST6Gal-I activity promotes cyclin D2 expression and pRb phosphorylation, which would be expected to facilitate entry into S phase.
Cells with High ST6Gal-I Expression Maintain a Greater Number of Cells in S Phase following Serum Deprivation
To determine the role of ST6Gal-I in regulating cell cycle progression, we quantified the percentage of cells in S phase. To this end, cells were incubated with EdU, a thymidine analog that incorporates into the DNA during DNA replication. EdU incorporation was monitored by flow cytometry. Upon serum deprivation, OV4 OE cells maintained a significantly greater percentage of S phase cells than the EV line. On the other hand, the number of S phase cells was equivalent for EV and OE cells grown in 10% FBS (a representative experiment is shown in Fig. 8A; an average of three independent experiments is shown in Fig. 8B). BxPC3 cells recapitulated this phenotype. Serum-starved BxPC3 EV cells had a greater abundance of S phase cells than serum-starved KD cells, whereas no differences were noted in the 10% FBS cultures (Fig. 8, C and D). These data suggest that ST6Gal-I does not significantly affect cell proliferation when serum growth factors are replete but, instead, acts to maintain cell proliferative capacity when growth factors are withdrawn.
FIGURE 8.

ST6Gal-I activity promotes cell cycle progression in serum-starved cells. To assay for cell cycle progression, cells in S phase were labeled with the Click-it EdU reagent (Thermo) and then quantified by flow cytometry. A, representative experiment showing a greater percentage of S phase cells in serum-starved OV OE compared with EV cells. B, three independent flow cytometry experiments measuring the percentage of cells in S phase. *, p < 0.05. C, representative experiment showing a greater percentage of S phase cells in serum-starved BxPC3 EV versus KD cells. D, three independent flow cytometry experiments measuring the percentage of cells in S phase. *, p < 0.05.
Discussion
The ability to survive and proliferate in the absence of growth factors is a defining feature of a tumor cell. Dysregulation of growth factor signaling networks comprises one of the principal characteristics of a transformed cell (30). One important mechanism tumor cells utilize to overcome growth factor deprivation is to up-regulate the expression of growth factors or growth factor receptors (31). Alternatively, cancer cells acquire activating mutations in growth factor receptors or downstream effector molecules, including ras, raf, and PI3K (30, 32). In addition to these established pathways, this study elucidates a novel glycosylation-dependent mechanism that protects tumor cells from the cytotoxic stress exerted by a growth factor-deficient environment.
More specifically, we show that the sialyltransferase ST6Gal-I is critical for maintaining prosurvival signaling cascades in cells exposed to serum starvation. Forced overexpression of ST6Gal-I enhances signaling through survival-associated molecules, including pAkt, p-p70S6K, pNFκB, and cIAP2, whereas ST6Gal-I knockdown inhibits these same proteins. Akt is a key downstream mediator of growth factor receptor activity, and this kinase plays a seminal role in protecting cells from a variety of stressors, including serum withdrawal (33–35). A positive association between ST6Gal-I and Akt activation has been reported previously, for example, during epithelial-to-mesenchymal transition, a process shown to be dependent on ST6Gal-I (36). As with Akt, the NFκB signaling axis is essential for cell survival (37), and, interestingly, Akt cross-talks with NFκB to confer protection against apoptosis (38, 39). NFκB directs the transcription of numerous molecules important for cell survival and proliferation (cIAP2), angiogenesis (IL-8), and inflammation (IL-6) (40). Our studies show that ST6Gal-I potentiates NFκB activation, leading to increased expression of cIAP2, IL-8, and IL-6. Of particular significance, cIAP2 is a potent inhibitor of apoptosis.
ST6Gal-I activity also promotes the expression of another major NFκB target, cyclin D2, a cell cycle gateway molecule that controls the transition between G1 and S phase. Cyclin D2 binds and activates cyclin-dependent kinases 4 and 6 (cdk4 and cdk6), and this complex then directs phosphorylation of pRb (29). pRb phosphorylation represses its normal function, which is to block progression into S phase. In accordance with ST6Gal-I-induced expression of cyclin D2, cells with high ST6Gal-I levels display increased phosphorylation of pRb and a corresponding enrichment in the proportion of cells that have entered S phase. These collective results point to a role for ST6Gal-I in enabling tumor cells to sustain proliferative capacity when challenged by a serum-depleted microenvironment. This concept is further supported by our finding that, after a 1-week exposure to serum deprivation, ST6Gal-I levels are elevated in the surviving population. We hypothesize that this reflects the selective survival and expansion of clonal variants with high ST6Gal-I expression.
The observation that ST6Gal-I protects cells against serum withdrawal adds to the growing body of literature suggesting that ST6Gal-I serves as a crucial tumor cell survival factor. ST6Gal-I expression is induced by oncogenes such as ras (41, 42) as well as the stem cell-associated transcription factor Sox2 (43). Up-regulation of ST6Gal-I in cancer cells functions to confer resistance to many antitumor treatments, including radiation (44) and the chemotherapeutic agents cisplatin (25), gemcitabine (16), and docetaxel (45). One likely mechanism by which ST6Gal-I fosters tumor cell survival is through imparting a CSC-like phenotype. CSCs are notoriously resistant to a multiplicity of death-inducing stimuli, including radiation, chemotherapy, and hypoxia (46). Relevant to this investigation, extensive literature has established that CSCs are better able to survive and proliferate under conditions of serum withdrawal than more differentiated tumor cells (47, 48). Reciprocally, long-term culture of cancer cells in serum-deficient medium is used as a method to isolate CSCs from a heterogeneous cell population (47).
Further studies will be needed to define the surface receptors that mediate ST6Gal-I-dependent resistance to serum deprivation. There are some established ST6Gal-I substrates that play fundamental roles in cell survival, including members of the integrin, death receptor, and growth factor receptor families. As an example, EGFR is known to be α2-6-sialylated, although, in this instance, sialylation appears to inhibit EGFR activation (14, 49). However, rather than a single, linear signaling pathway, it is possible that multiple differentially sialylated receptors act in concert to induce intracellular signaling events that confer the anti-apoptotic properties associated with CSCs. In essence, ST6Gal-I may function in an oncogene-like manner to coordinately regulate a select cohort of tumor cell receptors. The concept that certain glycosyltransferases could have an oncogene-like function has received limited attention despite the fact that an altered surface glycome was one of the earliest identified markers of a tumor cell. The results presented in this study provide an important advance by highlighting a new function for ST6Gal-I in promoting the viability of tumor cells exposed to the type of serum-depleted conditions often found in hypovascularized tumor microenvironments.
Experimental Procedures
Cell Culture
For routine propagation of cell lines, cells were grown in DMEM/F12 (OV4) or RPMI (BxPC3) medium containing 10% FBS and 1% antibiotic/antimycotic supplements. The antibiotic/antimycotic stock solution was purchased from Invitrogen and diluted 1:100 into medium. For studies of serum deprivation, cells were cultured for the indicated times using medium supplemented with either 10%, 1%, or 0% FBS. Stable polyclonal cell lines were created by transducing cells with a lentivirus encoding either the ST6Gal-I gene (Genecopoeia) or shRNA against ST6Gal-I (Sigma, TRCN00000035432, sequence CCGGCGTGTGCTACTACTACCAGAACTCGAGTTCTGGTAGTAGTAGCACACGTTTTTG), followed by selection with puromycin. ST6Gal-I overexpression or knockdown was verified by immunoblotting whole cell lysates with anti-ST6Gal-I goat polyclonal antibody (R&D Systems, AF5924) and by quantitative real-time PCR (Applied Biosystems). In addition, we conducted lectin staining to confirm differential ST6Gal-I activity. Cells were stained with either FITC-conjugated SNA lectin (EY Laboratories, F-6802-1), which is specific for α2-6 sialic acids, or FITC-MAA (EY Laboratories, F-7801-2), which binds to α2-3 sialic acids. Cells were stained for 40 min at 4 °C with a 1:200 dilution of SNA-FITC or a 1:80 dilution of MAA-FITC, and then binding of the lectin was quantified by flow cytometry.
Immunoblotting
Cells were serum-deprived for the indicated times and then lysed using radioimmune precipitation assay buffer supplemented with protease and phosphatase inhibitors (Sigma). Total protein concentration was measured by BCA (Pierce). Samples were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were incubated with 5% nonfat dry milk in TBS containing 0.1% Tween 20 (TBST). Immunoblots were probed with antibodies to ST6Gal-I (R&D Systems, AF5924), cIAP2 (Cell Signaling Technology, 3130, lot 6), pAkt (Ser-473, Cell Signaling Technology, 4060, lot 19), total Akt (Cell Signaling Technology, 4691, lot 20), AIF (Cell Signaling Technology, 4642, lot 3), Beclin (Cell Signaling Technology, 3495, lot 2), p-p70S6K (Thr-389, Cell Signaling Technology, 9234, lot 11), total p70S6K (Cell Signaling Technology, 2708, lot 7), pNFκB (p65, Ser-536, Cell Signaling Technology, 3033, lot 14), total NFκB (p65, Cell Signaling Technology, 8242, lot 4), cyclin D2 (Cell Signaling Technology, 3741, lot 4), p-pRb (Ser-809/811, Cell Signaling Technology, 8516, lot 4), or total pRb (Cell Signaling Technology, 9309, lot 9). Protein loading was verified using anti-β-actin (Abcam, ab20272) or anti-β-tubulin (Abcam, ab21058). Membranes were incubated with horseradish peroxidase-coupled secondary antibodies (Cell Signaling Technology), and protein was detected by enhanced chemiluminescence (Pierce). Immunoblotting for all targets was performed with at least three independently prepared cell lysates, and densitometric quantification of bands was accomplished using ImageJ software. All bands were normalized to their respective β-tubulin loading controls. Student's t test was employed to determine significance (p < 0.05).
qRT-PCR
RNA was extracted using the protocol for the RNeasy Plus mini kit (Qiagen). Total RNA concentration was measured, and cDNA was made using the SuperScript VILO cDNA synthesis kit (Thermo). qRT-PCR samples were prepared using TaqMan Fast Advanced Master Mix (Thermo). Primers for cIAP2 (Hs00985031_g1), IL-6 (Hs00174131_m1), IL-8 (Hs00174103_m1), and cyclin D2 (Hs00153380_m1) were acquired from Applied Biosystems. Data were normalized to GAPDH (Applied Biosystems, Hs02786624_gl), and significance was determined as p < 0.05 using Student's t test from at least four independent experiments, with each independent experiment performed in triplicate.
Flow Cytometry and Cell Cycle Analysis
S phase cells were measured using flow cytometry by following the protocol accompanying the Click-It EdU Alexa Fluor 488 flow cytometry assay kit (Thermo). Cells were incubated with 7.5 μm EdU for 2 h prior to cell labeling and analysis. For the EdU assays, OV4 cells were incubated in 0% FBS for 96 h, whereas BxPC3 cells were incubated in 1% serum for 24 h. These conditions were adopted because OV4 cells appear to have greater inherent resistance to serum deprivation.
Author Contributions
C. M. B. was responsible for the acquisition and analysis of the data, with oversight from S. L. B. K. A. D. aided with the collection and analysis of flow cytometry data. C. M. B. and S. L. B. were responsible for the concept and design of this study and together wrote the manuscript.
Acknowledgments
We thank the UAB Flow Cytometry Core Facility for assistance (supported by National Institutes of Health Grants P30AR048311 and P30AI027767).
This work was supported by National Institutes of Health Grants R01 GM111093 and R21 CA192629 (to S. L. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ST6Gal-I
- β-galactoside α-2,6-sialyltransferase 1
- EGFR
- EGF receptor
- CSC
- cancer stem cell
- OE
- overexpression
- EV
- empty vector
- KD
- knockdown
- AIF
- apoptosis-inducing factor
- Rb
- retinoblastoma protein
- qRT-PCR
- quantitative RT-PCR
- SNA
- Sambucus nigra agglutinin
- MAA
- Maackia amurensis agglutinin
- EdU
- 5-ethynyl-2′-deoxyuridine.
References
- 1. Munkley J., and Elliott D. J. (2016) Hallmarks of glycosylation in cancer. Oncotarget 7, 35478–35489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takahashi M., Kizuka Y., Ohtsubo K., Gu J., and Taniguchi N. (2016) Disease-associated glycans on cell surface proteins. Mol. Aspects Med. 51, 56–70 [DOI] [PubMed] [Google Scholar]
- 3. Stowell S. R., Ju T., and Cummings R. D. (2015) Protein glycosylation in cancer. Annu. Rev. Pathol. 10, 473–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinho S. S., and Reis C. A. (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555 [DOI] [PubMed] [Google Scholar]
- 5. Lu J., and Gu J. (2015) Significance of β-galactoside α2,6 sialyltranferase 1 in cancers. Molecules 20, 7509–7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schultz M. J., Swindall A. F., and Bellis S. L. (2012) Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. 31, 501–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Büll C., Stoel M. A., den Brok M. H., and Adema G. J. (2014) Sialic acids sweeten a tumor's life. Cancer Res. 74, 3199–3204 [DOI] [PubMed] [Google Scholar]
- 8. Dall'Olio F., Malagolini N., Trinchera M., and Chiricolo M. (2014) Sialosignaling: sialyltransferases as engines of self-fueling loops in cancer progression. Biochim. Biophys. Acta 1840, 2752–2764 [DOI] [PubMed] [Google Scholar]
- 9. Seales E. C., Jurado G. A., Brunson B. A., Wakefield J. K., Frost A. R., and Bellis S. L. (2005) Hypersialylation of β1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res. 65, 4645–4652 [DOI] [PubMed] [Google Scholar]
- 10. Hou S., Hang Q., Isaji T., Lu J., Fukuda T., and Gu J. (2016) Importance of membrane-proximal N-glycosylation on integrin β1 in its activation and complex formation. FASEB J. 30, 4120–4131 [DOI] [PubMed] [Google Scholar]
- 11. Ip C. K., Yung S., Chan T. M., Tsao S. W., and Wong A. S. (2014) p70 S6 kinase drives ovarian cancer metastasis through multicellular spheroid-peritoneum interaction and P-cadherin/β1 integrin signaling activation. Oncotarget 5, 9133–9149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swindall A. F., and Bellis S. L. (2011) Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 286, 22982–22990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Z., Swindall A. F., Kesterson R. A., Schoeb T. R., Bullard D. C., and Bellis S. L. (2011) ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 286, 39654–39662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park J. J., Yi J. Y., Jin Y. B., Lee Y. J., Lee J. S., Lee Y. S., Ko Y. G., and Lee M. (2012) Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem. Pharmacol. 83, 849–857 [DOI] [PubMed] [Google Scholar]
- 15. Swindall A. F., Londoño-Joshi A. I., Schultz M. J., Fineberg N., Buchsbaum D. J., and Bellis S. L. (2013) ST6Gal-I protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res. 73, 2368–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schultz M. J., Holdbrooks A. T., Chakraborty A., Grizzle W. E., Landen C. N., Buchsbaum D. J., Conner M. G., Arend R. C., Yoon K. J., Klug C. A., Bullard D. C., Kesterson R. A., Oliver P. G., O'Connor A. K., Yoder B. K., and Bellis S. L. (2016) The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 76, 3978–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lise M., Belluco C., Perera S. P., Patel R., Thomas P., and Ganguly A. (2000) Clinical correlations of α2,6-sialyltransferase expression in colorectal cancer patients. Hybridoma 19, 281–286 [DOI] [PubMed] [Google Scholar]
- 18. Wang Y. C., Stein J. W., Lynch C. L., Tran H. T., Lee C. Y., Coleman R., Hatch A., Antontsev V. G., Chy H. S., O'Brien C. M., Murthy S. K., Laslett A. L., Peterson S. E., and Loring J. F. (2015) Glycosyltransferase ST6GAL1 contributes to the regulation of pluripotency in human pluripotent stem cells. Sci. Rep. 5, 13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tateno H., Toyota M., Saito S., Onuma Y., Ito Y., Hiemori K., Fukumura M., Matsushima A., Nakanishi M., Ohnuma K., Akutsu H., Umezawa A., Horimoto K., Hirabayashi J., and Asashima M. (2011) Glycome diagnosis of human induced pluripotent stem cells using lectin microarray. J. Biol. Chem. 286, 20345–20353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nairn A. V., Aoki K., dela Rosa M., Porterfield M., Lim J. M., Kulik M., Pierce J. M., Wells L., Dalton S., Tiemeyer M., and Moremen K. W. (2012) Regulation of glycan structures in murine embryonic stem cells: combined transcript profiling of glycan-related genes and glycan structural analysis. J. Biol. Chem. 287, 37835–37856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhuo Y., Chammas R., and Bellis S. L. (2008) Sialylation of β1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J. Biol. Chem. 283, 22177–22185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukumori T., Takenaka Y., Yoshii T., Kim H. R., Hogan V., Inohara H., Kagawa S., and Raz A. (2003) CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 63, 8302–8311 [PubMed] [Google Scholar]
- 23. Amano M., Galvan M., He J., and Baum L. G. (2003) The ST6Gal I sialyltransferase selectively modifies N-glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J. Biol. Chem. 278, 7469–7475 [DOI] [PubMed] [Google Scholar]
- 24. Kitazume S., Imamaki R., Ogawa K., Komi Y., Futakawa S., Kojima S., Hashimoto Y., Marth J. D., Paulson J. C., and Taniguchi N. (2010) α2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J. Biol. Chem. 285, 6515–6521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schultz M. J., Swindall A. F., Wright J. W., Sztul E. S., Landen C. N., and Bellis S. L. (2013) ST6Gal-I sialyltransferase confers cisplatin resistance in ovarian tumor cells. J. Ovarian Res. 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berven L. A., and Crouch M. F. (2000) Cellular function of p70S6K: a role in regulating cell motility. Immunol. Cell Biol. 78, 447–451 [DOI] [PubMed] [Google Scholar]
- 27. Pearson R. B., and Thomas G. (1995) Regulation of p70s6k/p85s6k and its role in the cell cycle. Prog. Cell Cycle Res. 1, 21–32 [DOI] [PubMed] [Google Scholar]
- 28. Piva R., Belardo G., and Santoro M. G. (2006) NF-κB: a stress-regulated switch for cell survival. Antioxid. Redox. Signal 8, 478–486 [DOI] [PubMed] [Google Scholar]
- 29. Foster D. A., Yellen P., Xu L., and Saqcena M. (2010) Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1, 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 31. Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M. R., Carotenuto A., De Feo G., Caponigro F., and Salomon D. S. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 [DOI] [PubMed] [Google Scholar]
- 32. Regad T. (2015) Targeting RTK signaling pathways in cancer. Cancers 7, 1758–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eves E. M., Xiong W., Bellacosa A., Kennedy S. G., Tsichlis P. N., Rosner M. R., and Hay N. (1998) Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol. Cell Biol. 18, 2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clark A. S., West K., Streicher S., and Dennis P. A. (2002) Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 1, 707–717 [PubMed] [Google Scholar]
- 35. Kennedy S. G., Wagner A. J., Conzen S. D., Jordán J., Bellacosa A., Tsichlis P. N., and Hay N. (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11, 701–713 [DOI] [PubMed] [Google Scholar]
- 36. Lu J., Isaji T., Im S., Fukuda T., Hashii N., Takakura D., Kawasaki N., and Gu J. (2014) β-Galactoside α2,6-sialyltranferase 1 promotes transforming growth factor-β-mediated epithelial-mesenchymal transition. J. Biol. Chem. 289, 34627–34641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karin M., and Lin A. (2002) NF-κB at the crossroads of life and death. Nat. Immunol. 3, 221–227 [DOI] [PubMed] [Google Scholar]
- 38. Hussain A. R., Ahmed S. O., Ahmed M., Khan O. S., Al Abdulmohsen S., Platanias L. C., Al-Kuraya K. S., and Uddin S. (2012) Cross-talk between NFκB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS ONE 7, e39945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dan H. C., Cooper M. J., Cogswell P. C., Duncan J. A., Ting J. P., and Baldwin A. S. (2008) Akt-dependent regulation of NFκB is controlled by mTOR and Raptor in association with IκK. Genes Dev. 22, 1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baud V., and Karin M. (2009) Is NFκB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seales E. C., Jurado G. A., Singhal A., and Bellis S. L. (2003) Ras oncogene directs expression of a differentially sialylated, functionally altered β1 integrin. Oncogene 22, 7137–7145 [DOI] [PubMed] [Google Scholar]
- 42. Dalziel M., Dall'Olio F., Mungul A., Piller V., and Piller F. (2004) Ras oncogene induces β-galactoside α2,6-sialyltransferase (ST6Gal I) via a RalGEF-mediated signal to its housekeeping promoter. Eur. J. Biochem. 271, 3623–3634 [DOI] [PubMed] [Google Scholar]
- 43. Boumahdi S., Driessens G., Lapouge G., Rorive S., Nassar D., Le Mercier M., Delatte B., Caauwe A., Lenglez S., Nkusi E., Brohée S., Salmon I., Dubois C., del Marmol V., Fuks F., et al. (2014) SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250 [DOI] [PubMed] [Google Scholar]
- 44. Lee M., Park J. J., and Lee Y. S. (2010) Adhesion of ST6Gal I-mediated human colon cancer cells to fibronectin contributes to cell survival by integrin β1-mediated paxillin and AKT activation. Oncol. Rep. 23, 757–761 [PubMed] [Google Scholar]
- 45. Chen X., Wang L., Zhao Y., Yuan S., Wu Q., Zhu X., Niang B., Wang S., and Zhang J. (2016) ST6Gal-I modulates docetaxel sensitivity in human hepatocarcinoma cells via the p38 MAPK/caspase pathway. Oncotarget 7, 51955–51964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beck B., and Blanpain C. (2013) Unravelling cancer stem cell potential. Nat. Rev. Cancer 13, 727–738 [DOI] [PubMed] [Google Scholar]
- 47. Tavaluc R. T., Hart L. S., Dicker D. T., and El-Deiry W. S. (2007) Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle 6, 2554–2562 [DOI] [PubMed] [Google Scholar]
- 48. Lin S. P., Lee Y. T., Wang J. Y., Miller S. A., Chiou S. H., Hung M. C., and Hung S. C. (2012) Survival of cancer stem cells under hypoxia and serum depletion via decrease in PP2A activity and activation of p38-MAPKAPK2-Hsp27. PLoS ONE 7, e49605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu Y. C., Yen H. Y., Chen C. Y., Chen C. H., Cheng P. F., Juan Y. H., Chen C. H., Khoo K. H., Yu C. J., Yang P. C., Hsu T. L., and Wong C. H. (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. 108, 11332–11337 [DOI] [PMC free article] [PubMed] [Google Scholar]