

Abstract
The Schizosaccharomyces pombe shu1+ gene encodes a cell-surface protein required for assimilation of exogenous heme. In this study, shaving experiments showed that Shu1 is released from membrane preparations when spheroplast lysates are incubated with phosphoinositide-specific phospholipase C (PI-PLC). Shu1 cleavability by PI-PLC and its predicted hydropathy profile strongly suggested that Shu1 is a glycosylphosphatidylinositol-anchored protein. When heme biosynthesis is selectively blocked in hem1Δ mutant cells, the heme analog zinc mesoporphyrin IX (ZnMP) first accumulates into vacuoles and then subsequently, within the cytoplasm in a rapid and Shu1-dependent manner. An HA4-tagged shu1+ allele that retained wild-type function localizes to the cell surface in response to low hemin concentrations, but under high hemin concentrations, Shu1-HA4 re-localizes to the vacuolar membrane. Inactivation of abc3+, encoding a vacuolar membrane transporter, results in hem1Δ abc3Δ mutant cells being unable to grow in the presence of hemin as the sole iron source. In hem1Δ abc3Δ cells, ZnMP accumulates primarily in vacuoles and does not sequentially accumulate in the cytosol. Consistent with a role for Abc3 as vacuolar hemin exporter, results with hemin-agarose pulldown assays showed that Abc3 binds to hemin. In contrast, an Abc3 mutant in which an inverted Cys-Pro motif had been replaced with Ala residues fails to bind hemin with high affinity. Taken together, these results show that Shu1 undergoes rapid hemin-induced internalization from the cell surface to the vacuolar membrane and that the transporter Abc3 participates in the mobilization of stored heme from the vacuole to the cytosol.
Keywords: ABC transporter, glycosylphosphatidylinositol (GPI anchor), heme, metal homeostasis, yeast, yeast physiology
Introduction
The transition metal ion iron is well known to serve as catalytic cofactor for many enzymes that are intimately associated with essential cellular functions (1–3). Iron is also required to form heme, which is a prosthetic group that is composed of a protoporphyrin ring with an iron atom at its center (4). Hemoproteins such as cytochromes, hemoglobin, and catalases are required in fundamental biochemical processes, such as respiration, oxygen transport, and disproportionation of hydrogen peroxide, respectively. As a consequence of the importance to fulfill heme requirement for growth, several organisms have evolved with different means to obtain heme (5, 6). For instance, in the case of heme prototrophs, these organisms possess a heme biosynthetic pathway that involves eight conserved enzymes that are distributed in mitochondria and cytoplasm where their actions occur at specific steps of the biosynthetic pathway. A second strategy used by a number of organisms is to acquire heme from the environment. In these cases, molecules involved in acquisition of exogenous heme include Hrg-1 and Hrg-4 in Caenorhabditis elegans (7, 8), Lhr1 in Leishmania amazonensis (9), Hrg-1 in Dania rerio (zebrafish) (7), and Hrg-1, Hcp1, and FLVCR2 in mammals (8, 10, 11). Although the heme biosynthetic pathway has been extensively studied, the cellular components that are required for acquisition of exogenous heme and their mechanisms of action are much less well understood.
In many fungal species, a conserved 8 cysteine-containing domain has been found in cell-surface-anchored proteins (12, 13). Proteins containing this domain are members of the CFEM3 (common in fungal extracellular membranes) family (12). The CFEM domain is unique to fungal proteins and contains the following PXC(A/G)X2CX8–12CX1–3(X/T)DX2–5CXCX9–14CX3–4CX15–16C motif, where X is any amino acid residue (12, 13). In Candida albicans, studies have shown that four CFEM proteins, Rbt5, Rbt51 (or Pga10), Pga7, and Csa2, possess the ability to bind hemin (14–16). Genetic analyses of different C. albicans mutant strains have revealed that only Rbt5, Pga7, and Csa2 are required for optimal assimilation of exogenous hemin. In the case of Csa2, the crystal structure of the protein has revealed that the CFEM domain adopts a novel “helical basket”-fold that consists of six α-helices (15). Based on this three-dimensional structure of a CFEM protein, disulfide bonds formed by the 8 conserved Cys residues are predicted to stabilize the helical basket-fold, which itself would serve as a flat platform in which a planar heme molecule would sit at the top of the structure. Furthermore, a long N-terminal loop of Csa2 has been suggested to cover the heme prosthetic group and stabilize its association with the platform. It is also predicted that a conserved aspartic acid residue (Asp80) of the CFEM domain is exposed on the top of the platform to provide a critical contact point for heme binding (15). As a secreted hemophore-like protein, Csa2 is predicted to extract heme from hemoglobin and then behaves as carrier to deliver heme to the cell-surface-anchored CFEM proteins Rbt5 and Pga7, which are located in the cell wall and cell membrane (14, 16, 17).
Although Rbt5 plays an important role for C. albicans growth in the presence of hemoglobin or hemin as a sole source of iron, rbt5Δ/Δ cells are only partially defective in hemin assimilation (16). One possible explanation is that Pga7 also participates, along with Rbt5, in hemin acquisition (14). Studies have shown that Rbt5 and Pga7 exhibit distinct properties with respect to their cell-surface attachment, namely that Rbt5 is more accessible than Pga7 at the cell surface (14). Furthermore, Rbt5 exhibits a lower affinity for hemin than Pga7. This property may explain why pga7Δ/Δ inactivation attenuates virulence of C. albicans in a mouse model of systemic infection, although this is not the case for the loss of Rbt5 (14). According to a current model of hemin capture and uptake, Rbt5 would receive hemin from Csa2 at the cell surface and then would mediate its transfer to Pga7 through a relay pathway.
Utilization of hemin in C. albicans also depends of other cellular components, including heme oxygenase Hmx1 (18, 19), type I-myosin Myo5, proteins of the ESCRT (endosomal sorting complex required for transport) system, and vacuolar ATPase Vma11 (17). In the case of Hmx1, it would mediate the release of iron from the iron-protoporphyrin IX ring, producing the α isomer of biliverdin. In the case of Myo5 and ESCRT complex components, their involvement in heme assimilation suggests the existence of an endocytic pathway for heme internalization and delivery to the vacuole where it may undergo proteolytic processing (e.g. Vma11) and subsequent release into the cytoplasm (17).
Rbt5- and Pga7-like proteins are found in fungal species other than C. albicans. For instance, putative Rbt5 and Pga7 orthologs have been shown to be required for hemin assimilation in Candida parapsilosis and Paracoccidioides brasiliensis (20, 21). In Cryptococcus neoformans, the extracellular mannoprotein Cig1 binds hemin when produced as a recombinant protein in Escherichia coli (22). C. neoformans cells lacking Cig1 consistently exhibit delayed growth when hemin is the sole source of iron (22). Although Cig1 shares a similar hydropathy profile with Rbt5 and Pga7, it is not a member of the CFEM family. In fact, Cig1 is devoid of a canonical 8 Cys-containing CFEM domain. Interestingly, some downstream cellular components of the Cig1-dependent pathway involved in hemin acquisition are common to those identified in the C. albicans Rbt5/Pga7 pathway. For instance, Vps23, a component of the C. neoformans ESCRT-I complex that is required for intracellular trafficking of endocytic vesicles, is required for hemin acquisition as it is the case in C. albicans (17, 23, 24).
In Schizosaccharomyces pombe, iron-regulated cell-surface Shu1 protein (226 residues) is required for assimilation of exogenous hemin (25). Analysis of its sequence reveals that the N-terminal 20 amino acid residues correspond to a putative signal peptide that targets the protein to the secretory pathway. In addition, the C-terminal portion encompassing the last 26 amino acid residues corresponds to a hydrophobic region that contains Ser199, which may serve as an attachment site for a glycosylphosphatidylinositol (GPI) anchor. Although Shu1 contains 7 Cys residues, their arrangement does not correspond to that of a canonical 8 Cys-containing CFEM motif. However, 4 of these Cys residues (positions 72, 87, 92, and 101) exhibit an arrangement reminiscent of a partial CFEM motif, with a CX14CX4CX8C configuration in Shu1 as compared with a CX11CX4CX15C configuration in C. albicans Rbt5 and Pga7. Absorbance spectroscopy and hemin-agarose pulldown experiments have demonstrated that Shu1 binds to hemin (25). When iron levels are low and heme biosynthesis is selectively blocked, Shu1 localizes to the plasma membrane where it mediates acquisition of exogenous heme. Under similar conditions, shu1Δ mutant cells in which the heme biosynthetic pathway is selectively inactivated are unable to grow in the presence of exogenous hemin as a sole source of iron (25).
In C. albicans, studies have suggested that Rbt5-mediated heme assimilation involves an endocytic mechanism that results in the delivery of heme to the vacuole (17). Studies of the yeast vacuole have shown that this organelle can serve as a storage compartment for micronutrients, including metal ions (26–28). In the case of redox active metal ions, vacuolar storage could prevent their accumulation to toxic levels in the cytosol. Moreover, assuming that vacuolar metal ions are present in a usable form, the vacuole represents a reservoir of metals that could be redistributed throughout the cell under metal-deficient conditions (29–34). In the case of S. pombe, knowledge is lacking concerning transport systems capable of extracting metal or heme from the vacuole. With respect to iron homeostasis, transcriptional analyses have revealed that the expression of the ABC-type transporter Abc3 is induced under low-iron conditions (35). Under these conditions, Abc3 localizes to the membrane vacuole where it may participate in the redistribution of stored inorganic iron or organic iron conjugates from the vacuole to the cytosol (35).
In the present report, we show that Shu1 is a membrane-bound protein that is sensitive to the action of phosphoinositide-specific phospholipase C (PI-PLC). Under low hemin conditions, Shu1 localizes at the cell surface. However, in the presence of high concentrations of hemin, Shu1 undergoes internalization from the cell surface to the vacuole. Disruption of the iron-regulated vacuolar transporter abc3+ in cells lacking a functional heme biosynthetic pathway (hem1Δ) results in an inability of these mutant cells (hem1Δ abc3Δ) to grow in the presence of hemin as a sole source of iron. Microscopic analyses of hem1Δ abc3Δ mutant cells in the presence of ZnMP showed that this heme analog accumulates within the cell but is restricted to vacuoles in comparison with wild-type cells in which case ZnMP successively accumulates in the vacuoles and then the cytoplasm. Analysis by hemin-agarose pulldown assays showed that Abc3 interacts with hemin. Site-directed mutagenesis identified an inverted Cys-Pro motif (residues 151–152) within a hydrophilic loop region of Abc3 that is required for its association with hemin. Collectively, these results describe novel features of the Shu1-dependent heme acquisition pathway, including the discovery of the role of Abc3 in the mobilization of vacuolar heme stores.
Results
Shu1 Is a Membrane Protein That Exhibits PI-PLC Sensitivity
Microscopic analysis using spheroplasts have previously shown that Shu1 is a plasma membrane protein (25). Based on protein structure predictions, Shu1 (226 residues) contains a GPI-anchoring signal that comprises a GPI-attachment site (or ω-site; Ser199) followed by 2 amino acid residues with relatively short side chains (Ser200 and Ala201), a spacer region of 13 amino acid residues, and a hydrophobic carboxyl-terminal tail of 12 amino acid residues. We performed experiments to test the validity of these predicted properties. We initially conducted protein shaving experiments using treatment with PI-PLC. hem1Δ shu1Δ abc3Δ cells co-expressing shu1+-HA4 and abc3+-GFP alleles were grown in ALA-free medium containing hemin (0.075 μm) and 2,2′-dipyridyl (Dip) (250 μm). Spheroplasts were produced and a membrane fraction was isolated by ultracentrifugation. This fraction was divided into two aliquots of equal sizes, one was left untreated, whereas the other one was treated with PI-PLC. Results showed that in the absence of PI-PLC treatment, Shu1-HA4 and Abc3-GFP were present only in the pellet fraction (containing membrane proteins) (Fig. 1). However, membrane treatment with PI-PLC resulted in the release of a large proportion of Shu1-HA4 in the supernatant fraction (Fig. 1). Only a small proportion of Shu1-HA4 remained associated with the pellet fraction. In the case of Abc3-GFP that is predicted to possess membrane spans, results showed that the protein was resistant to PI-PLC treatment and was exclusively detected in the pellet fraction (Fig. 1). In conclusion, considering a previous demonstration that Shu1 localizes to the plasma membrane (25), these results strongly suggested that Shu1 is attached to the membrane via a GPI anchor that is sensitive to the activity of PI-PLC, an enzyme that removes the GPI moiety from GPI-anchored proteins.
FIGURE 1.
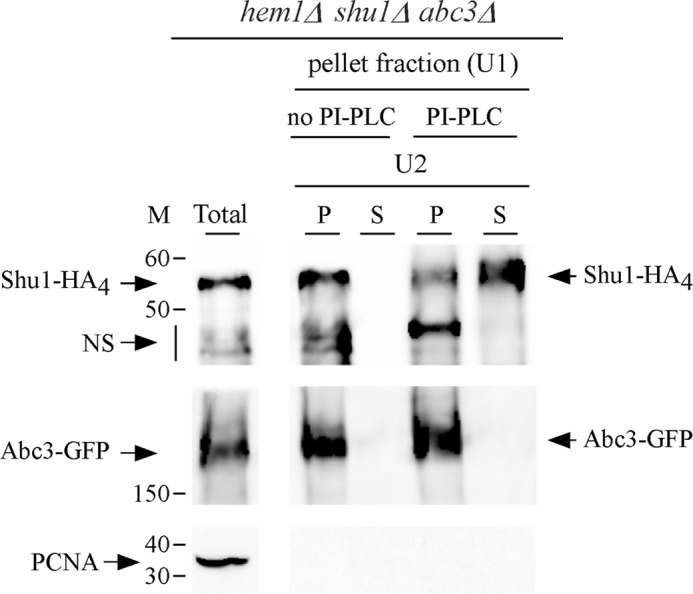
PI-PLC-mediated release of Shu1 from the membrane fraction to the supernatant. A triple-disruption strain (hem1Δ shu1Δ abc3Δ) was co-transformed with functional shu1+-HA4 and abc3+-GFP alleles and precultured in the presence of Dip (50 μm) and ALA (200 μm). Mid-logarithmic phase cells were incubated in ALA-free medium containing Dip (250 μm) for 6 h. Whole cell extract was prepared and ultracentrifuged (U1) to obtain a pellet fraction (P) of membrane proteins. The membrane-containing pellet fraction was treated with PI-PLC phospholipase or left untreated, and then ultracentrifuged (U2) a second time. The supernatant (S) and pellet (P) fractions were resolved on SDS-polyacrylamide gels and analyzed by immunoblotting using anti-HA, anti-GFP, and anti-PCNA antibodies. Abc3-GFP, a PI-PLC-resistant transmembrane protein was used as control. The positions of the molecular mass (M) of protein standards (in kDa) are indicated on the left-hand side. NS, nonspecific signal. Results are representative of three independent experiments.
Shu1-mediated Pathway Involves Delivery of the Heme Analog ZnMP into Vacuoles, followed by Its Transfer within the Cytoplasm
We have previously reported that the presence of Shu1 led to cellular accumulation of fluorescent ZnMP, a heme analog (25). To gain insight into the pathway by which ZnMP was assimilated, hem1Δ shu1Δ cells expressing a wild-type shu1+ allele were treated for 6 h in the presence of Dip (250 μm) or iron (100 μm) in the absence of δ-aminolevulinate (ALA) and hemin. Subsequently, monochlorobimane (100 μm) was first added for 3 h and then ZnMP (2 μm) was added for 0, 10, and 30 min. Under low-iron conditions and after 10 min in the presence of ZnMP, hem1Δ shu1Δ cells expressing Shu1 produced a ZnMP fluorescence signal that was primarily located in vacuoles (Fig. 2). To further confirm that ZnMP-associated fluorescence was detected in vacuoles, the vacuolar-sequestered fluorescent compound bimane-GS was used as a marker (36). At the 10-min time point, ZnMP and bimane-GS exhibited equivalent subcellular localization patterns within the cells, as well as fluorescence signals that corresponded to vacuoles (Fig. 2). At the 30-min time point, ZnMP-associated fluorescence was predominantly located in the cytoplasm and was, in most cells, largely excluded from the nucleus (Fig. 2). In the case of bimane-GS, its fluorescence signal remained in vacuoles (30-min time point). Although a hem1Δ shu1Δ double mutant harboring an empty integrating plasmid accumulated bimane-GS in vacuoles, this mutant failed to significantly increase ZnMP concentrations within the cell. In fact, its fluorescence signal was negligible as compared with a hem1Δ shu1Δ double mutant in which a wild-type shu1+ allele was expressed (Fig. 2). There was an absence of ZnMP fluorescent signal in hem1Δ shu1Δ or hem1Δ shu1Δ + shu1+ cells when they had been incubated in the presence of high iron concentrations (Fig. 2). Taken together, these results revealed that Shu1-mediated cellular accumulation of the heme analog ZnMP occurs sequentially, starting in the vacuoles and migrating later to the cytoplasm.
FIGURE 2.

Assimilation of ZnMP is initially detected in vacuoles and then within the cytoplasm. hem1Δ shu1Δ mutant cells expressing Shu1 were precultured in the presence of Dip (50 μm) or FeCl3 (100 μm), and ALA (200 μm). Cells were washed and incubated in ALA-free medium containing Dip (250 μm) or FeCl3 (100 μm) for 6 h. In the final 3 h of treatment, monochlorobimane (100 μm) was added and then ZnMP (2 μm) was added for the indicated times. A hem1Δ shu1Δ double mutant strain in which an empty plasmid was reintegrated (bottom row) was cultured and treated in an identical manner. A, iron-starved (Dip) cells were analyzed by fluorescence microscopy for accumulation of fluorescent ZnMP (far left) and bimane-GS (center left). The merged images are shown in the center right panels. Normarski optics (far right) was used to examine cell morphology. B, as negative controls, iron-treated (Fe) cells are shown because the shu1+ gene is known to be repressed under iron-replete conditions. White arrows indicate examples of vacuoles, whereas yellow arrows depict the cytoplasm where ZnMP was detected after 30 min. Results of microscopy are representative of five independent experiments.
Hemin Triggers the Traffic of Shu1 from the Cell Surface to the Vacuole
We next sought to determine whether Shu1 permanently resides at the plasma membrane or whether it could undergo cellular relocation in response to hemin. This question was addressed using the nmt+41x inducible/repressible promoter system (37). An integrative plasmid harboring shu1+-HA4 or the shu1-C72A/C87A/C92A/C101A-HA4 mutant gene expressed under control of the nmt+41x promoter was cotransformed with pSP1abc3+-GFP in hem1Δ shu1Δ abc3Δ cells. Cultures were grown in a thiamine-free medium containing Dip (50 μm) to create a pool of either Shu1-HA4 or Shu1-C72A/C87A/C92A/C101A-HA4 protein and to ensure the biosynthesis of Abc3-GFP (under the control of its own promoter) that was used as a vacuolar transmembrane resident marker (35). Cotransformed cells were then transferred to thiamine-replete and ALA-free medium for 0 and 30 min. After addition of thiamine to repress further synthesis of Shu1-HA4 or Shu1-C72A/C87A/C92A/C101A-HA4, results showed that the pool of Shu1-HA4 or its mutant derivative was localized at the plasma membrane (Fig. 3A). Results further showed that shu1+-HA4 or shu1-C72A/C87A/C92A/C101A-HA4 mRNA levels were extinguished after addition of thiamine compared with their levels of expression observed in cells at the zero time point (supplemental Fig. S1). The effect of hemin on the subcellular localization of Shu1-HA4 or Shu1-C72A/C87A/C92A/C101A-HA4 was assessed as follows. Cells were transferred in the same thiamine-replete and ALA-free medium, except that the medium was supplemented with hemin (50 μm) for 0 and 30 min. Results showed that after 30 min in the presence of hemin, Shu1-HA4 underwent relocalization from the plasma membrane to the vacuolar membrane (Fig. 3B). In contrast, the Shu1-C72A/C87A/C92A/C101A-HA4 mutant remained located at the cell surface under the same conditions that were used for the wild-type protein (Fig. 3B). To gain insight into the hemin concentrations that were required for triggering trafficking of Shu1-HA4, cells that had been transferred to thiamine-replete and ALA-free medium were treated with different concentrations of hemin (0.05, 1, 5, 10, and 50 μm) for 30 min. Results showed that 10 and 50 μm hemin fostered an effective translocation of Shu1-HA4 from the plasma membrane to the vacuolar membrane (Fig. 3C). Although 1 and 5 μm hemin altered Shu1-HA4 localization pattern as compared with that of untreated cells (Fig. 3A), it was unclear whether Shu1-HA4 accumulated at the vacuolar membrane with reference to the vacuolar transmembrane resident marker Abc3 (Fig. 3C). Results showed that 0.05 μm hemin did not alter the Shu1-HA4 localization pattern and was, for the most part, detected at the plasma membrane (Fig. 3C). Taken together, the results indicated that exposure of S. pombe to a concentration of hemin higher than 10 μm induces internalization of Shu1 from the cell surface to the vacuolar membrane.
FIGURE 3.

Hemin-dependent internalization of Shu1 from the plasma membrane to the vacuole. A, hem1Δ shu1Δ abc3Δ mutant cells were co-transformed with pSP1abc3+-GFP + pBPnmt41x-shu1+-HA4 or pSP1abc3+-GFP + pBPnmt41x-shu1C72A/C87A/C92A/C101A-HA4 plasmids and precultured in medium containing ALA (200 μm) and thiamine (5 μm). After washes, cultures were grown in thiamine-free medium containing Dip (50 μm) for 18 h. The cells were then transferred to thiamine-replete and ALA-free medium for 0 and 30 min. Indirect immunofluorescence microscopy was performed to visualize cellular location of Shu1-HA4 and its mutant derivative. Cell morphology was examined using Nomarski optics. B, co-transformed cells were cultured as described for panel A, except that they were treated with 50 μm hemin for 0 and 30 min. Cellular location of Abc3-GFP was determined by indirect immunofluorescence microscopy (Alexa Fluor Red dye as a vacuolar membrane protein marker). C, hem1Δ shu1Δ abc3Δ cells co-transformed with pSP1abc3+-GFP and pBPnmt41x-shu1+-HA4 were grown under the same conditions as described for panel A, except that they were treated with 50, 10, 5, 1, and 0.05 μm hemin for 0 and 30 min. Blue arrows indicate the plasma membrane, whereas white arrows show examples of vacuoles. Results of microscopy are representative of five independent experiments.
To further assess vacuolar localization of Shu1 in response to high hemin concentrations, yeast vacuoles were isolated from hem1Δ shu1Δ abc3Δ cells co-expressing Shu1-HA4 and Abc3-GFP. In these experiments, the expression of an integrated shu1+-HA4 allele was under the control of the nmt+41x promoter, whereas abc3+-GFP was under the control of its own promoter. Thiamine- and ALA-replete cells were initially incubated in the presence of Dip (50 μm) and monochlorobimane that allowed Abc3-GFP expression and vacuolar bimane-GS accumulation, respectively. Cells were subsequently divided into four treatment groups as follows: ALA, thiamine, and absence of hemin (group 1); ALA, absence of thiamine and hemin (group 2); absence of ALA but addition of thiamine and hemin (group 3); and, absence of ALA and thiamine but addition of hemin (group 4). After 18 h, analyses of cells by fluorescence microscopy revealed that bimane-GS was sequestered in vacuoles of cells in each experimental group (Fig. 4A). Vacuoles were purified from these different cultures and their integrity was analyzed in two ways. First, vacuole morphology was assessed by differential interference contrast (Nomarski) (Fig. 4B). Second, because of the fact that cells were cultured in the presence of monochlorobimane, which resulted in vacuolar accumulation of its fluorescent derivative bimane-GS, we observed that vacuoles maintained their integrity throughout the purification procedure as the bimane-GS-associated fluorescence was retained by the vast majority of the isolated vacuoles (Fig. 4B). Immunoblotting analysis of vacuole preparations from experimental group 4 showed that Shu1-HA4 co-fractionated with Abc3-GFP, a known resident vacuolar membrane protein (Fig. 4C). These observations led to the conclusion that removal of thiamine fostered Shu1-HA4 expression, whereas the presence of hemin triggered its trafficking from the plasma membrane to the vacuole, thereby explaining its detection in vacuoles (Fig. 4C). Furthermore, the data supported previous observations of indirect fluorescence microscopy of vacuolar localization (Fig. 3, B and C). Immunoblotting analysis of experimental groups 1 and 3 in which thiamine was present to shut off shu1+-HA4 expression, consistently failed to show the presence of Shu1-HA4 in vacuole and whole cell extract preparations (Fig. 4C). Immunoblotting analysis of vacuole preparations from experimental group 2 showed that these vacuolar preparations were negative for the presence of Shu1-HA4 (Fig. 4C), confirming that in the absence of hemin Shu1-HA4 did not undergo relocalization from the cell surface to the vacuole (Fig. 3A). However, the absence of thiamine allowed expression and therefore detection of Shu1-HA4 in whole cell extracts (Fig. 4C).
FIGURE 4.

Shu1 co-purifies with yeast vacuoles in response to hemin. A, a strain harboring a hem1Δ shu1Δ abc3Δ triple deletion was co-transformed with pBPnmt41x-shu1+-HA4 and pSP1abc3+-GFP plasmids. Co-transformed cells were precultured in medium containing ALA (200 μm) and thiamine (5 μm). After washing, cells incubated in the presence of Dip (50 μm) and monochlorobimane (100 μm) were divided into four treatment groups: ALA (200 μm), thiamine (15 μm) and no hemin (group 1); ALA (200 μm), no thiamine and no hemin (group 2); hemin (10 μm), thiamine (15 μm), and no ALA (group 3); hemin (10 μm), no thiamine and no ALA (group 4). After 18 h, cells were analyzed by fluorescence microscopy to visualize their morphology and to detect bimane-GS fluorescence (vacuole marker). B, vacuoles were purified from each group of cultures described in panel A and visualized by microscopy to observe bimane-GS fluorescence. White arrows indicate examples of vacuoles. Results of microscopy are representative of five independent experiments. C, vacuole preparations from each group of cultures described in panel A were analyzed by immunoblotting using anti-HA and anti-GFP antibodies. Abc3-GFP was used as vacuolar membrane protein marker and loading control for proteins. The positions of molecular weight standards are indicated on the left. Shu1-HA4 and Abc3-GFP are indicated with arrows.
Shu1 and Abc3 Are Required for Assimilation of Exogenous Hemin
In a previous study and results reported here, we showed that the S. pombe hem1Δ mutant strain could be maintained alive by adding exogenous ALA, which is the metabolic product that normally generates the Hem1 protein when present and functional (Fig. 5A) (25). The presence of ALA allows heme biosynthesis to start at the second step of the biosynthetic pathway. A second way to keep hem1Δ cells alive is to supplement the mutant cells with exogenous hemin (Fig. 5B). This strategy fosters hem1Δ cells to use their own heme uptake system. This experimental design (hem1Δ + hemin) ensures that heme biosynthesis is blocked, setting conditions to investigate heme acquisition by these cells. Results showed that under these experimental conditions, the heme analog ZnMP was first delivered into the vacuole and then redistributed within the cytoplasm (Fig. 2). Based on the knowledge that Abc3 is an iron-regulated vacuolar transporter in S. pombe and that its expression is required under low iron concentrations (35), we created a hem1Δ abc3Δ double mutant strain to investigate whether Abc3 may function in heme acquisition. hem1Δ abc3Δ cells were incubated in the absence of ALA and in the presence of hemin. Under these conditions, hem1Δ abc3Δ cells exhibited poor growth as compared with hem1Δ cells containing an endogenous abc3+ gene or hem1Δ abc3Δ cells in which functional untagged abc3+ and GFP-tagged abc3+ alleles were re-integrated (Fig. 5B). After 73 h, the hem1Δ abc3Δ strain displayed ∼7–8-fold less growth compared with strains that expressed functional abc3+ alleles (Fig. 5B). This phenotype (poor growth) of hem1Δ cells lacking abc3 (abc3Δ) in the absence of ALA and presence of hemin was reminiscent to the phenotype previously reported for hem1Δ shu1Δ cells (Fig. 5B) (25). When growth assays were performed using hem1Δ shu1Δ and hem1Δ abc3Δ cells containing HA4-tagged shu1-C72A/C87A/C92A/C101A and GFP-tagged abc3-P151A/C152A mutant alleles, respectively, results showed that these mutant strains exhibited poor growth as compared with hem1Δ shu1Δ and hem1Δ abc3Δ cells expressing functional shu1+-HA4 and abc3+-GFP alleles (Fig. 5B).
FIGURE 5.

Abc3 loss of function phenocopies the effect of Shu1 deletion. A, growth of the indicated yeast strains was assessed in YES medium that was supplemented with exogenous ALA (200 μm). In the case of the parental (WT) strain and a hem1Δ mutant (last strain at the bottom depicted with pink diamond forms), no exogenous ALA was added. Strain color curve codes are: wild-type (WT) in blue; hem1Δ in green (+ALA) and pink (no ALA); hem1Δ shu1Δ in red (ALA); and, hem1Δ abc3Δ in violet (ALA). B, isogenic hem1Δ, hem1Δ shu1Δ, and hem1Δ abc3Δ strains were assessed in ALA-free medium. In the case of hem1Δ shu1Δ cells, they were transformed with an empty plasmid or a plasmid harboring a HA4-tagged shu1+ or HA4-tagged shu1-C72A/C87A/C92A/C101A allele. In the case of hem1Δ abc3Δ cells, they were transformed with an empty plasmid or a plasmid harboring an untagged abc3+, GFP-tagged abc3+, or GFP-tagged abc3-P151A/C152A allele. Growth of strains was assessed in the presence of hemin (0.075 μm) but in the absence of ALA. Strain color codes were: hem1Δ in green; hem1Δ shu1Δ in red; hem1Δ abc3Δ in violet; hem1Δ shu1Δ expressing shu1+-HA4 in blue (unfilled circle); hem1Δ shu1Δ expressing shu1-C72A/C87A/C92A/C101A-HA4 in gray (empty square); hem1Δ abc3Δ expressing abc3+ in orange; hem1Δ abc3Δ expressing abc3+-GFP in black; and hem1Δ abc3Δ expressing abc3-P151A/C152A-GFP in pale purple (unfilled triangle).
Given the similar growth phenotypes of the respective hem1Δ abc3Δ and hem1Δ shu1Δ mutants in a medium lacking ALA and supplemented with low hemin concentrations (0.075 μm), we tested a potential relationship between Shu1 and Abc3 to ascertain whether they could operate in the same hemin mobilization pathway. The following isogenic strains were used for that purpose: hem1Δ shu1Δ expressing shu1+ and abc3+, hem1Δ shu1Δ harboring an empty plasmid and abc3+, hem1Δ abc3Δ expressing shu1+ and abc3+, and hem1Δ abc3Δ containing an empty plasmid and shu1+. When these strains had grown to mid-logarithmic phase, they were washed to remove ALA and then incubated in the presence of Dip (250 μm) or iron (100 μm) for 6 h. The two treatment groups were subsequently incubated in the presence of monochlorobimane (100 μm) for 3 h and then ZnMP (2 μm) for 30 min. Results consistently showed an absence of ZnMP fluorescence signal in a strain lacking shu1 (hem1Δ shu1Δ with plasmid alone and abc3+) (Fig. 6A). In contrast, when abc3 was deleted (hem1Δ abc3Δ with plasmid alone and shu1+), there was cellular accumulation of fluorescent ZnMP but it was restricted to vacuoles even after 30 min (Fig. 6A). In these cells, both ZnMP and bimane-GS fluorescent compounds were selectively accumulated in vacuoles as there was superimposition of their respective fluorescent signals (Fig. 6A). These data were consistent with Abc3 acting downstream of Shu1 and suggested that, in the absence of Abc3, there was vacuolar accumulation of ZnMP due to the inability of yeast cells to mobilize stored ZnMP from the vacuole to replenish the cytoplasm. In the case of hem1Δ shu1Δ shu1+ abc3+ and hem1Δ abc3Δ shu1+ abc3+ cells that had been incubated in the presence of Dip, intracellular ZnMP fluorescent signal was primarily located in the cytoplasm over a time period of 30 min (Fig. 6A). There was an absence of ZnMP fluorescent signal in all of the above-mentioned strains when they had been treated under iron-replete conditions (Fig. 6B). Taken together, these data suggested that Abc3 is required to provide bioavailable heme analog ZnMP via its mobilization from the vacuole to the cytoplasm.
FIGURE 6.

Deletion of abc3+ leads to vacuolar accumulation of ZnMP. The indicated isogenic yeast strains were precultured in the presence of Dip (50 μm) or FeCl3 (Fe, 100 μm), and ALA (200 μm). Cells were washed and incubated in ALA-free medium and treated with Dip (250 μm) or FeCl3 (100 μm) for 6 h. Monochlorobimane (100 μm) was then added for the last 3 h of culture. Subsequently, ZnMP (2 μm) was added for 30 min. A, iron-starved (Dip) cells were analyzed by fluorescence microscopy for accumulation of fluorescent ZnMP (far left) and bimane-GS (center left). The merged images are shown in the center right panels. Normarski optics (far right) was used to ascertain cell morphology. B, as negative controls, iron-treated strains are shown because shu1+ and abc3+ genes are known to be repressed under iron-replete conditions. White arrows indicate examples of vacuoles where bimane-GS was sequestered. Yellow arrows depict cytoplasmic accumulation of ZnMP. Results of microscopy are representative of five independent experiments.
Binding of Abc3 to Hemin-agarose
To determine whether Abc3 was able to interact with hemin, wild-type shu1+-HA4 and abc3+-GFP fusion alleles were co-transformed in a hem1Δ shu1Δ abc3Δ triple mutant strain. When cells had grown to mid-log phase, they were washed to remove ALA and then incubated in the presence of Dip (250 μm) or FeCl3 (100 μm) for 6 h. Whole protein extracts from these cells were prepared and cell membranes were fractionated and isolated by ultracentrifugation. Membrane preparations were solubilized with Triton X-100 and then re-fractionated by performing a second ultracentrifugation. Dissolved membrane protein fraction that contained released Shu1-HA4 and Abc3-GFP proteins was mixed with hemin-agarose or agarose (control) beads. Results showed that Abc3-GFP isolated from iron-starved cells was retained on hemin-agarose beads (Fig. 7, bound). Only a small fraction of Abc3-GFP was detected in the flow-through fraction (Fig. 7, unbound). As a control of known heme-binding protein, Shu1-HA4 was retained on hemin-agarose beads (Fig. 7, bound). Only a minor fraction of Shu1-HA4 was detected in the unbound fraction (Fig. 7). On the other hand, Abc3-GFP and Shu1-HA4 were primarily detected in the unbound fraction (flow-through) when agarose beads (control) were used (Fig. 7). Consistent with iron-mediated repression of abc3+-GFP and shu1+-HA4 mRNA levels (under the control of abc3+ and shu1+ promoters, respectively), Abc3-GFP and Shu1-HA4 proteins were barely or not detected in membrane protein fractions that had been prepared from iron-replete cells (Fig. 7). Taken together, these results showed that Abc3 expressed in S. pombe interacts with hemin, supporting the proposed model that Abc3 is required as a means to mobilize stored heme from the vacuole to the cytoplasm.
FIGURE 7.

Abc3 is a hemin-binding protein. A, hem1Δ shu1Δ abc3Δ cells expressing HA4-tagged Shu1 and GFP-tagged Abc3 were precultured in the presence of ALA (200 μm) and Dip (50 μm). After washing, cells were transferred to ALA-free medium. At this point, they were treated with Dip (250 μm) or FeCl3 (100 μm) for 6 h and then whole cell extracts were prepared. Cell membranes were obtained by ultracentrifugation of whole cell extracts and treated with Triton X-100 to release membrane proteins. Triton X-100-solubilized membrane proteins (input) were subjected to hemin pulldown assays using hemin-agarose or agarose alone. Unbound and bound protein fractions were analyzed by immunoblot assays using anti-GFP, anti-HA, and anti-PCNA antibodies. Soluble fractions contain proteins found in the supernatant after ultracentrifugation of whole cell extract preparations. The positions of molecular weight standards are indicated on the left. Abc3-GFP, Shu1-HA4, and PCNA are indicated with arrows.
An inverted CP motif (Pro151-Cys152) is present within the sequence of a predicted loop region of Abc3. CP motif and some cases of inverted CP motif (Pro-Cys) have been reported to exhibit heme-binding propensity (38–43). We therefore assessed whether Pro151 and Cys152 residues were involved in the ability of Abc3 to interact with hemin. Pro151 and Cys152 residues were replaced with alanine residues and the mutated protein expressed in hem1Δ shu1Δ abc3Δ shu1+-HA4 cells. Cultures were washed to remove ALA and then incubated for 6 h in the presence of Dip (250 μm) or iron (100 μm). Under low iron conditions, the Abc3-P151A/C152A-GFP mutant protein was located on the membrane of vacuoles, as observed in the case of the wild-type Abc3-GFP protein (Fig. 8A). However, there was an absence of fluorescent signal in the case of wild-type or mutant form of GFP-tagged Abc3 in iron-treated cells (Fig. 8A). Membrane fractions collected after ultracentrifugation were treated with Triton X-100 and the solubilized membrane proteins were subjected to pulldown assays using hemin-agarose beads. Results showed that Abc3-P151A/C152A-GFP was almost exclusively detected in the unbound (flow-through) fraction, consistent with a loss of affinity for hemin (Fig. 8B). There was undetectable amount of Abc3-P151A/C152A-GFP in the dissolved membrane fractions of iron-treated cells (Fig. 8B). As a control, Shu1-HA4 was retained on hemin-agarose beads (Fig. 8B, bound). Only a minor fraction of Shu1-HA4 was detected in the unbound fraction (Fig. 8B). As an additional control for specificity of the resin, Abc3-GFP and Shu1-HA4 were mainly detected in the unbound fraction when agarose beads were used (Fig. 8B). Together, these results strongly suggested that the inverted CP motif (residues 151–152) of Abc3 is required for its ability to bind hemin.
FIGURE 8.

Subcellular localization and hemin-agarose pulldown assays with a mutant version of Abc3. A, fluorescence microscopy of subcellular location of Abc3-GFP or Abc3-P151A/C152A-GFP that was co-expressed with Shu1-HA4 in hem1Δ shu1Δ abc3Δ cells in the presence of Dip (250 μm) or FeCl3 (Fe, 100 μm). Nomarski optics were used to monitor cell morphology. Results of microscopy are representative of five independent experiments. B, whole cell extracts were prepared from cells expressing the mutant version of Abc3 under conditions described above for panel A. Supernatant (soluble proteins) and pellet (membrane proteins) fractions were prepared by ultracentrifugation from whole cell extracts. Triton X-100-solubilized proteins (input) were subjected to hemin pulldown assays using hemin-agarose or agarose. Unbound and bound fractions were analyzed by Western blots. Proteins were revealed using an anti-GFP, anti-HA, or anti-PCNA antibody. The positions of molecular weight standards are indicated on the left. Abc3-GFP, Shu1-HA4, and PCNA are indicated with arrows.
To further assess the effect of the P151A and C152A mutations on Abc3 function, a plasmid harboring the wild-type abc3+ or abc3-P151A/C152A-GFP mutant or an empty plasmid was transformed into a hem1Δ abc3Δ shu1+ strain. Mid-logarithmic cell cultures were washed to remove ALA and then treated with Dip (250 μm) or iron (100 μm) for 6 h. During the last 3 h of growth, cultures were incubated in the presence of monochlorobimane (100 μm) and then treated with ZnMP (2 μm) for 30 min. Results showed that in the case of cells expressing wild-type abc3+ allele under low-iron conditions, ZnMP-associated fluorescence was predominantly detected in the cytosol 30 min after ZnMP treatment (Fig. 9A). Under the same conditions, hem1Δ abc3Δ shu1+ cells expressing abc3-P151A/C152A-GFP mutant allele or an empty plasmid exhibited cellular accumulation of fluorescent ZnMP in vacuoles (Fig. 9A). The resulting fluorescent ZnMP pattern was identical to that of the bimane-GS fluorescent compound, which is known to selectively accumulate in vacuoles (Fig. 9A). As controls, hem1Δ shu1Δ abc3+ cells were transformed with a plasmid expressing shu1+ or an empty plasmid and were subsequently cultured under conditions identical to those described above for hem1Δ abc3Δ shu1+ cells. In the case of cells expressing wild-type shu1+, intracellular ZnMP fluorescent signal was seen primarily in the cytoplasm after 30 min of ZnMP treatment (Fig. 9A). In contrast, in the case of cells lacking Shu1 and harboring an empty plasmid, they failed to accumulate the ZnMP fluorescent compound in comparison with hem1Δ shu1Δ abc3+ cells expressing a wild-type shu1+ that was re-integrated (Fig. 9A). There was an absence of ZnMP fluorescent signal in all the transformed strains when they had been pretreated with high concentrations of iron (Fig. 9B). Taken together, these results suggested that mobilization of a heme analog (ZnMP) from the vacuole to the cytosol requires Abc3 and involves an inverted CP motif (residues 151–152) within its amino-terminal region.
FIGURE 9.

Expression of Abc3-P151A/C152A mutant protein alters ZnMP cellular distribution, leading to its vacuolar accumulation. A, the indicated isogenic yeast strains were cultured in the same manner as described in the legend to Fig. 6. After a 30-min treatment with ZnMP, iron-starved cells were examined by fluorescence microscopy to visualize cellular distribution of fluorescent ZnMP (far left). Vacuoles were detected by bimane-GS staining and cell morphology by Nomarski optics (Nomarski). Merged images of ZnMP and bimane-GS fluorescent signals (center right) are shown next to Nomarski microscope images. B, as negative controls, iron-treated strains are shown because shu1+ and abc3+ genes are known to be repressed under iron-replete conditions. White arrows indicate examples of vacuoles where bimane-GS was sequestered, whereas yellow arrows depict cytoplasmic accumulation of ZnMP. Results of microscopy are representative of five independent experiments.
Discussion
Although heme is an essential prosthetic group for several proteins and can serve as a source of iron, cellular components that are required for heme assimilation and iron acquisition from heme are still poorly understood. Using the model organism S. pombe, our previous studies have identified Shu1 as a novel cell membrane protein involved in assimilation of exogenous heme (25). Shu1 is predicted to contain a GPI anchor that attaches the protein to the membrane (44). If this were the case, exposing the cell membranes to exogenous PI-PLC should release Shu1 from its anchor. Here, results showed that a PI-PLC treatment triggered the release of Shu1, suggesting removal of membrane-bound Shu1 from its GPI anchor. These results lent support to the prediction of a Shu1-associated GPI-attachment site (ω-site) corresponding to Ser199. In the case of Shu1, the ω-site is followed by 2 relatively short side chain amino acid residues (Ser200 and Ala201), especially in the case of ω+2 position (Ala201). This motif is known to favor the addition of a GPI anchor at a Ser residue. One additional favorable feature is that the polypeptide region downstream of the ω, ω+1, and ω+2 residues of Shu1 is constituted of a putative spacer region of ∼13 amino acid residues, followed by a predicted hydrophobic tail of 12 residues. Overall, the predicted Shu1 GPI attachment site and its downstream region exhibit characteristics that are commonly found in GPI-anchored proteins.
Our previous studies have shown that shu1+ is regulated at the level of gene transcription (25). It is induced under conditions of iron starvation and turned off under iron-replete conditions. To investigate whether Shu1 underwent post-translational regulation, we studied changes in Shu1 localization in the presence of low (0.075 μm) and high (50 μm) hemin concentrations. A functional shu1+-HA4 allele was expressed under the control of a thiamine-sensitive nmt+ promoter to address this question. Biosynthesis of Shu1-HA4 occurred in the absence of thiamine but the addition of thiamine repressed further synthesis. We then investigated the effect of hemin on the subcellular localization of Shu1-HA4. When cells were treated with low concentrations of hemin, Shu1 localized at the cell surface, whereas under conditions of high concentrations of hemin, Shu1 was detected on vacuolar membrane. Consistent with these results, we observed that the activity of Shu1 led to vacuolar accumulation of ZnMP after 10 min. Subsequently, ZnMP fluorescent signal was progressively found in the cytoplasm after 30 min. These results were reminiscent of findings for C. albicans Rbt5. Using fluorescently rhodamine-tagged hemoglobin, C. albicans cells expressing Rbt5 accumulate rhodamine-tagged hemoglobin in the vacuoles and then to a lesser extent in the cytosol (17).
After it has reached the plasma membrane, Shu1 then internalized in response to heme. The mechanism of Shu1 internalization remains unclear at this point because its anchoring to the membrane by a GPI structure yields a protein that lacks a cytoplasmic domain that could be used as a target by an endocytic pathway. One possible explanation is that Shu1 is involved in lateral interactions with other plasma membrane components, which could serve as direct targets to be modified and taken up as a heteromeric plasma membrane complex in endocytic vesicles. Alternatively, there are a few reported cases of GPI-anchored proteins as in the case of the prion protein that leave plasma membrane lipid “raft” domains to enter clathrin-coated pits and then undergo endocytosis in coated vesicles (45). In the case of the prion protein, its amino-terminal domain is sufficient to trigger internalization within the cell (46). Although most cell-surface proteins appear to be internalized by dynamin-dependent pathways that include the clathrin-coated mediated pathway, a large proportion of GPI-anchored proteins are selectively internalized through a dynamin-independent pathway (45, 47). These findings have led to the proposal that several GPI-anchored proteins are endocytosed by pinocytosis through specialized early endosomal compartments known as GEECs (GPI-anchored protein-enriched early endosomal compartments) (45, 48). Because GEECs result from fusion of primary uncoated clathrin-independent vesicular carriers called CLICs (derived from the cell surface), this process is called the CLIC/GEEC pathway (47). However, the CLIC/GEEC pathway has not yet been described in fungi, including S. pombe, which makes this process of internalization speculative to explain endocytic internalization Shu1.
Although the mechanism by which heme-dependent GPI-anchored CFEM-like proteins undergo cellular internalization remains unclear, it appears that the endocytic process may involve the endosomal sorting complex required for transport (ESCRT) machinery (49, 50). The ESCRT machinery is composed of four complexes, ESCRT-0, -I, -II, and -III, and other cellular components such as the Vps4 complex (51). ESCRT complexes capture endosomal membrane proteins that are monoubiquitinated and deliver them to the multivesicular body compartment and, from there, to the vacuole (yeast) or lysosome (mammalian cell), where ubiquitinated cargos are degraded. In C. albicans, the cell-surface GPI-anchored Rbt5 and Rbt51 proteins bind hemin and hemoglobin (16). These proteins are absent in Saccharomyces cerevisiae, which is unable to take up exogenous hemin or hemoglobin. Interestingly, heterologous expression of C. albicans Rbt51 in S. cerevisiae enables acquisition and utilization of exogenous hemoglobin (17). However, S. cerevisiae mutants lacking ESCRT-II and -III components (such as Vps22 and Vps32) are defective in assimilation of exogenous hemoglobin (17). Furthermore, C. albicans ESCRT complex mutants exhibit significant growth defects in media containing hemoglobin as the sole iron source (17). Proteins of the ESCRT pathway have also been found to be required for hemin use in C. neoformans (23, 24). In addition to ESCRT-II (Vps22) and -III (Vps20 and Vps32) components, the subunit Vps23 of the ESCRT-I complex contributes to hemin uptake (24). However, there is yet absence of evidence that Rbt5/51 and Cig1 represent ubiquitinated cargos of the C. albicans and C. neoformans ESCRT proteins, respectively. Given that the ESCRT machinery is committed to targeting of membrane proteins to the vacuole and the fact that Shu1 undergoes relocalization from the cell surface to the vacuole in response to hemin, it is likely that components of the ESCRT machinery are required for Shu1 internalization. If this were the case, then Shu1 may become ubiquitinated upon its association with hemin. However, it is far from clear how Shu1 would become ubiquitinated. In S. pombe, an alternative route for cargos to reach the ESCRT pathway is through an association with Nbr1 (52, 53). Upon formation of a cargo-Nbr1 complex, there is ubiquitination of Nbr1 and that results in the sequestration of the cargo-Nbr1 complex inside of MBVs in a ESCRT-dependent manner. Subsequently, the cargo-Nbr1 complex is delivered to the vacuole where the proteins that are bound by Nbr1 are released into the vacuole (52).
Our results revealed that Shu1 is required for internalization of hemin or ZnMP, which is then delivered to the vacuole. Once in vacuoles, hemin (or ZnMP) may undergo further degradation, resulting in the release of iron (or zinc) from the protoporphyrin ring. This series of events would constitute a specialized pathway and make the vacuole an important site for intracellular hemin and iron stores. S. pombe is one of the yeast species that does not express homologs of S. cerevisiae vacuolar iron transporters (Smf3, Fth1, and Fet5) or heme oxygenase (Hmx1) like that found in C. albicans. Instead, we have previously shown that when S. pombe undergoes a transition from high to low iron concentrations, the vacuolar transmembrane protein Abc3 is highly induced and participates in the mobilization of stored iron sources from the vacuole (35). However, the nature of the usable form of iron that can be transported by Abc3 remains unclear. Results of pulldown assays using hemin-agarose showed that Abc3-GFP expressed in iron-starved cells was specifically retained on the beads. This observation was in agreement with the fact that Abc3 possesses an inverted CP motif that is predicted to be located at the beginning of an exposed hydrophilic linker region (denoted L0), which separates the N-terminal NTE domain of Abc3 from its ABC core domain. According to a predicted three-dimensional model by I-TASSER (54), the inverted CP motif (Pro151 and Cys152) of Abc3 is oriented toward the vacuolar side. In several hemoproteins such as FeoB (E. coli), IRP2 (human), and DP8 (human), the CP motif is part of a central heme-binding domain due to the presence of the Pro side chain that fosters a distinct conformation of the Cys thiol group. This property enables Cys to coordinate heme through its thiol group. Other examples of hemoproteins that include GlpF (E. coli) and DGCR8 (human) exhibit an inverted CP motif that is sufficient to confer heme binding capacity (42). Here, when the inverted CP motif (Pro151-Cys152) was mutated in Abc3, cells expressing the abc3-P151A/C152A allele selectively accumulated the heme analog ZnMP in vacuoles and were unable to subsequently trigger its redistribution in the cytoplasm in comparison to cells expressing a wild-type abc3+ allele. Furthermore, pulldown assays using protein lysates prepared from cells expressing Abc3-P151A/C152A showed that substitutions of Pro151-Cys152 residues (in Ala) strongly decreased Abc3 binding to hemin-agarose. These results suggested that the inverted CP motif (Pro151-Cys152) participates in hemin coordination in Abc3. However, whether one residue contributes more than another or whether there is influence of additional amino acid residues flanking the motif or elsewhere will require additional studies.
Data reported here concerning Abc3 are reminiscent of those observed in the case of the human ABCG2 protein, which is an ABC transporter known to be involved in the export of heme, hemin, and ZnMP (38, 55). Although Abc3 and ABCG2 are members of different ABC protein subfamilies, they possess a large loop that contains an inverted CP motif. In the case of ABCG2, the ECL3 loop located between transmembrane domains 5 and 6 contains an inverted CP motif (Pro602-Cys603) (38). Studies showed that a single-point mutation C603A inside ECL3 dramatically decreases the binding of hemin to ABCG2 (38). Given the fact that results described in this report as well as previous studies (17) have implicated the vacuole as a player in heme assimilation and assuming that vacuolar heme is present in a usable form, we suggest that Abc3 is an intracellular transporter that mobilizes stores of heme from this organelle for redistribution within the cytoplasm when cells face heme/iron deprivation.
Experimental Procedures
Strains and Growth Conditions
Genotypes of S. pombe strains used in this study are listed in Table 1. Under nonselective conditions, all strains were grown on yeast extract medium (YES) containing 0.5% yeast extract, 3% glucose, and 225 mg/liter of adenine, histidine, leucine, uracil, and lysine. Strains for which plasmid integration or transformation was required were grown on synthetic Edinburgh minimal medium (EMM) missing specific nutrients that allowed selection and maintenance of the integrated DNA fragment or plasmid (56). In the case of hem1Δ deletion strains, they were supplemented with ALA (200 μm) to allow heme biosynthesis starting at the second step of the biosynthetic pathway. An alternative way to maintain hem1Δ cells alive in the absence of ALA was to add exogenous hemin (heme chloride, 0.075 μm) to foster cells to use their heme uptake system instead of their own heme biosynthesis pathway. For monitoring cell growth, precultures of cells were carried out in YES medium containing Dip (50 μm) to chelate iron and, at the same time, fostering up-regulation of iron-dependent genes, including shu1+ and abc3+. During precultures of cells, ALA (200 μm) was added to ensure biosynthesis of heme. Once precultures reached mid-logarithmic phase, cells were washed twice and then diluted 1000-fold in YES containing Dip (250 μm) and hemin (0.075 μm) but in the absence of ALA (unless otherwise stated). Cell cultures were then initiated and monitored (A600) at each of the indicated times.
TABLE 1.
S. pombe strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| FY435 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 | 60 |
| TMY1 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::KANr | 25 |
| TMY2 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | 25 |
| shu1Δ::KANr | ||
| TMY4 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | 25 |
| shu1Δ::loxP abc3Δ::KANr | ||
| TMY5 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | 25 |
| shu1Δ::KANr shu1+::ade6+ | ||
| TMY6 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | 25 |
| shu1Δ::KANr shu1+-HA4::ade6+ | ||
| TMY7 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | 25 |
| shu1Δ::loxP abc3Δ::KANr shu1+-HA4::ade6+ abc3+-GFP::leu+ | ||
| TMY9 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | This study |
| shu1Δ::loxP abc3Δ::KANr shu1-C72A/C87A/C92A/C101A-HA4::ade6+ | ||
| abc3+-GFP::leu+ | ||
| TMY10 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP | This study |
| shu1Δ::loxP abc3Δ::KANr nmt41x-shu1+-HA4::ade6+ abc3+-GFP::leu+ | ||
| TMY11 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP | This study |
| abc3Δ::KANr nmt41x-shu1-C72A/C87A/C92A/C101A-HA4::ade6+ | ||
| abc3+-GFP::leu+ | ||
| TMY12 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP abc3Δ::KANr | This study |
| TMY13 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP abc3Δ::KANr | This study |
| abc3+::leu+ | ||
| TMY14 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP abc3Δ::KANr | This study |
| abc3+-GFP::leu+ | ||
| TMY15 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP | This study |
| abc3Δ::KANr shu1+-HA4::ade6+ abc3-P151A/C152A-GFP::leu+ | ||
| TMY16 | h+ his7–366 leu1–32 ura4-Δ18 ade6-M210 hem1Δ::loxP abc3Δ::KANr | This study |
| abc3-P151A/C152A-GFP::leu+ |
Plasmids
Plasmids pBP-1317shu1+-HA4, pBP-1317shu1-C72A/C87A/C92A/C101A-HA4, pSP1abc3+, and pSP1abc3+-GFP have been described previously (25, 35). The nmt1+ promoter region up to position −1178 from the initiator codon of the nmt1+ gene was isolated from pREP41x by PCR (37). PCR amplification was performed using primers designed to generate ApaI and XmaI sites at the 5′ and 3′ termini of the promoter region. Subsequently, the ApaI-XmaI nmt1+ promoter fragment was exchanged to replace the shu1+ promoter using the ApaI and XmaI restriction sites found in plasmids pBP-1317shu1+-HA4 and pBP-1317shu1-C72A/C87A/C92A/C101A-HA4. The resulting plasmids were denoted pBPnmt41x-shu1+-HA4 and pBPnmt41x-shu1-C72A/C87A/C92A/C101A-HA4, respectively. Plasmid pSP1abc3+-GFP was used to introduce mutations in the coding sequence of abc3+. Codons corresponding to Pro151 and Cys152 were replaced by nucleotide triplets that encode alanine. These site-specific mutations were created by a PCR overlap extension method (57). The resulting PCR fragment containing the abc3 mutant allele with P151A and C152A mutations was then swapped for the equivalent SpeI-XhoI DNA fragment in pSP1abc3+-GFP, creating pSP1abc3P151A/C152A-GFP plasmid.
PI-PLC Treatment
hem1Δ shu1Δ abc3Δ cells co-expressing shu1+-HA4 and abc3+-GFP alleles were treated with Dip (250 μm) for 6 h. After centrifugation, cells were resuspended in buffer F (100 mm Tris-HCl, pH 9.4, and 20 mm β-mercaptoethanol) as a pretreatment. Cells were harvested and incubated in the presence of zymolyase (1 mg/ml) and novozyme (3 mg/ml) using a buffer containing 10 mm β-mercaptoethanol, 1.2 m sorbitol, and 20 mm K2HPO4/KH2PO4, pH 7.5. Spheroplast preparations were washed, resuspended in HEGN100 buffer (20 mm HEPES, pH 7.9, 100 mm NaCl, 1 mm EDTA, 10% glycerol, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, and protease inhibitors), and disrupted using FastPrep lysing glass beads. Lysates were ultracentrifuged (100,000 × g) for 30 min at 4 °C. The supernatant containing soluble proteins was set aside, whereas the pellet fraction was divided into two equal fractions that were, respectively, used for PI-PLC treatment (0.5 units/100 μg of protein) and control (lack of enzyme treatment). Following incubation at 30 °C for 2.5 h, PI-PLC-treated and untreated fractions were centrifuged (100,000 × g) to obtain a supernatant containing proteins that had been released from the membrane. In the case of untreated fractions, proteins were still membrane-bound. Samples were added to 2× SDS loading buffer (100 mm Tris-HCl, pH 8.0, 5 mm EDTA, 15% SDS, 0.01% bromphenol blue) containing 8.0 m urea and 4% β-mercaptoethanol, unless otherwise indicated. After incubation for 30 min at 37 °C, samples were resolved by electrophoresis on 10 (Shu1-HA4) and 6% (Abc3-GFP) SDS-polyacrylamide gels and transferred on membrane for Western blot analysis. The following antibodies were used for immunodetection of Shu1-HA4, Abc3-GFP, α-tubulin, and PCNA: monoclonal anti-HA antibody F-7; monoclonal anti-GFP antibody B-2 (Santa Cruz Biotechnology); monoclonal anti-α-tubulin antibody (clone B-5-1–2, Sigma); and monoclonal anti-PCNA antibody PC10 (Sigma). Following incubation, membranes were washed and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences), developed with enhanced chemiluminescence (ECL) reagents (Amersham Biosciences), and visualized by chemiluminescence using an ImageQuant LAS 4000 instrument (GE Healthcare) equipped with a Fujifilm High Sensitivity F0.85 43-mm camera.
Fluorescence Microscopy
For detection of fluorescent ZnMP and bimane-GS accumulation within cells, liquid cultures of the indicated strains were seeded to an A600 of ∼0.3. The cultures were then incubated in ALA-deficient medium containing Dip (250 μm) or FeCl3 (100 μm) for 6 h. At mid-logarithmic phase, monochlorobimane (100 μm) was first added to cells for 3 h and then ZnMP (2 μm) was added for 0, 10, and 30 min, unless otherwise indicated. ZnMP accumulation was stopped by adding 5 volumes of ice-cold 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). After centrifugation, cells were resuspended in ice-cold 2% BSA in PBS and examined by fluorescence microscopy using a ×1,000 magnification and concanavalin A-coated glass slides. Fluorescence and differential interference contrast images (Nomarski) of cells were obtained using a Nikon Eclipse E800 epifluorescent microscope (Nikon, Melville, NY) equipped with a Hamamatsu ORCA-ER digital cooled camera (Hamamatsu, Bridgewater, NJ). Fields of cells shown in this study correspond to a minimum of five independent experiments.
Indirect Immunofluorescence Microscopy
hem1Δ shu1Δ abc3Δ cells were co-transformed with pBPnmt41x-shu1+-HA4 + pSP1abc3+-GFP or pBPnmt41x-shu1-C72A/C87A/C92A/C101A-HA4 + pSP1abc3+-GFP. Co-transformed cells were precultivated in the presence of thiamine (5 μm) and ALA (200 μm) to an A600 of 1.0. At this growth point, the cells were washed twice to remove thiamine and diluted 10-fold in EMM containing Dip (50 μm). After 18 h, cells were transferred to ALA-free and thiamine-replete medium to stop shu1+-HA4 or shu1-C72A/C87A/C92A/C101A-HA4 gene expression. At this time point, the pool of plasma membrane Shu1-HA4 or Shu1-C72A/C87A/C92A/C101A-HA4 was analyzed in the absence or presence of exogenous hemin (0.05, 1, 5, 10, or 50 μm) for 0 and 30 min. Cells were fixed by adding formaldehyde (3.7%) as described previously (58). Fixed cells were harvested and washed with 0.1 m K2HPO4/KH2PO4, pH 6.5, buffer, containing 1.2 m sorbitol. Cells were spheroplasted using zymolase (30 mg/ml), β-mercaptoethanol (10 mm), and Triton X-100 (1%) as described previously (25). Spheroplasts were adsorbed on poly-l-lysine-coated (0.1%) multiwall slides as described previously (25). After a 30-min block with TBS (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% BSA, and 0.02% sodium azide), cells were incubated with anti-HA antibody F-7 (for Shu1-HA4) or anti-GFP antibody B-2 (for Abc3-GFP) diluted 1:250 in TBS. After a 18-h reaction, cells were washed with TBS and incubated for 90 min with goat anti-mouse Alexa Fluor 546 conjugate diluted 1:250 in TBS. The cells were viewed with a Nikon Eclipse E800 epifluorescent microscope. Fields of cells shown in this study correspond to a minimum of five independent experiments.
Isolation of Intact Vacuoles
Early logarithmic phase hem1Δ shu1Δ abc3Δ cells harboring pBPnmt41x-shu1+-HA4 and pSP1abc3+-GFP plasmids were incubated in the presence of Dip (50 μm) and monochlorobimane (100 μm). When an A600 value of 0.025 was reached, cells were divided into four aliquots for the following treatments: ALA (200 μm), thiamine (15 μm) and no hemin (group 1); ALA (200 μm), no thiamine and no hemin (group 2); hemin (10 μm), thiamine (15 μm) and no ALA (group 3); hemin (10 μm), no thiamine and no ALA (group 4). After 18 h, cells were subjected to cell wall digestion, disruption, and fractionation by differential centrifugation as described previously (59). After the second Percoll step gradient (50%, v/v), which is one of the last steps of vacuole isolation, the integrity of vacuole preparations was assessed by monitoring their ability to retain the fluorescent glutathione S-conjugate of monochlorobimane (bimane-GS). This approach is based on the fact that free, unsubstituted monochlorobimane is membrane permeant and nonfluorescent. However, when it is S-conjugated to glutathione by cytosolic glutathione S-transferases, the bimane-GS product becomes fluorescent and is actively transported into and sequestered within the vacuole of intact cells.
Hemin-agarose Pulldown Assays
To investigate the capacity of Abc3 to bind heme, hem1Δ shu1Δ abc3Δ cells expressing shu1+-HA4 + abc3+-GFP or shu1+-HA4 + abc3-P151A/C152A-GFP were grown to early logarithmic phase. At this point, cells were washed to remove ALA and then incubated in the presence of Dip (250 μm) or FeCl3 (100 μm) for 6 h. Whole extracts were prepared with glass beads using a RIPA buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 1% SDS, 8 m urea, 0.1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, and a complete protease inhibitor mixture (Sigma; P8340). Whole cell extracts were ultracentrifuged (100,000 × g) for 30 min at 4 °C. The supernatant containing soluble proteins was kept, whereas the pellet fraction was resuspended in buffer A (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol and protease inhibitors) and treated with Triton X-100 (1%). Once resuspended, the pellet fraction was kept on ice and then re-fractionated by performing a second ultracentrifugation. In the presence of Triton X-100, a nonionic detergent that solubilizes membranes, Shu1-HA4, Abc3-GFP, and Abc3-P151A/C152A-GFP-GFP, were largely released from membranes, and the supernatant preparation was treated with hemin-agarose or agarose beads as described previously (25). The immunoprecipitates and unbound material were resuspended and mixed, respectively, with loading buffer (100 mm Tris-HCl, pH 8.0, 1.4 m β-mercaptoethanol, 140 mm SDS, 5 mm EDTA, 8 m urea, and 0.72 mm bromphenol blue) and heated for 30 min at 37 °C. Samples were resolved by electrophoresis on 10 (Shu1-HA4) and 6% (Abc3-GFP and Abc3-P151A/C152A-GFP-GFP) SDS-polyacrylamide gels and were transferred to membrane for Western blot analysis.
Author Contributions
T. M. planned, designed, and performed most of the experiments. V. N. produced several DNA constructs, including Abc3-GFP mutant plasmids and performed immunoblotting experiments. T. M., V. N., and S. L. analyzed data. T. M. and S. L. conceptualized research and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Dr. Gilles Dupuis for critical reading of the manuscript and valuable comments. We thank Ariane Millette-St-Hilaire for excellent technical assistance.
Note Added in Proof
In the version of this article that was published as a Paper in Press on February 13, 2017, the PCNA immunoblot shown for the pellet fraction in Fig. 1 was inadvertently shifted. Additionally, an incorrect Nomarski image was inserted into the Abc3-GFP column in Fig. 3C. These errors have been corrected and do not affect the results or conclusions of this work.
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Grant RGPIN-2015/2020-04878 (to S. L.). The authors declare that they have no conflict of interest with the content of this article.

This article contains supplemental Fig. S1.
- CFEM
- common in fungal extracellular membranes
- ALA
- δ-aminolevulinate
- Dip
- 2,2′-dipyridyl
- EMM
- Edinburgh minimal medium
- GPI
- glycosylphophatidylinositol
- PCNA
- proliferating cell nuclear antigen
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- YES
- yeast extract plus supplements
- ZnMP
- zinc(II) mesoporphyrin IX
- GEEC
- GPI-anchored protein-enriched early endosomal compartments
- CLIC
- clathrin-independent vesicular carriers
- ESCRT
- endosomal sorting complex required for transport.
References
- 1. Kaplan J., and Ward D. M. (2013) The essential nature of iron usage and regulation. Curr. Biol. 23, R642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paul V. D., and Lill R. (2015) Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 1853, 1528–1539 [DOI] [PubMed] [Google Scholar]
- 3. Philpott C. C., Leidgens S., and Frey A. G. (2012) Metabolic remodeling in iron-deficient fungi. Biochim. Biophys. Acta 1823, 1509–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Severance S., and Hamza I. (2009) Trafficking of heme and porphyrins in metazoa. Chem. Rev. 109, 4596–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hamza I., and Dailey H. A. (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 1823, 1617–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reddi A. R., and Hamza I. (2016) Heme mobilization in animals: a Metallolipid's journey. Acc. Chem. Res. 49, 1104–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rajagopal A., Rao A. U., Amigo J., Tian M., Upadhyay S. K., Hall C., Uhm S., Mathew M. K., Fleming M. D., Paw B. H., Krause M., and Hamza I. (2008) Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453, 1127–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White C., Yuan X., Schmidt P. J., Bresciani E., Samuel T. K., Campagna D., Hall C., Bishop K., Calicchio M. L., Lapierre A., Ward D. M., Liu P., Fleming M. D., and Hamza I. (2013) HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 17, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huynh C., Yuan X., Miguel D. C., Renberg R. L., Protchenko O., Philpott C. C., Hamza I., and Andrews N. W. (2012) Heme uptake by Leishmania amazonensis is mediated by the transmembrane protein LHR1. PLoS Pathog. 8, e1002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duffy S. P., Shing J., Saraon P., Berger L. C., Eiden M. V., Wilde A., and Tailor C. S. (2010) The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell. Biol. 30, 5318–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shayeghi M., Latunde-Dada G. O., Oakhill J. S., Laftah A. H., Takeuchi K., Halliday N., Khan Y., Warley A., McCann F. E., Hider R. C., Frazer D. M., Anderson G. J., Vulpe C. D., Simpson R. J., and McKie A. T. (2005) Identification of an intestinal heme transporter. Cell 122, 789–801 [DOI] [PubMed] [Google Scholar]
- 12. Kulkarni R. D., Kelkar H. S., and Dean R. A. (2003) An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem. Sci. 28, 118–121 [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z. N., Wu Q. Y., Zhang G. Z., Zhu Y. Y., Murphy R. W., Liu Z., and Zou C. G. (2015) Systematic analyses reveal uniqueness and origin of the CFEM domain in fungi. Sci. Rep. 5, 13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuznets G., Vigonsky E., Weissman Z., Lalli D., Gildor T., Kauffman S. J., Turano P., Becker J., Lewinson O., and Kornitzer D. (2014) A relay network of extracellular heme-binding proteins drives C. albicans iron acquisition from hemoglobin. PLoS Pathog. 10, e1004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nasser L., Weissman Z., Pinsky M., Amartely H., Dvir H., and Kornitzer D. (2016) Structural basis of haem-iron acquisition by fungal pathogens. Nat. Microbiol. 1, 16156. [DOI] [PubMed] [Google Scholar]
- 16. Weissman Z., and Kornitzer D. (2004) A family of Candida cell surface haem-binding proteins involved in haemin and haemoglobin-iron utilization. Mol. Microbiol. 53, 1209–1220 [DOI] [PubMed] [Google Scholar]
- 17. Weissman Z., Shemer R., Conibear E., and Kornitzer D. (2008) An endocytic mechanism for haemoglobin-iron acquisition in Candida albicans. Mol. Microbiol. 69, 201–217 [DOI] [PubMed] [Google Scholar]
- 18. Pendrak M. L., Chao M. P., Yan S. S., and Roberts D. D. (2004) Heme oxygenase in Candida albicans is regulated by hemoglobin and is necessary for metabolism of exogenous heme and hemoglobin to α-biliverdin. J. Biol. Chem. 279, 3426–3433 [DOI] [PubMed] [Google Scholar]
- 19. Santos R., Buisson N., Knight S., Dancis A., Camadro J. M., and Lesuisse E. (2003) Haemin uptake and use as an iron source by Candida albicans: role of CaHMX1-encoded haem oxygenase. Microbiology 149, 579–588 [DOI] [PubMed] [Google Scholar]
- 20. Bailão E. F., Parente J. A., Pigosso L. L., de Castro K. P., Fonseca F. L., Silva-Bailão M. G., Báo S. N., Bailão A. M., Rodrigues M. L., Hernandez O., McEwen J. G., and Soares C. M. (2014) Hemoglobin uptake by Paracoccidioides spp. is receptor-mediated. PLoS Negl. Trop. Dis. 8, e2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ding C., Vidanes G. M., Maguire S. L., Guida A., Synnott J. M., Andes D. R., and Butler G. (2011) Conserved and divergent roles of Bcr1 and CFEM proteins in Candida parapsilosis and Candida albicans. PloS One 6, e28151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cadieux B., Lian T., Hu G., Wang J., Biondo C., Teti G., Liu V., Murphy M. E., Creagh A. L., and Kronstad J. W. (2013) The Mannoprotein Cig1 supports iron acquisition from heme and virulence in the pathogenic fungus Cryptococcus neoformans. J. Infect. Dis. 207, 1339–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu G., Caza M., Cadieux B., Bakkeren E., Do E., Jung W. H., and Kronstad J. W. (2015) The endosomal sorting complex required for transport machinery influences haem uptake and capsule elaboration in Cryptococcus neoformans. Mol. Microbiol. 96, 973–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu G., Caza M., Cadieux B., Chan V., Liu V., and Kronstad J. (2013) Cryptococcus neoformans requires the ESCRT protein Vps23 for iron acquisition from heme, for capsule formation, and for virulence. Infect. Immun. 81, 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mourer T., Jacques J. F., Brault A., Bisaillon M., and Labbé S. (2015) Shu1 is a cell-surface protein involved in iron acquisition from heme in Schizosaccharomyces pombe. J. Biol. Chem. 290, 10176–10190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li L., Chen O. S., McVey Ward D., and Kaplan J. (2001) CCC1 is a transporter that mediates vacuolar iron storage in yeast. J. Biol. Chem. 276, 29515–29519 [DOI] [PubMed] [Google Scholar]
- 27. Ramsay L. M., and Gadd G. M. (1997) Mutants of Saccharomyces cerevisiae defective in vacuolar function confirm a role for the vacuole in toxic metal ion detoxification. FEMS Microbiol. Lett. 152, 293–298 [DOI] [PubMed] [Google Scholar]
- 28. Simm C., Lahner B., Salt D., LeFurgey A., Ingram P., Yandell B., and Eide D. J. (2007) Saccharomyces cerevisiae vacuole in zinc storage and intracellular zinc distribution. Eukaryot. Cell 6, 1166–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bellemare D. R., Shaner L., Morano K. A., Beaudoin J., Langlois R., and Labbé S. (2002) Ctr6, a vacuolar membrane copper transporter in Schizosaccharomyces pombe. J. Biol. Chem. 277, 46676–46686 [DOI] [PubMed] [Google Scholar]
- 30. MacDiarmid C. W., Gaither L. A., and Eide D. (2000) Zinc transporters that regulate vacuolar zinc storage in Saccharomyces cerevisiae. EMBO J. 19, 2845–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rees E. M., Lee J., and Thiele D. J. (2004) Mobilization of intracellular copper stores by the Ctr2 vacuolar copper transporter. J. Biol. Chem. 279, 54221–54229 [DOI] [PubMed] [Google Scholar]
- 32. Rees E. M., and Thiele D. J. (2007) Identification of a vacuole-associated metalloreductase and its role in Ctr2-mediated intracellular copper mobilization. J. Biol. Chem. 282, 21629–21638 [DOI] [PubMed] [Google Scholar]
- 33. Singh A., Kaur N., and Kosman D. J. (2007) The metalloreductase Fre6p in Fe-efflux from the yeast vacuole. J. Biol. Chem. 282, 28619–28626 [DOI] [PubMed] [Google Scholar]
- 34. Urbanowski J. L., and Piper R. C. (1999) The iron transporter Fth1p forms a complex with the Fet5 iron oxidase and resides on the vacuolar membrane. J. Biol. Chem. 274, 38061–38070 [DOI] [PubMed] [Google Scholar]
- 35. Pouliot B., Jbel M., Mercier A., and Labbé S. (2010) abc3+ encodes an iron-regulated vacuolar ABC-type transporter in Schizosaccharomyces pombe. Eukaryotic Cell 9, 59–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sarry J. E., Chen S., Collum R. P., Liang S., Peng M., Lang A., Naumann B., Dzierszinski F., Yuan C. X., Hippler M., and Rea P. A. (2007) Analysis of the vacuolar luminal proteome of Saccharomyces cerevisiae. FEBS J. 274, 4287–4305 [DOI] [PubMed] [Google Scholar]
- 37. Forsburg S. L. (1993) Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res. 21, 2955–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Desuzinges-Mandon E., Arnaud O., Martinez L., Huché F., Di Pietro A., and Falson P. (2010) ABCG2 transports and transfers heme to albumin through its large extracellular loop. J. Biol. Chem. 285, 33123–33133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Igarashi J., Murase M., Iizuka A., Pichierri F., Martinkova M., and Shimizu T. (2008) Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2α kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J. Biol. Chem. 283, 18782–18791 [DOI] [PubMed] [Google Scholar]
- 40. Ishikawa H., Kato M., Hori H., Ishimori K., Kirisako T., Tokunaga F., and Iwai K. (2005) Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol. Cell 19, 171–181 [DOI] [PubMed] [Google Scholar]
- 41. Kühl T., Wißbrock A., Goradia N., Sahoo N., Galler K., Neugebauer U., Popp J., Heinemann S. H., Ohlenschläger O., and Imhof D. (2013) Analysis of Fe(III) heme binding to cysteine-containing heme-regulatory motifs in proteins. ACS Chem. Biol. 8, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 42. Schubert E., Florin N., Duthie F., Henning Brewitz H., Kühl T., Imhof D., Hagelueken G., and Schiemann O. (2015) Spectroscopic studies on peptides and proteins with cysteine-containing heme regulatory motifs (HRM). J. Inorg. Biochem. 148, 49–56 [DOI] [PubMed] [Google Scholar]
- 43. Yang J., Kim K. D., Lucas A., Drahos K. E., Santos C. S., Mury S. P., Capelluto D. G., and Finkielstein C. V. (2008) A novel heme-regulatory motif mediates heme-dependent degradation of the circadian factor period 2. Mol. Cell. Biol. 28, 4697–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Groot P. W., Hellingwerf K. J., and Klis F. M. (2003) Genome-wide identification of fungal GPI proteins. Yeast 20, 781–796 [DOI] [PubMed] [Google Scholar]
- 45. Mayor S., and Riezman H. (2004) Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol. 5, 110–120 [DOI] [PubMed] [Google Scholar]
- 46. Sunyach C., Jen A., Deng J., Fitzgerald K. T., Frobert Y., Grassi J., McCaffrey M. W., and Morris R. (2003) The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 22, 3591–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mayor S., Parton R. G., and Donaldson J. G. (2014) Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 6, a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nichols B. (2009) Endocytosis of lipid-anchored proteins: excluding GEECs from the crowd. J. Cell Biol. 186, 457–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Henne W. M., Buchkovich N. J., and Emr S. D. (2011) The ESCRT pathway. Dev. Cell 21, 77–91 [DOI] [PubMed] [Google Scholar]
- 50. Raiborg C., and Stenmark H. (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452 [DOI] [PubMed] [Google Scholar]
- 51. Christ L., Raiborg C., Wenzel E. M., Campsteijn C., and Stenmark H. (2017) Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 42, 42–56 [DOI] [PubMed] [Google Scholar]
- 52. Liu X. M., Sun L. L., Hu W., Ding Y. H., Dong M. Q., and Du L. L. (2015) ESCRTs cooperate with a selective autophagy receptor to mediate vacuolar targeting of soluble cargos. Mol. Cell 59, 1035–1042 [DOI] [PubMed] [Google Scholar]
- 53. Mizushima N. (2015) Nbr1, a receptor for ESCRT-dependent endosomal microautophagy in fission yeast. Mol. Cell 59, 887–889 [DOI] [PubMed] [Google Scholar]
- 54. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krishnamurthy P., Ross D. D., Nakanishi T., Bailey-Dell K., Zhou S., Mercer K. E., Sarkadi B., Sorrentino B. P., and Schuetz J. D. (2004) The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 279, 24218–24225 [DOI] [PubMed] [Google Scholar]
- 56. Sabatinos S. A., and Forsburg S. L. (2010) Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 470, 759–795 [DOI] [PubMed] [Google Scholar]
- 57. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 58. Beaudoin J., Laliberté J., and Labbé S. (2006) Functional dissection of Ctr4 and Ctr5 amino-terminal regions reveals motifs with redundant roles in copper transport. Microbiology 152, 209–222 [DOI] [PubMed] [Google Scholar]
- 59. Sooksa-Nguan T., Yakubov B., Kozlovskyy V. I., Barkume C. M., Howe K. J., Thannhauser T. W., Rutzke M. A., Hart J. J., Kochian L. V., Rea P. A., and Vatamaniuk O. K. (2009) Drosophila ABC transporter, DmHMT-1, confers tolerance to cadmium: DmHMT-1 and its yeast homolog, SpHMT-1, are not essential for vacuolar phytochelatin sequestration. J. Biol. Chem. 284, 354–362 [DOI] [PubMed] [Google Scholar]
- 60. Pelletier B., Beaudoin J., Mukai Y., and Labbé S. (2002) Fep1, an iron sensor regulating iron transporter gene expression in Schizosaccharomyces pombe. J. Biol. Chem. 277, 22950–22958 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.