

Abstract
Caspases are cysteine aspartate proteases that are major players in key cellular processes, including apoptosis and inflammation. Specifically, caspase-6 has also been implicated in playing a unique and critical role in neurodegeneration; however, structural similarities between caspase-6 and other caspase active sites have hampered precise targeting of caspase-6. All caspases can exist in a canonical conformation, in which the substrate binds atop a β-strand platform in the 130's region. This caspase-6 region can also adopt a helical conformation that has not been seen in any other caspases. Understanding the dynamics and interconversion between the helical and strand conformations in caspase-6 is critical to fully assess its unique function and regulation. Here, hydrogen/deuterium exchange mass spectrometry indicated that caspase-6 is inherently and dramatically more conformationally dynamic than closely related caspase-7. In contrast to caspase-7, which rests constitutively in the strand conformation before and after substrate binding, the hydrogen/deuterium exchange data in the L2′ and 130's regions suggested that before substrate binding, caspase-6 exists in a dynamic equilibrium between the helix and strand conformations. Caspase-6 transitions exclusively to the canonical strand conformation only upon substrate binding. Glu-135, which showed noticeably different calculated pKa values in the helix and strand conformations, appears to play a key role in the interconversion between the helix and strand conformations. Because caspase-6 has roles in several neurodegenerative diseases, exploiting the unique structural features and conformational changes identified here may provide new avenues for regulating specific caspase-6 functions for therapeutic purposes.
Keywords: apoptosis, cysteine protease, hydrogen exchange mass spectrometry, molecular dynamics, neurodegeneration
Introduction
Caspases are cysteine proteases that recognize aspartate-containing substrates and are major players in key cellular processes, including apoptosis and inflammation. Caspase active sites contain a Cys-His dyad required for cleavage of peptide bonds adjacent to aspartate residues in select protein substrates. There are two main classes of caspases, initiator and executioner caspases, classified based on their cellular function and domain organization. Initiator caspases (caspase-2, -8, and -9) function upstream in the apoptotic pathway and activate the downstream executioner caspases (caspase-3, -6, and -7) by proteolytic cleavage at an intersubunit linker between large and small subunits. Executioner caspases then cleave a select group of protein targets to promote apoptosis. Initiator caspases generally exist as monomers and are subsequently activated by dimerization mediated by interaction with a molecular platform (e.g. apoptosome) (1). In contrast, executioner caspases exist as dimeric inactive zymogens and upon activation by limited proteolysis become functional (2). Significantly, classification of caspase-6 as an executioner has not been unequivocal. Caspase-6 is weakly apoptotic, although overexpression of caspase-6 in mammalian cells does result in apoptosis (3). As an executioner, caspase-6 has been identified as the only caspase that cleaves the nuclear lamellar protein, lamin A/C, during apoptosis (4–6). Caspase-6 has been shown to cleave caspase-8 (7) and be activated by caspase-3 (8) and caspase-1 (9), suggesting that caspase-6 assumes simultaneous roles as an executioner, initiator, and inflammatory caspase.
Unlike other executioner caspases, which necessitate processing by initiator caspases for activation (10), caspase-6 is capable of self-cleavage and activation (11, 12). Although self-cleavage does not lead to apoptosis, this caspase-6 self-cleavage may account for some nonapoptotic roles of caspase-6, including axonal pruning in development (13–16) and in adult brains (17) as well as in B cell activation and differentiation (18). In the context of neurodegeneration, caspase-6 cleaves a compelling set of neuronal substrates, including microtubule-associated protein Tau (19), amyloid precursor protein (20), presenilin I and II (20), polyglutamine-expanded and native huntingtin protein (21), and Parkinson disease protein 7 (PARK7), also known as protein deglycase DJ-1 (22). Caspase-6 is considered a promising molecular target for neurodegeneration treatments because cleavage of these neuronal substrates plays key roles in the pathophysiological outcome in Alzheimer's (23–33), Huntington's (21, 34–40), and Parkinson's diseases (22). Thus, a full understanding of caspase-6 structure and its relation to function is central to achieving caspase-6-specific regulation in neurodegeneration.
Perhaps the most distinctive feature of caspase-6 is that it is the only caspase that can adopt two very different structural conformations. These two conformations stem from structural changes in the 130's region, which generates the platform upon which substrate binds (Fig. 1). Structures of the procaspase-6 zymogen, in which the intersubunit linker occupies and orients the active site loops (12, 41), and mature caspase-6 bound to a substrate-like inhibitor (12, 42, 43) are similar to other caspases and show the canonical strand conformation. These structures underscore the observation that caspase-6 is fully capable of adopting the canonical caspase active site conformation. The mature unliganded caspase-6 is also capable of adopting a distinctive and noncanonical conformation, with extended helices in the 60's region (residues 57–70) and 130's region (residues 125–142) (44, 45). These conformational changes are also accompanied by a 21° outward rotation of the 90's helix (Fig. 1). This helical conformation of unliganded caspase-6 is not seen in crystal structures of other caspases. In fact, the helical form is disallowed in all other caspases by the presence of helix-breaking residues in this region (45, 46). In caspase-6, when the 130's region is in the extended helical conformation, the Cys-His catalytic dyad, whose reactive centers must be within 1.8–3.5 Å of one another to achieve catalysis (47), are pushed away from each other by 9 Å. In addition, the top of the 130's helix prevents substrate binding. As a consequence, caspase-6 would be catalytically inactive if locked in this conformation. For caspase-6 to accommodate a substrate in the active site, a conformational rearrangement has to occur in the 60's, 90's, and 130's helices and loops 1–4. This transition is thought to occur through a low stability/high energy intermediate strand-containing state in the 130's region (46).
FIGURE 1.

Caspase-6 undergoes helix-strand transition upon substrate binding. The overall fold of canonical caspases before and after substrate binding is represented by the superimposition of the unliganded (orange; PDB code 1K86) and the peptide-based substrate mimic DEVD-bound (green; PDB code 1F1J) structures of caspase-7 (middle). Highlighted regions are the active site cysteine (blue), the dimer interface, and the substrate binding loops 1–4 (L1–L4). Caspase-7, like all other caspases, adopts a canonical strand conformation in its 130's region in both the unliganded state (orange; PDB code 1K86) and the peptide-based substrate mimic DEVD-bound (green; PDB code 1F1J) states (right). In contrast, caspase-6 can adopt a noncanonical extended helical conformation in its 130's region in the unliganded state (orange, PDB code 2WDP) but recovers the canonical strand conformation upon binding to a peptide-based substrate mimic VEID-aldehyde (green; PDB code 3OD5) (left).
Unliganded caspase-6 has also been crystallized in the canonical strand conformation with a more properly positioned Cys-His dyad (separated only by 3.6 Å) capable of binding substrate (48). This structure of the mature unliganded form of caspase-6 is similar to all other caspases. Thus, it is clear that caspase-6 is capable of adopting both helical and strand conformation of the 130's region in the absence of substrate. These structures underscore the remaining question: What is the physiologically relevant structural ensemble for unliganded caspase-6? The goal of this work is to understand the relative populations of the helical and strand conformations of mature caspase-6 before substrate binding.
The crystal structures of caspase-6 are static molecular snapshots. A complementary study to depict temporal dynamic changes is needed to define the conformational ensemble of caspase-6 before substrate binding. Hydrogen/deuterium exchange mass spectrometry (H/DX-MS)2 is a powerful technique to study protein conformation and dynamics (for a review, see Ref. 49) and has been applied to several systems, including protein-ligand complexes (50), chaperones (51), amyloid fibrils (52), viral proteins (53), antibody-drug conjugates (54), and even peripheral (55) and transmembrane (56) proteins. H/DX-MS reports the extent of hydrogen bonding and relative solvent exposure of the backbone amide hydrogens and is well suited for extrapolating different protein conformations (57).
In this study, H/DX-MS, combined with molecular dynamics simulations, was used to probe the distinct conformational dynamics in caspase-6 relative to caspase-7 upon peptide-based substrate binding. Caspase-7 serves as an ideal control, possessing the same overall fold, but maintaining constitutively the strand form before and after substrate binding. Our results show that caspase-6 displays unique backbone dynamics in the 130's region compared with caspase-7, consistent with the helix-strand interconversion upon substrate binding. Moreover, the protonation of Glu-135 in this region was found to be an important contributor to the stability of the helical state of unliganded caspase-6. Exploiting these unique structural features may provide new avenues for regulating specific caspase-6 functions.
Results
Overall H/DX-MS Profiles of Caspase-6 and Caspase-7
To explore the differences in the backbone dynamics and conformational flexibility of caspases, caspase-6 and caspase-7 were profiled using H/DX-MS in two states: mature unliganded and bound to a substrate-like inhibitor. The substrate-bound states were achieved by binding inhibitors built from the tetrapeptide recognition substrates, VEID-aldehyde for caspase-6 and DEVD-aldehyde for caspase-7. The H/D exchange rate of the backbone amide hydrogen with deuterium in the solvent is interpreted as reporting the local fluctuations of the conformational states of proteins (58). Thus, fast rates of H/D exchange indicate higher solvent accessibility and flexibility of associated protein region, whereas those regions that show slow rates of H/D exchange are characterized as being buried and rigid (57). Peptide heat maps show the extent of the relative deuterium uptake of the peptic peptides of caspase-6 and caspase-7 in both the unliganded and inhibitor-bound forms after being subjected to between 10 s and 2 h of H/D exchange (Fig. 2). The corresponding relative deuterium uptake profiles were mapped onto the crystal structures of caspase-6 (PDB code 2WDP) and caspase-7 (PDB code 1K86) (supplemental Fig. S1). The percentage difference in the relative deuterium uptake of the unliganded and inhibitor-bound states of caspase-6 and caspase-7 were also mapped onto the linear amino acid sequences (supplemental Fig. S2) and onto the corresponding crystal structures (supplemental Fig. S3). All deuterium uptake plots for both unliganded and bound states of caspase-7 and caspase-6 were also prepared (supplemental Figs. S4 and S5). Overall, peptides covering 91 and 93% of the linear sequences of caspase-6 and caspase-7, respectively, were observed (supplemental Fig. S6). Approximately 70% of the backbone amide hydrogens in both caspase-6 and caspase-7 underwent less than 30% H/D exchange within 10 s, suggesting well folded and dynamically stable proteins overall. In contrast, the other 30% of the backbone amide hydrogens were rapidly deuterated. These rapidly deuterated regions are predominantly observed in the highly flexible regions, including the loop bundles (L2, L2′, L3, and L4), which form the substrate-binding groove of caspases (Fig. 1). The reproducibility of the H/DX-MS experiment was extremely high across the majority of the sequence, and even small differences in the susceptibility to H/D exchange between the unliganded and liganded states of caspases could be accurately quantified, facilitating identification of regions in which H/D exchange was altered in the presence of a peptide-based substrate mimic (supplemental Figs. S4 and S5).
FIGURE 2.

H/D exchange heat map of the relative deuterium incorporation. For each peptic peptide of the unliganded and the peptide-based substrate mimic-bound states of caspase-7 (A) and caspase-6 (B), the percentage relative deuterium level for each H/D exchange incubation time (0.17, 1, 10, 60, and 120 min) is mapped onto its corresponding linear sequence. The percentage relative deuterium incorporation is calculated by dividing the observed deuterium uptake by the theoretical maximum deuterium uptake for each peptide. The H/DX-MS experiments followed 64 peptides common to both unliganded and DEVD-bound caspase-7 that covers 93% of the linear sequence. Likewise, H/DX-MS experiments followed 70 peptides common to both unliganded and VEID-bound caspase-6 that covers 91% of the linear sequence. Peptic peptides with no H/D exchange data at any given incubation time are colored white. All caspase-6 and caspase-7 variants used in the H/DX-MS experiments were cleaved, active forms lacking both the prodomain (residues 1–23 in both caspase-6 and caspase-7) and linker (residues 180–193 in caspase-6; residues 199–206 in caspase-7). The secondary structural elements are also shown above the caspase-6 and caspase-7 sequences. The percentage relative deuterium level of each peptic peptide represents the average values of duplicate experiments performed on two separate days.
Caspase-7 Adopts the Canonical Strand Conformation before and after Substrate Binding
Caspase-7 serves as an ideal control for observing conformational changes and dynamics in a canonical caspase before and after binding of a substrate-like inhibitor. All crystal structures of caspase-7 to date show the canonical (130's strand) structure before and after binding of a substrate-like peptide-based inhibitor, DEVD (Fig. 1). During H/DX-MS on caspase-7, only slight changes in the overall H/D exchange occurred upon binding of the substrate-like inhibitor (DEVD) (Fig. 2A). The percentage of relative deuterium incorporation is calculated by dividing the observed deuterium uptake by the theoretical maximum deuterium uptake for each peptide. As expected, the most significant differences in the deuterium uptake in the unliganded versus the DEVD-bound states mapped onto peptides from within the active site loop 2 (L2, peptides 181–191 and 182–192). There were no significant changes in the deuterium uptake between the unliganded and the DEVD-bound states in the 60's and 90's helices of caspase-7, which were covered by peptides 85–97 and 117–136, respectively (Fig. 3, A, C, and D). The 130's is the region that undergoes a helix-strand interconversion in caspase-6. The 130's region comprises residues 125–142 for caspase-6 and 148–165 for caspase-7. In caspase-7, the corresponding 130's region was covered by five peptides: 148–161, 148–166, 150–166, 151–166, and 152–166. In particular, peptide 150–166 within the 130's region of caspase-7 (Fig. 3, A and C, and supplemental Fig. S7A) showed changes in deuterium incorporation that are observable from the earliest time point and persisted throughout the deuterium labeling time with almost the same difference between the unliganded and the DEVD-bound states. The shape of this deuterium incorporation curve indicates (59) that one rapidly exchanging amide hydrogen at Ile-150 and one at another residue between 152 and 166 are deuterated before the earliest time point of the H/D exchange experiment (Fig. 3C). Nevertheless, these data showed only a very slight difference (2–6%) in the deuterium uptake in the unliganded versus the DEVD-bound state (supplemental Fig. S2A), suggesting that the 130's region of caspase-7 is partially protected and moderately dynamic but that binding of DEVD does not impact the overall structure. The minor changes in conformational dynamics in the 60's and 130's regions with virtually no changes in the 90's helix of caspase-7 in the unliganded versus the DEVD-bound states are consistent with the reported invariantly canonical strand structures of caspase-7 (10, 60) (Fig. 1). Importantly, this demonstrates that the magnitude of change in the theoretical maximum deuterium uptake expected to occur directly from substrate-like inhibitor binding to a caspase active site should be in the range of 2–6%, if not accompanied by changes in protein conformational states or their dynamics.
FIGURE 3.

Caspase-6 shows distinctive conformational dynamics in its 130's region. Shown is the difference in deuterium uptake (Da) of the corresponding peptic peptides in the unliganded and the peptide-based substrate mimic-bound states of caspase-7 (A) and caspase-6 (B) at the indicated time points of exposure to deuterium in solution. The residue numbers for each peptic peptide are listed with corresponding secondary structural elements. For these data, a deuterium uptake difference of >0.6 Da is considered significant at a 98% confidence interval. The intensity of the blue color represents the peptides that undergo a significant decrease in H/D exchange (less solvent-exposed, less flexible) upon peptide-based substrate mimic binding. The intensity of the red color represents the peptides that undergo significant increase in H/D exchange (more solvent-exposed, more flexible) upon peptide-based substrate mimic binding. C, representative deuterium incorporation plots for peptic peptides covering the 60's, 90's, and 130's regions of caspase-7 (top) and caspase-6 (bottom) in both unliganded (black lines) and peptide-based substrate mimic-bound (blue lines) states. The representative MS spectra of the highlighted peptic peptides are shown in supplemental Fig. S7. Error bars, S.D. of duplicate H/DX-MS measurements done on two separate days. The residue numbering is listed for the homologous regions in caspase-7 and caspase-6, which have different numbering for the structurally homologous regions due to differences in the lengths of their respective subunits. D, difference in deuterium uptake between the unliganded and the DEVD-bound states of caspase-7 after 2-h incubation mapped onto the structure of caspase-7 (PDB code 1K86) shown in both ribbon and surface representations. E, difference in deuterium uptake between the unliganded and the VEID-bound states of caspase-6 after 2-h incubation mapped onto the structure of caspase-6 (PDB code 2WDP) shown in both ribbon and surface representations.
Caspase-6 Shows Different H/D Exchange Dynamics in the 130's Region Compared with Caspase-7
In terms of conformational flexibility, caspase-6 might be expected to differ from all other caspases. Caspase-6 has been observed to exist in both the canonical (strand) or helical conformations in the 130's region (Fig. 1), suggesting that interchange between two conformations could be involved in its function. We anticipated that H/DX-MS would allow identification of regions of caspase-6 that undergo significant conformational interchange, particularly in the native state, and are therefore more highly susceptible to H/D exchange. H/DX-MS on caspase-6 in the unliganded state or bound to an inhibitor built from the cognate caspase-6 tetrapeptide substrate (VEID-aldehyde) revealed dramatic changes in the deuterium uptake levels in several regions of the protein (Fig. 3, B and E). H/D exchange in the substrate-binding loops (L2, L2′, L3, and L4), which are engaged in accommodating the tetrapeptide in the active site, significantly decreased by 12–30% upon VEID binding (Fig. 3B and supplemental Fig. S2B). In particular, the L2′ region of caspase-6 is the most exchangeable region of the protein before VEID binding but becomes much less susceptible to exchange after binding. This is in contrast to caspase-7. In the canonical conformation of both caspase-6 and -7, L2′ is engaged in interactions with L2. In caspase-7, L2′ does not show nearly the magnitude in changes in H/D exchange that L2′ shows in caspase-6. In the helical form of caspase-6, L2′ is not engaged in any stabilizing interactions (supplemental Fig. S7B). Thus, H/D exchange is consistent with the model of L2′ being exposed in caspase-6 before VEID binding, as is observed in the helical conformation.
In addition to significant changes in L2′, H/D exchange in the 130's region (peptide 119–130) significantly decreased by 12–16% upon VEID binding (Fig. 3 (B and C) and supplemental Fig. S2B), which suggests that this region also becomes substantially more ordered when substrate binds. The deuterium uptake profile of caspase-6 peptide 119–130 (Fig. 3C) showed that in the absence of VEID binding, there is significant protection at earlier H/D exchange time points, but it takes up deuterium over the course of the H/D exchange incubation time. The shape of this deuterium uptake curve indicates (59) that this region of unliganded caspase-6 visits the H/D exchange-competent state much more frequently than after VEID binding. This suggests that upon substrate binding, this region becomes less dynamic. VEID- or DEVD-bound caspase-6 (12, 61) is in the same canonical conformation as DEVD-bound caspase-7 (60). The dispersed and significant differences in H/D exchange that are observed for caspase-6 (Fig. 3, B and E, and supplemental Fig. S2B) indicate that there are conformational differences that cannot be explained just by the protection from substrate binding changes. If unliganded caspase-6 rested in the canonical strand conformation, we would expect to see changes in the H/D exchange for unliganded versus peptide-based substrate mimic-bound similar to those for caspase-7 (Fig. 3). If caspase-6 rested exclusively in the helical form, we would also expect to see relatively small changes in deuterium uptake because residues in a stable helix should be resistant to H/D exchange. Only during the transient unfolding of either the strand or helix conformations during interconversion of the 130's region would we expect increased susceptibility to H/D exchange. Moreover, the total percentage of H/D exchange was the same between caspase-6 and caspase-7 (Table 1), making it possible to conclude that observed local differences in H/D exchange behavior reflect bona fide conformational differences between caspase-6 and -7. The much more significant changes in susceptibility to H/D exchange that we observed for caspase-6 in both the L2′ and the 130's regions are consistent with the model that in the unliganded form caspase-6 is in a dynamic ensemble with states intermediate between the helical and strand conformations.
TABLE 1.
Percentage of total amide hydrogen exchanged with deuterium
The number of amide hydrogens exchanged in 2 h, relative to the total number of exchangeable amide hydrogens in each peptide and corrected for percentage of sequence coverage achieved in each analysis is listed.
| Unliganded (%) | Ligand-bound (%) | |
|---|---|---|
| Caspase-6 | 22.4 | 18.0 |
| Caspase-7 | 23.3 | 21.6 |
Local pKa Values of Key Amino Acid Residues within the 130's Region Vary between the Unliganded (Helical) and the VEID-bound (Strand) States of Caspase-6
The H/D exchange data suggest that caspase-6 exists in a dynamic equilibrium that transitions between the helical and canonical strand conformations, but the molecular basis of this interconversion was not defined. Inspection of the 130's region in both unliganded and VEID-bound states of caspase-6 revealed an altered set of interactions among key residues in this region (Fig. 4A). In particular, residues His-52, Glu-63, Glu-135, and Glu-221 have distinct molecular contacts in the unliganded (helical) compared with the VEID-bound (strand) conformations. For example, His-52 makes hydrogen-bonding contacts with Asp-90, Tyr-128, and Glu-135 in the helical state but loses these contacts upon its transition to strand state upon substrate binding. The change in the microenvironment of these residues is reflected in the changes in their calculated microenvironment pKa values between the helical and strand conformations (Fig. 4B). The difference in the pKa values ranges from 0.9 to 6.6 pH units and generally transitions from a basic to a more acidic microenvironment following helix-strand interconversion. In fact, the helical structures of caspase-6 were all trapped during low pH crystallization (44, 45), whereas the strand conformation of unliganded caspase-6 was trapped during neutral pH crystallization (48). The H/D exchange reported here was performed at neutral pH but strongly suggests that caspase-6 is constantly undergoing an interconversion between the helical and strand states even at neutral pH. On the basis of the local pKa changes between the two-state conformations of caspase-6, we proposed that the protonation of key residues within the 130's region would impact the dynamics of this region.
FIGURE 4.

Protonation state of Glu-135 is critical to the dynamics of the 130's region. A, the unliganded (orange) and VEID-bound (green) states of caspase-6 present different local charge states of four identified residues in close proximity and within the 130's region, which are deprotonated upon substrate binding (shown in black). B, calculated local pKa values of the four amino acid residues that undergo differential protonation states between the unliganded (helical) and the VEID-bound (strand) states of caspase-6. C, plot of RMSF (Å) of the backbone atoms of each amino acid in caspase-6. A 1.2-ns-long simulated annealing-based molecular dynamics simulation was performed in all capase-6 protonation variants. The unliganded helical protonation variant represents the control ensemble of the triply protonated state (HisH-52, GluH-135, GluH-221) of caspase-6. The VEID-bound (strand) protonation variant represents the control ensemble of the triply deprotonated state (His-52, Glu-135, Glu-221) of caspase-6. Unliganded with deprotonated E135, protonation variant of caspase-6 that has deprotonated Glu-135 but remains protonated at the other two residues (HisH-52 and GluH-221). Residue-by-residue fluctuations are shown for caspase-6 in the unliganded (helical) state (black), VEID-bound (strand) state (red), and the unliganded caspase-6 with deprotonated Glu-135 (blue). The RMSF profile of unliganded caspase-6 with deprotonated Glu-135 behaves similarly to the VEID-bound (strand) state in the 130's region (inset). D, variation of the RMSF (Å) along key regions of caspase-6 mapped onto the structure of unliganded caspase-6 (PDB code 2WDP) highlighting only the 60's, 90's, and 130's regions.
Protonation State of Glu-135 Is Key to Conformational Changes in the Caspase-6 130's Region
We performed 1.2-ns-long simulated annealing-based molecular dynamics simulations on the unliganded (PDB code 2WDP) and VEID-bound (PBD code 3OD5) caspase-6 to determine the key factors that contribute to the helix-strand transition. In these studies, protonation states of His-52, Glu-135, and Glu-221 were varied from fully protonated (optimal for the unliganded structure) to fully deprotonated (optimal for the VEID-bound structure). A simulation plot of the annealing studies (Fig. 4C) shows pronounced difference in the root mean square fluctuation (RMSF) profile between the control ensembles of the triply protonated and triply deprotonated states, particularly in the amino acid residues within the 130's region (inset). Altering the protonation states of His-52 and Glu-221 did not result in significant changes in the RMSF profile compared with the control ensembles of caspase-6. Significantly, the protonation state of a single residue, Glu-135, appeared to control the conformational dynamics of the 130's region, with Glu-135 and protonated Glu (GluH)-135 reproducing to a large extent the RMSF profiles of triply deprotonated and triply protonated states, respectively. This observation suggests that the protonation state of Glu-135 is critical to the transition between the unliganded helical and the canonical strand conformations of caspase-6. Mapping the displacement observed during the molecular dynamics simulations onto the structure of caspase-6 underscores the propensity of the 130's helix to undergo a structural transition (Fig. 4D). This transition could be significantly facilitated by the deprotonation of a single residue, Glu-135, in the helical state. On the other hand, when caspase-6 in the canonical strand conformation with deprotonated Glu-135 transitions to the helical state, the dramatic change in the microenvironment should strongly favor protonation, which would be easier to achieve at a lower pH. Once protonated, the helical conformation is stabilized. Thus, the protonation state of Glu-135 is probably one of the major contributing factors in stabilizing the helical form of caspase-6.
E135Q, a Mimic of Protonated Glu-135, Stabilizes Caspase-6
Because protonation of Glu-135 appears to be a critical factor for stabilizing the helical state of caspase-6, we generated a variant, E135Q, which maintains the size of Glu but mimics the protonated (GluH) state. The kinetic parameters of E135Q are similar to wild type (WT), suggesting that the E135Q substitution does not negatively impact caspase-6 in processing of a peptide-based substrate, N-acetyl-Val-Glu-Ile-Asp-(7-amino-4-methyl-coumarin) (VEID-AMC) (Fig. 5A). We expected that the E135Q mutation would have a more significant impact on basal caspase-6 function, if there were a large energy barrier to the interconversion between the helical and strand states. The activity profile of E135Q as a function of pH is also similar to that of WT caspase-6, suggesting that increasing the helical fraction does not prevent conversion to the strand form in the presence of substrate (Fig. 5B), suggesting a low energy barrier between the two conformational states in the presence of substrate. WT and E135Q did not show significant differences in their CD spectra, suggesting that the secondary structures are similar in the unliganded conformation (Fig. 6A). In contrast, E135Q showed a pH-dependent thermal denaturation profile distinct from that of WT caspase-6 (Fig. 6, B and E), consistent with the calculated fraction of Glu-135 protonated in the helical state (Fig. 4C). E135Q is more stable than WT caspase-6, particularly at pH 7.5 and 8.5 (Fig. 6E). Although this is a small stabilization, it is of the same magnitude as the stability increase when WT caspase-6 binds an active site ligand (45), suggesting that stabilization is relevant. At a lower pH, helical WT caspase-6 should be predominantly protonated at the Glu-135 position (Fig. 6C), and hydrogen-bonding interactions with neighboring residues Arg-54 and Glu-131 will be favored (Fig. 6D). Meanwhile, at a higher pH, deprotonation of Glu-135 is expected to occur, and a stronger negative charge of the side chain carboxylate group of Glu-135 will be enhanced, leading to electrostatic interactions as the dominant interaction with neighboring Arg-54 and His-52 in the vicinity. In addition, just 3.8 Å above Glu-135 sits the negatively charged Glu-131 residue, which will probably also be deprotonated at a higher pH, promoting electrostatic repulsion that should destabilize the helical structure in the 130's region. Conversely, the GluH-135-mimicking E135Q will probably engage in hydrogen bonding with neighboring residues Arg-54 and Glu-131 and avoid the helix-destabilizing electrostatic repulsion. At a more basic pH, the stabilizing effect of E135Q is more pronounced due to the deprotonation of Glu-135 in the helical state and the sustained hydrogen-bonding interaction with Arg-54 and Glu-131 that results in higher stability of the E135Q compared with caspase-6 WT. In summary, using E135Q as a surrogate for GluH-135 suggests that protonation of this residue leads to overall stabilization of caspase-6. This further suggests that a significant fraction of caspase-6 exists in the helical state, because deprotonation, which should favor conversion to the strand conformation, leads to a destabilization relative to the E135Q variant.
FIGURE 5.
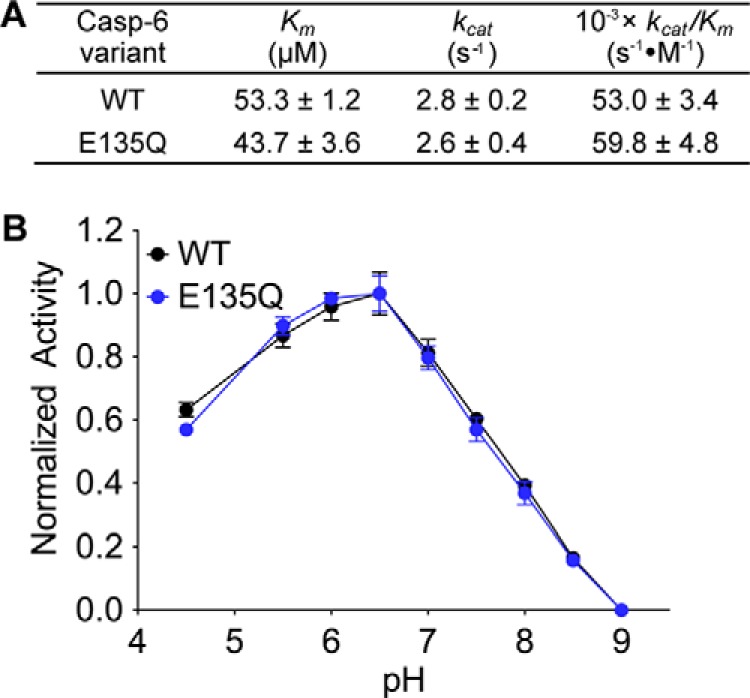
E135Q has kinetics and pH profile similar to those of WT caspase-6. A, the Michaelis-Menten kinetic parameters of caspase-6 WT and the “constantly protonated” variant, E135Q. The values are reported as mean ± S.E. of three independent trials performed on three separate days. B, the activity of caspase-6 WT (black line) and E135Q (blue line) variants as a function of pH. For normalization, the highest and the lowest relative fluorescence response for each data set was set to 100 and 0%, respectively, and reported as fractions. Error bars, S.D. of duplicate measurements on two separate days. All caspase-6 activity assays used fluorescence-based measurements following cleavage of a peptide-based substrate mimic, VEID-AMC, by caspase-6.
FIGURE 6.
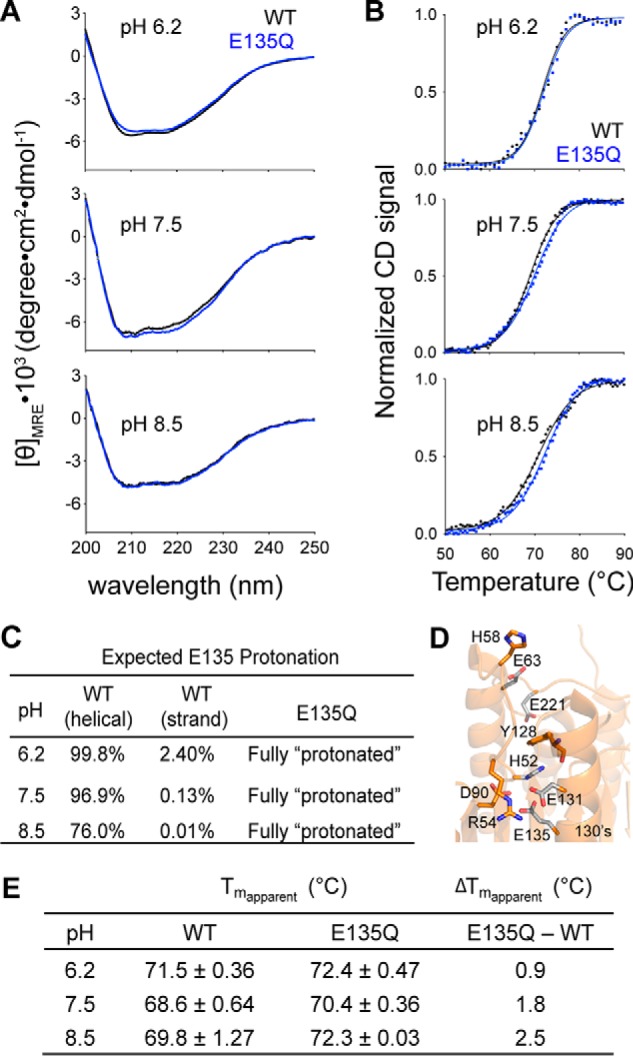
The stabilizing effect of constantly protonated E135Q variant is more pronounced at higher pH. A, the CD spectra of caspase-6 WT (black line) and the E135Q variant (blue line) measured at different pH levels. B, CD thermal denaturation profiles of caspase-6 WT (black dotted line) and E135Q (blue dotted line) at the indicated pH levels. Normalization of the CD signal was achieved by setting the highest and the lowest values for each data set as 100 and 0%, respectively. The thermal denaturation data were then fitted to the Boltzmann sigmoidal equation (black solid line in WT and blue solid line in E135Q), where the midpoint of the curve was determined to be the apparent Tm. C, the expected percentage protonation of Glu-135 at different pH was determined based on the calculated pKa values in Fig. 4B following the Henderson-Hasselbalch equation. D, interactions between Glu-135 and adjacent residues that impact the helix-strand interconversion. E, the individual apparent Tm values and the differences in the apparent Tm values of caspase-6 WT and E135Q at different pH are tabulated. The apparent Tm values are reported as mean ± S.D. of duplicate measurements on two independently prepared samples performed on two different days.
Discussion
This study reveals that caspase-6 has local backbone dynamics in L2′ and in the 130's region consistent with the helix-strand interconversion. These data showing a helix-strand interconversion are in contrast to previous work on unliganded caspase-6 determined at the physiological pH 7.4, which showed a canonical strand structure in the 130's region of unliganded caspase-6. The authors concluded that the earlier two helical structures of unliganded caspase-6 assumed a pH-inactivated form as it was crystallized at pH 4.5 (48). If that was true, and in solution caspase-6 existed exclusively in the canonical strand conformation, the H/D exchange for caspase-6 should mirror that of caspase-7. The fact that the H/D exchange for caspase-6 with and without a substrate mimic is so strikingly different from that of caspase-7 with and without a substrate mimic strongly supports the helix-strand interconversion model as governing caspase-6 structure and dynamics before substrate binding.
All caspases have mobile loops critical for substrate binding and catalysis. Caspase-6 is different because it has additional mobile regions. Two distinct conformational states have been identified in the 130's region by crystallography, and H/DX-MS has identified a unique interconversion probably between those two states. Although caspase-6 is the only caspase known to adopt the helical conformation and can interconvert to the canonical form upon substrate binding, this type of secondary structural shift is not confined to caspases but has also been observed in other protein systems. Helix-strand interconversion is exploited in nature to allow GTP hydrolysis by EF-Tu (62, 63), susceptibility to conformational diseases (64), and membrane association (65–67). For example, the influenza A M2 protein, a proton channel that facilitates viral assembly and budding, is found to interconvert between α-helical and β-sheet conformations dependent on the membrane composition (65). The binding of M2 protein to 1,2-dimyristoyl-sn-glycero-3-phosphocholine promotes the β-sheet conformation, whereas its binding to cholesterol-rich membrane enriched the α-helical conformation. Thus, the biological role of the secondary structure switch in caspase-6 is intriguing. One could imagine such a change in structure to play roles in other as yet unidentified functions, such as substrate recognition and specificity, cellular signaling and transport, or even binding of natural regulators in the cell.
Our work suggests that the helix-strand interconversion is dependent on the microenvironment pKa values of a specific residue in the 130's region and, correspondingly, on the pH. Protonation-conformation coupling is important for the function of several protein systems, including the nitric oxide carrier heme protein, Nitrophorin 4 (68), β-lactoglobulin (69), and the Escherichia coli multidrug efflux transporter AcrB (70). Specific titratable amino acids in a region influence the conformation and function of the protein. These amino acids are expected to have different protonation states in the helical and strand conformations resulting from two sets of distinct pKa values. In caspase-6, Glu-135 within the 130's region displays two distinct microenvironment pKa values, which are 4.4 pH units apart between the helical and the strand conformations. In our simulations, the change in the protonation state of the Glu-135 impacts the relative stability of the helical state of caspase-6. The relevance of this finding may be explained in the context of the pH of the environment that caspase-6 experiences. The pH may dictate the preference of one conformation of caspase-6 over the other (i.e. helical or strand). It has been suggested that pH fluctuation happens in the cellular context, where oxidative stress results in cytosolic acidification that can ultimately lead to neurodegeneration (for a review, see Ref. 71). Oxidative stress, a trigger for apoptosis, induced by the addition of hydrogen peroxide in HEK 293/Tau cells, increases active caspase-6 and caspase-3 activity (72). An increase in caspase-6 activity has also been documented in global brain ischemia (73) and acute ischemic stroke (74), where slight intracellular acidification has been observed to acidify slightly to pH 6.43 (75). Although the precise role of the two-state conformations of caspase-6 is still not known, pH changes in the context of neurodegeneration could conformationally enrich caspase-6 in either helical or strand conformations. The pH-induced conformational change may explain the propensity of caspase-6 to recognize different substrates during various cellular processes, including apoptosis and neurodegeneration. Future studies on determining the substrate preference of caspase-6 in the context of varying intracellular pH condition may shed light on the importance of the unique two-state conformation of the protein.
In addition to having strikingly different secondary structures, the helical and the strand states of caspase-6 have different electrostatic surface potentials (supplemental Fig. S8). Interestingly, the caspase-6 helical conformation exposes an aromatic-rich hydrophobic patch at the top of the 130's helix (Fig. 7). Hydrophobic patches are often involved in substrate recognition (76). The differences in electrostatics and hydrophobic exposure might be expected to influence substrate recognition and function. Critical inspection of the helical and the strand structures of caspase-6 revealed that the top of the 130's helix as well as the surrounding residues within loop 1 are flocked with an exposed hydrophobic patch that contains Phe-55, Phe-56, Trp-57, and Tyr-128. One might imagine that this cluster of aromatic residues could potentially function as an exosite for initial substrate recruitment by helical caspase-6. Accommodation of the substrate near the active site might then facilitate transition of helical caspase-6 to the substrate binding-competent strand state for efficient catalysis. Upon the eventual release of the substrate, caspase-6 may then return to continuous helix-strand interconversion. This potential role of the hydrophobic patch on caspase-6 is at present unexplored and untapped. Future studies dissecting potential exosites for cellular and neuronal substrates of caspase-6 (e.g. Tau and huntingtin proteins) could shed light on protein-protein interactions that might fruitfully be exploited for novel therapeutics in the future.
FIGURE 7.
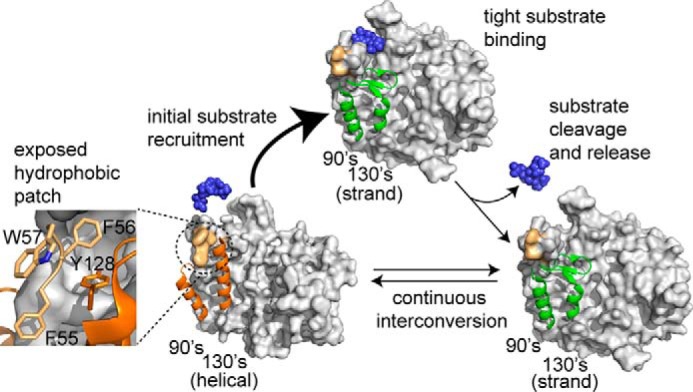
A model showing caspase-6 undergoes a helix-strand transition upon substrate binding. Before substrate binding, caspase-6 continuously interconverts between the noncanonical (helical) conformation and the canonical strand conformation and fully transitions to the strand conformation upon substrate binding. This transition may be facilitated by the presence of a dynamic hydrophobic patch (light orange, inset view) that could also function as an exosite for substrate binding. After substrate cleavage and release, caspase-6 exists in a dynamic equilibrium between the helical and strand states.
Caspase-6 has been predicted to be the only caspase capable of undergoing a helix-strand interconversion (45, 46); however, this assertion has been debated (48). Our H/D exchange data convincingly confirm that the structural dynamics of caspase-6 are fundamentally different from that of other caspases, which are always in the canonical strand conformation. These data further support strongly a model where unliganded caspase-6 interconverts between the helical and strand conformations. Studies on caspase-6 structure and dynamics are particularly relevant because caspase-6 is implicated in several neurodegenerative disorders, including Alzheimer's and Huntington's diseases. Thus, if trapping the helical conformation is indeed inactivating, then stabilizing the helical conformation may be a promising mode of achieving caspase-6-specific inhibition. On the other hand, cleavage of the neuronal substrate DJ-1 (22) appears to be protective against neurodegeneration in Parkinson's disease. If the exposed hydrophobic patch does serve as a substrate-recruiting exosite (Fig. 7), promoting the caspase-6 helical conformation might increase interaction with substrates like DJ-1, for which cleavage is neuroprotective.
Experimental Procedures
Generation of Caspase Variants
The WT caspase-6 used in this study was a constitutive two-chain form of caspase-6 derived from the synthetic, E. coli codon-optimized (His)6 C-terminally tagged caspase-6 gene (Celtek Bioscience) that was ligated into the NdeI/BamHI sites of pET11a vector (45). The wild-type caspase-7 used was a constitutively two-chain corrected version in pET23b that is designed to independently express the large and small subunits of caspase-7 with a His6 tag at the C terminus (77). In both caspase-6 and caspase-7, the protein constructs were designed to exclude the prodomain (residues 1–23 in both caspase-6 and caspase-7) and linker (residues 180–193 in caspase-6; residues 199–206 in caspase-7). Phusion® site-directed mutagenesis (Thermo Scientific) was used to introduce the E135Q mutation in the caspase-6 constitutive two-chain construct.
Caspase Protein Expression and Purification
The caspase-6 and caspase-7 constructs were transformed into the BL21(DE3) T7 express strain of E. coli (New England Biolabs). Overnight seed cultures were initially grown in 2× YT media supplemented with 0.1 mg/ml ampicillin (Sigma) at 37 °C. Dense cultures were then diluted 1,000-fold with 2× YT containing 0.1 mg/ml ampicillin and shaken at 37 °C until A600 reached 0.6. Protein expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside at 20 °C for 18 h. Cells were centrifuged at 4,700 × g for 10 min at 4 °C and stored at −20 °C until use. Freeze-thawed cells were lysed using a microfluidizer (Microfuidics, Inc.) in lysis buffer (50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, 50 mm imidazole) and centrifuged at 30,600 × g for 1 h at 4 °C. The supernatant was loaded into a 5-ml HiTrap nickel affinity column (GE Healthcare) and washed with lysis buffer until the absorbance returned to baseline. The protein was eluted with elution buffer (50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, 250 mm imidazole) and diluted 5-fold with buffer A (20 mm Tris, pH 8.5, 2 mm DTT) to reduce the salt concentration. This protein sample was then loaded into a 5-ml HiTrap Q HP column (GE Healthcare). The column was developed with a linear NaCl gradient, and the protein was eluted in 20 mm Tris, pH 8.5, 200 mm NaCl, 2 mm DTT. This eluted protein was stored at −80 °C until use. The purified caspases were analyzed by SDS-PAGE to confirm identity and purity.
Caspase Activity Assays
To measure caspase activity, 100 nm purified caspase was assayed over 7 min at 37 °C in caspase-6 activity assay buffer (100 mm HEPES, 120 mm NaCl, 0.1% CHAPS, 10% sucrose, 5 mm DTT) or in caspase-7 activity assay buffer (100 mm HEPES, pH 7.5, 5 mm CaCl2, 10% PEG 400, 0.1% CHAPS, 5 mm DTT). For substrate titration, a range of 0–500 μm fluorogenic substrate VEID-AMC (Enzo Life Sciences Inc.) was used for caspase-6, and a range of 0–200 μm fluorogenic substrate N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (DEVD-AMC; Enzo Life Sciences Inc.) was used for caspase-7. Fluorescence kinetic measurements (λex/λem, 365 nm/495 nm) were performed in three independent trials on three different days in 100-μl reactions in a 96-well format using a microplate reader (SpectraMax M5, Molecular Devices). Initial velocities versus substrate concentration were fit to a rectangular hyperbola using GraphPad Prism (GraphPad Software, La Jolla, CA) to determine the kinetic parameters Km and kcat. Enzyme concentrations were determined by active site titration with the quantitative covalent inhibitor N-acetyl-Val-Glu-Ile-Asp-aldehyde (VEID-CHO; Enzo Life Sciences Inc.) for caspase-6 or N-acetyl-Asp-Glu-Val-Asp-aldehyde (DEVD-CHO; Enzo Life Sciences Inc.) for caspase-7. Protein was added to inhibitor solvated in DMSO in 96-well V-bottom plates at room temperature for 1.5 h in caspase activity assay buffer. Aliquots (90 μl) were transferred in duplicate to black well plates and assayed with a 50-fold molar excess of substrate. The protein concentration was determined to be the lowest concentration at which full inhibition was observed and was thus used to calculate kcat.
Hydrogen/Deuterium Exchange Mass Spectrometry
H/D exchange experiments were performed using an initial stock of 15 μm caspase-6 or caspase-7 in 20 mm Tris, pH 8.5, 200 mm NaCl, and 2 mm DTT in H2O. For samples without substrate-like inhibitor, 1 μl of DMSO was added to a final volume of 200 μl, and for substrate-like inhibitor-bound protein samples, 1 μl of 30 μm peptide substrate-like inhibitor was added (VEID-CHO for caspase-6; DEVD-CHO for caspase-7) to a final volume of 200 μl. The protein samples were incubated at room temperature for 1 h and then introduced into the nanoACQUITY system equipped with H/D exchange technology for UPLC separation (78) (Waters Corp., Milford, MA), which performed all subsequent manipulations for the H/D exchange. H/D exchange was initiated by dilution of each sample 15-fold with D2O exchange buffer (10 mm phosphate pD 7.5, 200 mm NaCl), and the mixture was incubated at predetermined H/D exchange time points (0.17, 1, 10, 60, and 120 min) at 25 °C. At the indicated H/D exchange time point, an aliquot from the exchange reaction was removed, and deuterium labeling was quenched by adding equal volume of quench buffer (100 mm phosphate, pH 2.5, 200 mm NaCl) at 3 °C. For the non-deuterated samples, the same procedure was performed in H2O buffers. Quenched samples were introduced into a 5-μm BEH 2.1 × 30-mm EnzymateTM immobilized pepsin column (Waters Corp., Milford, MA) at 100 μl/min in 0.1% formic acid at 10 °C and then incubated for 4.5 min to allow on-column digestion. Peptide fragments were collected at 0 °C on a C18 VanGuard trap column (1.7 μm × 30 mm) (Waters) for desalting with 0.1% formic acid in H2O and were then separated using a 1.8-μm HSS T3 C18 2.1 × 30-mm nanoACQUITY UPLC® column (Waters) for a 10-min gradient from 0.1% formic acid to acetonitrile (7 min, 5–35%; 1 min, 35–85%; 2-min hold, 85% acetonitrile) at 40 μl/min at 0 °C. Fragments were subsequently mass-analyzed using a Synapt G2Si ESI-Q-ToF mass spectrometer (Waters). Between sample injections, a wash step was performed to minimize peptide carry-over. Accurate mass and collision-induced dissociation in data-independent acquisition mode (MSE) (79) and ProteinLynx Global Server (PLGS) version 3.0 software (Waters) were used to determine the peptic peptides in the undeuterated protein samples analyzed on the same UPLC-ESI-Q-ToF system used for H/DX-MS experiments. Peptic peptides generated from PLGS were imported into DynamX version 3.0 (Waters) with peptide quality thresholds of MS1 signal intensity ≥5,000, maximum sequence length of 25 amino acids, maximum mass error of 1 ppm, and minimum products per amino acid of ≥0.3. Automated results were manually inspected to ensure that the corresponding m/z and isotopic distributions at various charge states were properly assigned to the appropriate peptic peptide. DynamX 3.0 was then used to generate the relative deuterium incorporation plot and H/DX heat map for each peptic peptide. The relative deuterium incorporation of each peptide was determined by subtracting the weight-averaged centroid mass of the isotopic distribution of undeuterated control sample from that of the weight-averaged centroid mass of the isotopic distribution of deuterium-labeled samples at each labeling time point. All comparisons were performed under identical experimental conditions, thus negating the need for back exchange correction in the determination of the deuterium incorporation. Thus, H/D exchange levels are reported as relative (57). The fractional relative deuterium uptake was calculated by dividing the relative deuterium uptake of each peptic peptide by its theoretical maximum uptake. All H/DX-MS experiments were performed in duplicate on two separate days, and a 98% confidence limit for the uncertainty of the mean relative deuterium uptake of ±0.6 Da was calculated as described (80). Differences in deuterium uptake between two states that exceed 0.6 Da were considered significant.
Circular Dichroism Spectroscopy
Thermal stability of caspase-6 variants at various pH levels was monitored by loss of circular dichroism signal at 222 nm over a range of 20–90 °C. CD spectra (250 to 190 nm) were measured on a J-1500 CD spectrometer (Jasco) with Peltier temperature controller. Appropriate buffers for effective buffering capacity at various pH levels (20 mm MES, pH 6.2, 120 mm NaCl; 20 mm phosphate buffer, pH 7.5, 120 mm NaCl; or 20 mm Tris, pH 8.5, 120 mm NaCl) were used. Sample proteins were buffer-exchanged three times into each appropriate buffer using an Amicon Ultra 0.5-ml centrifugal filter (molecular weight cut-off 10,000) (Millipore), and the protein concentration (5 μm) was determined by absorbance at 280 nm (Nanodrop 2000C spectrophotometer). All data were collected at least in duplicate on different days. The melting curves were plotted using GraphPad Prism (GraphPad software) and fitted to a Boltzmann sigmoidal plot to determine the melting temperature, Tm.
Caspase Activity Versus pH
The activity of caspase-6 variants were assessed as a function of pH using 20 nm protein and 60 μm VEID-AMC as a substrate in appropriate buffer at various pH levels (pH 4.5–9.0). The common components for all buffer at different pH levels contained 120 mm NaCl, 0.1% CHAPS, 10% sucrose, 5% glycerol, and 5 mm DTT. The following buffers were used at 100 mm concentration with its corresponding pH range: acetate (pH 4.5–5.0), MES (pH 5.5–6.5), HEPES (pH 7.0–7.5), and Tris base (pH 8.0–9.0). Fluorescence kinetic measurements (λex/λem, 365 nm/495 nm) were performed in duplicate in 100-μl reactions in a 96-well format using a SpectraMax M5 microplate reader (Molecular Devices).
In Silico Model Preparation
The initial model for the mature dimeric human caspase-6 was generated by producing an all-atom structure of the truncated enzyme by using the coordinates of C (residues 31–165 and 201–293) and D (residues 31–165 and 199–293) chains in the ligand-free structure (PDB code 2WDP) and Maestro's Protein Preparation Wizard (Schrödinger, LLC, New York). Next, the C terminus of the cleaved intersubunit linker in each protomer was rebuilt by (i) grafting residues 166–175 from the procaspase-6 structure (PDB code 4N5D, chain A) using the Prime's Protein Splicing tool (version 3.8, Schrödinger, LLC) and (ii) attaching the pentapeptide terminus (residues 175–179) in its extended conformation to the resulting chimeric structures. The N-terminal residues of the linker (residues 194–201), missing in the 2WDP chains, were reconstructed using the coordinates from a Zn(II)-bound caspase-6 structure (PDB code 4FXO). The resulting chimeric dimer was subjected to a protonation state assignment algorithm (Epik, Schrödinger, LLC) and restrained minimization (force field, OPLS3) sequence within Maestro's Protein Preparation Wizard. The model was further refined via a simulated annealing sequence within Desmond (version 4.0, D.E. Shaw Research and Schrödinger, LLC). Thus, an NVT ensemble was built with a neutralized (by 8 Cl− ions) system and further Na+ and Cl− ions to simulate 150 mm concentration in an explicit SPC solvent model (16,783 water molecules), using an orthorhombic simulation box (10 Å buffer) with periodic boundary conditions. The ensemble was then subjected to the following seven-stage schedule: (i) incubation at 10 K for 30 ps; (ii) heating to 100 K for 70 ps; (iii) heating to 300 K for 100 ps; (iv) heating to 400 K for 100 ps; (v) incubation at 400K for 200 ps; (vi) cool down to 300 K for 500 ps; (vii) incubation at 300 K for 200 ps. The structure from the final frame of the 1.2-ns simulation was reminimized (Prime, OPLS3) and selected as the starting point for all subsequent simulations.
Simulated Annealing Studies of Protonation Variants
Protonation states of the residues in the two forms of caspase-6 were obtained using the Epik algorithm (Schrödinger, LLC) and the all-atom models derived from unliganded (PDB code 2WDP) and VEID-bound (PDB code 3OD5) forms. Using the refined model obtained above, several protonation state variants at Glu and His residues were produced: (i) “2WDP-like” or the triply protonated helical caspase-6 variant (HisH-52, GluH-135, and GluH-221); (ii) “3OD5-like” or the triply deprotonated strand caspase-6 variant (His-52, Glu-135, and Glu-221); (iii) “His-52” (His-52, GluH-135, and GluH-221); (iv) “Glu-135” (HisH-52, Glu-135, and GluH-221); and (v) “Glu-221” (HisH-52, GluH-135, and Glu-221). The resulting models were neutralized with the corresponding number of Cl− ions and treated as described above to produce starting NVT ensembles for simulated annealing experiments. The 1.2-ns-long seven-stage schedule was reapplied on the protonation variants, and the RMSF analysis was performed using the Simulation Event Analysis tool within Desmond.
Author Contributions
K. B. D. initiated and performed all experimental aspects of the study, performed the extensive final analysis of H/DX-MS data, prepared all final figures, and was the principal author for the manuscript. N. B.-C. worked with K. B. D. to prepare the samples for H/DX-MS and perform H/D exchange analysis and performed initial analysis of H/DX-MS peptides and wrote an early draft of the manuscript. S. N. S. built and analyzed molecular models of caspase-6, performed molecular dynamics simulation, and helped to prepare the manuscript. J. A. H conceptualized the project, secured funding, directed the research project, wrote parts of the manuscript, and edited the manuscript.
Supplementary Material
Acknowledgment
We thank Stephen J. Eyles, director of the UMass Institute of Applied Life Sciences Mass Spectrometry core facility for abundant assistance with H/D exchange mass spectrometry data collection and processing.
This work was supported by National Institutes of Health Grant GM080532. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S8.
- H/DX-MS
- hydrogen/deuterium exchange mass spectrometry
- H/D
- hydrogen/deuterium
- L1
- loop 1
- L2
- loop 2
- L2′
- loop 2′
- L3
- loop 3
- L4
- loop 4
- PDB
- Protein Data Bank
- RMSF
- root mean square fluctuation
- GluH
- protonated GluH
- HisH
- protonated His
- AMC
- 7-amino-4-methyl-coumarin
- CHO
- aldehyde.
References
- 1. Boatright K. M., Renatus M., Scott F. L., Sperandio S., Shin H., Pedersen I. M., Ricci J. E., Edris W. A., Sutherlin D. P., Green D. R., and Salvesen G. S. (2003) A unified model for apical caspase activation. Mol. Cell 11, 529–541 [DOI] [PubMed] [Google Scholar]
- 2. Pop C., and Salvesen G. S. (2009) Human caspases: activation, specificity, and regulation. J. Biol. Chem. 284, 21777–21781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suzuki A., Kusakai G., Kishimoto A., Shimojo Y., Miyamoto S., Ogura T., Ochiai A., and Esumi H. (2004) Regulation of caspase-6 and FLIP by the AMPK family member ARK5. Oncogene 23, 7067–7075 [DOI] [PubMed] [Google Scholar]
- 4. Orth K., Chinnaiyan A. M., Garg M., Froelich C. J., and Dixit V. M. (1996) The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J. Biol. Chem. 271, 16443–16446 [PubMed] [Google Scholar]
- 5. Srinivasula S. M., Fernandes-Alnemri T., Zangrilli J., Robertson N., Armstrong R. C., Wang L., Trapani J. A., Tomaselli K. J., Litwack G., and Alnemri E. S. (1996) The Ced-3:interleukin 1b converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2a are substrates for the apoptotic mediator CPP32. J. Biol. Chem. 271, 27099–27106 [DOI] [PubMed] [Google Scholar]
- 6. Takahashi A., Alnemri E. S., Lazebnik Y. A., Fernandes-Alnemri T., Litwack G., Moir R. D., Goldman R. D., Poirier G. G., Kaufmann S. H., and Earnshaw W. C. (1996) Cleavage of lamin A by Mch2 α but not CPP32: multiple interleukin 1 β-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 8395–8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cowling V., and Downward J. (2002) Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ. 9, 1046–1056 [DOI] [PubMed] [Google Scholar]
- 8. Srinivasula S. M., Ahmad M., MacFarlane M., Luo Z., Huang Z., Fernandes-Alnemri T., and Alnemri E. S. (1998) Generation of constitutively active recombinant caspases-3 and -6 by rearrangement of their subunits. J. Biol. Chem. 273, 10107–10111 [DOI] [PubMed] [Google Scholar]
- 9. Guo H., Pétrin D., Zhang Y., Bergeron C., Goodyer C. G., and LeBlanc A. C. (2006) Caspase-1 activation of caspase-6 in human apoptotic neurons. Cell Death Differ. 13, 285–292 [DOI] [PubMed] [Google Scholar]
- 10. Chai J., Wu Q., Shiozaki E., Srinivasula S. M., Alnemri E. S., and Shi Y. (2001) Crystal structure of a procaspase-7 zymogen: mechanisms of activation and substrate binding. Cell 107, 399–407 [DOI] [PubMed] [Google Scholar]
- 11. Klaiman G., Champagne N., and LeBlanc A. C. (2009) Self-activation of Caspase-6 in vitro and in vivo: caspase-6 activation does not induce cell death in HEK293T cells. Biochim. Biophys. Acta 1793, 592–601 [DOI] [PubMed] [Google Scholar]
- 12. Wang X. J., Cao Q., Liu X., Wang K. T., Mi W., Zhang Y., Li L. F., LeBlanc A. C., and Su X. D. (2010) Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 11, 841–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harrington E. P., Zhao C., Fancy S. P., Kaing S., Franklin R. J., and Rowitch D. H. (2010) Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann. Neurol. 68, 703–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nikolaev A., McLaughlin T., O'Leary D. D., and Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15. Schoenmann Z., Assa-Kunik E., Tiomny S., Minis A., Haklai-Topper L., Arama E., and Yaron A. (2010) Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J. Neurosci. 30, 6375–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simon D. J., Weimer R. M., McLaughlin T., Kallop D., Stanger K., Yang J., O'Leary D. D., Hannoush R. N., and Tessier-Lavigne M. (2012) A caspase cascade regulating developmental axon degeneration. J. Neurosci. 32, 17540–17553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park K. J., Grosso C. A., Aubert I., Kaplan D. R., and Miller F. D. (2010) p75NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat. Neurosci. 13, 559–566 [DOI] [PubMed] [Google Scholar]
- 18. Watanabe C., Shu G. L., Zheng T. S., Flavell R. A., and Clark E. A. (2008) Caspase 6 regulates B cell activation and differentiation into plasma cells. J. Immunol. 181, 6810–6819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horowitz P. M., Patterson K. R., Guillozet-Bongaarts A. L., Reynolds M. R., Carroll C. A., Weintraub S. T., Bennett D. A., Cryns V. L., Berry R. W., and Binder L. I. (2004) Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J. Neurosci. 24, 7895–7902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Albrecht S., Bogdanovic N., Ghetti B., Winblad B., and LeBlanc A. C. (2009) Caspase-6 activation in familial Alzheimer disease brains carrying amyloid precursor protein or presenilin i or presenilin II mutations. J. Neuropathol. Exp. Neurol. 68, 1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Graham R. K., Deng Y., Slow E. J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., Warby S. C., Doty C. N., Roy S., Wellington C. L., Leavitt B. R., et al. (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125, 1179–1191 [DOI] [PubMed] [Google Scholar]
- 22. Giaime E., Sunyach C., Druon C., Scarzello S., Robert G., Grosso S., Auberger P., Goldberg M. S., Shen J., Heutink P., Pouysségur J., Pagès G., Checler F., and Alves da Costa C. (2010) Loss of function of DJ-1 triggered by Parkinson's disease-associated mutation is due to proteolytic resistance to caspase-6. Cell Death Differ. 17, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Albrecht S., Bourdeau M., Bennett D., Mufson E. J., Bhattacharjee M., and LeBlanc A. C. (2007) Activation of caspase-6 in aging and mild cognitive impairment. Am. J. Pathol. 170, 1200–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galvan V., Chen S., Lu D., Logvinova A., Goldsmith P., Koo E. H., and Bredesen D. E. (2002) Caspase cleavage of members of the amyloid precursor family of proteins. J. Neurochem. 82, 283–294 [DOI] [PubMed] [Google Scholar]
- 25. Galvan V., Gorostiza O. F., Banwait S., Ataie M., Logvinova A. V., Sitaraman S., Carlson E., Sagi S. A., Chevallier N., Jin K., Greenberg D. A., and Bredesen D. E. (2006) Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. U.S.A. 103, 7130–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gervais F. G., Xu D., Robertson G. S., Vaillancourt J. P., Zhu Y., Huang J., LeBlanc A., Smith D., Rigby M., Shearman M. S., Clarke E. E., Zheng H., Van Der Ploeg L. H. T., Ruffolo S. C., Thornberry N. A., et al. (1999) Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell 97, 395–406 [DOI] [PubMed] [Google Scholar]
- 27. Guo H., Albrecht S., Bourdeau M., Petzke T., Bergeron C., and LeBlanc A. C. (2004) Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am. J. Pathol. 165, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klaiman G., Petzke T. L., Hammond J., and Leblanc A. C. (2008) Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol. Cell Proteomics 7, 1541–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. LeBlanc A. C. (2013) Caspase-6 as a novel early target in the treatment of Alzheimer's disease. Eur. J. Neurosci. 37, 2005–2018 [DOI] [PubMed] [Google Scholar]
- 30. Lu D. C., Rabizadeh S., Chandra S., Shayya R. F., Ellerby L. M., Ye X., Salvesen G. S., Koo E. H., and Bredesen D. E. (2000) A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat. Med. 6, 397–404 [DOI] [PubMed] [Google Scholar]
- 31. Nguyen T. V., Galvan V., Huang W., Banwait S., Tang H., Zhang J., and Bredesen D. E. (2008) Signal transduction in Alzheimer disease: p21-activated kinase signaling requires C-terminal cleavage of APP at Asp664. J. Neurochem. 104, 1065–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saganich M. J., Schroeder B. E., Galvan V., Bredesen D. E., Koo E. H., and Heinemann S. F. (2006) Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J. Neurosci. 26, 13428–13436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao M., Su J., Head E., and Cotman C. W. (2003) Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer's disease. Neurobiol. Dis. 14, 391–403 [DOI] [PubMed] [Google Scholar]
- 34. Aharony I., Ehrnhoefer D. E., Shruster A., Qiu X., Franciosi S., Hayden M. R., and Offen D. (2015) A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum. Mol. Genet. 24, 2604–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Graham R. K., Deng Y., Carroll J., Vaid K., Cowan C., Pouladi M. A., Metzler M., Bissada N., Wang L., Faull R. L., Gray M., Yang X. W., Raymond L. A., and Hayden M. R. (2010) Cleavage at the 586 amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J. Neurosci. 30, 15019–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hermel E., Gafni J., Propp S. S., Leavitt B. R., Wellington C. L., Young J. E., Hackam A. S., Logvinova A. V., Peel A. L., Chen S. F., Hook V., Singaraja R., Krajewski S., Goldsmith P. C., Ellerby H. M., et al. (2004) Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 11, 424–438 [DOI] [PubMed] [Google Scholar]
- 37. Milnerwood A. J., Gladding C. M., Pouladi M. A., Kaufman A. M., Hines R. M., Boyd J. D., Ko R. W., Vasuta O. C., Graham R. K., Hayden M. R., Murphy T. H., and Raymond L. A. (2010) Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron 65, 178–190 [DOI] [PubMed] [Google Scholar]
- 38. Riechers S. P., Butland S., Deng Y., Skotte N., Ehrnhoefer D. E., Russ J., Laine J., Laroche M., Pouladi M. A., Wanker E. E., Hayden M. R., and Graham R. K. (2016) Interactome network analysis identifies multiple caspase-6 interactors involved in the pathogenesis of HD. Hum. Mol. Genet. 25, 1600–1618 [DOI] [PubMed] [Google Scholar]
- 39. Warby S. C., Doty C. N., Graham R. K., Carroll J. B., Yang Y. Z., Singaraja R. R., Overall C. M., and Hayden M. R. (2008) Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum. Mol. Genet. 17, 2390–2404 [DOI] [PubMed] [Google Scholar]
- 40. Wong B. K., Ehrnhoefer D. E., Graham R. K., Martin D. D., Ladha S., Uribe V., Stanek L. M., Franciosi S., Qiu X., Deng Y., Kovalik V., Zhang W., Pouladi M. A., Shihabuddin L. S., and Hayden M. R. (2015) Partial rescue of some features of Huntington disease in the genetic absence of caspase-6 in YAC128 mice. Neurobiol. Dis. 76, 24–36 [DOI] [PubMed] [Google Scholar]
- 41. Cao Q., Wang X. J., Li L. F., and Su X. D. (2014) The regulatory mechanism of the caspase 6 pro-domain revealed by crystal structure and biochemical assays. Acta Crystallogr. D Biol. Crystallogr. 70, 58–67 [DOI] [PubMed] [Google Scholar]
- 42. Müller I., Lamers M. B., Ritchie A. J., Dominguez C., Munoz-Sanjuan I., and Kiselyov A. (2011) Structure of human caspase-6 in complex with Z-VAD-FMK: new peptide binding mode observed for the non-canonical caspase conformation. Bioorg. Med. Chem. Lett. 21, 5244–5247 [DOI] [PubMed] [Google Scholar]
- 43. Liu X., Zhang H., Wang X. J., Li L. F., and Su X. D. (2011) Get phases from arsenic anomalous scattering: de novo SAD phasing of two protein structures crystallized in cacodylate buffer. PLoS One 6, e24227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baumgartner R., Meder G., Briand C., Decock A., D'arcy A., Hassiepen U., Morse R., and Renatus M. (2009) The crystal structure of caspase-6, a selective effector of axonal degeneration. Biochem. J. 423, 429–439 [DOI] [PubMed] [Google Scholar]
- 45. Vaidya S., Velázquez-Delgado E. M., Abbruzzese G., and Hardy J. A. (2011) Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 406, 75–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vaidya S., and Hardy J. A. (2011) Caspase-6 latent state stability relies on helical propensity. Biochemistry 50, 3282–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Štrajbl M, Florián J and Warshel A. (2001) Ab initio evaluation of the free energy surfaces for the general base/acid catalyzed thiolysis of formamide and the hydrolysis of methyl thiolformate: a reference solution reaction for studies of cysteine proteases. J. Phys. Chem. B 105, 4471–4484 [Google Scholar]
- 48. Müller I., Lamers M. B., Ritchie A. J., Park H., Dominguez C., Munoz-Sanjuan I., Maillard M., and Kiselyov A. (2011) A new apo-caspase-6 crystal form reveals the active conformation of the apoenzyme. J. Mol. Biol. 410, 307–315 [DOI] [PubMed] [Google Scholar]
- 49. Pirrone G. F., Iacob R. E., and Engen J. R. (2015) Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem. 87, 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Landgraf R. R., Goswami D., Rajamohan F., Harris M. S., Calabrese M. F., Hoth L. R., Magyar R., Pascal B. D., Chalmers M. J., Busby S. A., Kurumbail R. G., and Griffin P. R. (2013) Activation of AMP-activated protein kinase revealed by hydrogen/deuterium exchange mass spectrometry. Structure 21, 1942–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kirschke E., Goswami D., Southworth D., Griffin P. R., and Agard D. A. (2014) Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hodkinson J. P., Radford S. E., and Ashcroft A. E. (2012) The role of conformational flexibility in β2-microglobulin amyloid fibril formation at neutral pH. Rapid Commun. Mass Spectrom. 26, 1783–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Snijder J., Benevento M., Moyer C. L., Reddy V., Nemerow G. R., and Heck A. J. (2014) The cleaved N-terminus of pVI binds peripentonal hexons in mature adenovirus. J. Mol. Biol. 426, 1971–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pan L. Y., Salas-Solano O., and Valliere-Douglass J. F. (2014) Conformation and dynamics of interchain cysteine-linked antibody-drug conjugates as revealed by hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 86, 2657–2664 [DOI] [PubMed] [Google Scholar]
- 55. Burke J. E., Perisic O., Masson G. R., Vadas O., and Williams R. L. (2012) Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc. Natl. Acad. Sci. U.S.A. 109, 15259–15264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mehmood S., Domene C., Forest E., and Jault J. M. (2012) Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc. Natl. Acad. Sci. U.S.A. 109, 10832–10836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 58. Englander S. W., and Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655 [DOI] [PubMed] [Google Scholar]
- 59. Morgan C. R., and Engen J. R. (2009) Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr. Protoc. Protein Sci. 10.1002/0471140864.ps1706s58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wei Y., Fox T., Chambers S. P., Sintchak J., Coll J. T., Golec J. M., Swenson L., Wilson K. P., and Charifson P. S. (2000) The structures of caspases-1, -3, -7 and -8 reveal the basis for substrate and inhibitor selectivity. Chem. Biol. 7, 423–432 [DOI] [PubMed] [Google Scholar]
- 61. Hill M. E., MacPherson D. J., Wu P., Julien O., Wells J. A., and Hardy J. A. (2016) Reprogramming caspase-7 specificity by regio-specific mutations and selection provides alternate solutions for substrate recognition. ACS Chem. Biol. 11, 1603–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abel K., Yoder M. D., Hilgenfeld R., and Jurnak F. (1996) An α to β conformational switch in EF-Tu. Structure 4, 1153–1159 [DOI] [PubMed] [Google Scholar]
- 63. Polekhina G., Thirup S., Kjeldgaard M., Nissen P., Lippmann C., and Nyborg J. (1996) Helix unwinding in the effector region of elongation factor EF-Tu-GDP. Structure 4, 1141–1151 [DOI] [PubMed] [Google Scholar]
- 64. Pan K. M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., and Cohen F. E. (1993) Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liao S. Y., Fritzsching K. J., and Hong M. (2013) Conformational analysis of the full-length M2 protein of the influenza A virus using solid-state NMR. Protein Sci. 22, 1623–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shepard L. A., Heuck A. P., Hamman B. D., Rossjohn J., Parker M. W., Ryan K. R., Johnson A. E., and Tweten R. K. (1998) Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37, 14563–14574 [DOI] [PubMed] [Google Scholar]
- 67. Yassine W., Taib N., Federman S., Milochau A., Castano S., Sbi W., Manigand C., Laguerre M., Desbat B., Oda R., and Lang J. (2009) Reversible transition between α-helix and β-sheet conformation of a transmembrane domain. Biochim. Biophys. Acta 1788, 1722–1730 [DOI] [PubMed] [Google Scholar]
- 68. Di Russo N. V., Estrin D. A., Martí M. A., and Roitberg A. E. (2012) pH-dependent conformational changes in proteins and their effect on experimental pK(a)s: the case of Nitrophorin 4. PLoS Comput. Biol. 8, e1002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Qin B. Y., Bewley M. C., Creamer L. K., Baker H. M., Baker E. N., and Jameson G. B. (1998) Structural basis of the Tanford transition of bovine β-lactoglobulin. Biochemistry 37, 14014–14023 [DOI] [PubMed] [Google Scholar]
- 70. Murakami S., Nakashima R., Yamashita E., Matsumoto T., and Yamaguchi A. (2006) Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179 [DOI] [PubMed] [Google Scholar]
- 71. Majdi A., Mahmoudi J., Sadigh-Eteghad S., Golzari S. E., Sabermarouf B., and Reyhani-Rad S. (2016) Permissive role of cytosolic pH acidification in neurodegeneration: a closer look at its causes and consequences. J. Neurosci. Res. 94, 879–887 [DOI] [PubMed] [Google Scholar]
- 72. Zhao H., Zhao W., Lok K., Wang Z., and Yin M. (2014) A synergic role of caspase-6 and caspase-3 in Tau truncation at D421 induced by H2O 2. Cell Mol. Neurobiol. 34, 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ding Z. M., Wu B., Zhang W. Q., Lu X. J., Lin Y. C., Geng Y. J., and Miao Y. F. (2012) Neuroprotective effects of ischemic preconditioning and postconditioning on global brain ischemia in rats through the same effect on inhibition of apoptosis. Int. J. Mol. Sci. 13, 6089–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Akpan N., Serrano-Saiz E., Zacharia B. E., Otten M. L., Ducruet A. F., Snipas S. J., Liu W., Velloza J., Cohen G., Sosunov S. A., Frey W. H. 2nd, Salvesen G. S., Connolly E. S. Jr, and Troy C. M. (2011) Intranasal delivery of caspase-9 inhibitor reduces caspase-6-dependent axon/neuron loss and improves neurological function after stroke. J. Neurosci. 31, 8894–8904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nedergaard M., Kraig R. P., Tanabe J., and Pulsinelli W. A. (1991) Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 260, R581–R588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schulman B. A., Lindstrom D. L., and Harlow E. (1998) Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. U.S.A. 95, 10453–10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Witkowski W. A., and Hardy J. A. (2011) A designed redox-controlled caspase. Protein Sci. 20, 1421–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wales T. E., Fadgen K. E., Gerhardt G. C., and Engen J. R. (2008) High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 80, 6815–6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Geromanos S. J., Vissers J. P., Silva J. C., Dorschel C. A., Li G. Z., Gorenstein M. V., Bateman R. H., and Langridge J. I. (2009) The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependent LC-MS/MS. Proteomics 9, 1683–1695 [DOI] [PubMed] [Google Scholar]
- 80. Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.