

Abstract
Protein phosphatase 2A (PP2A) plays important roles in controlling mitosis in all eukaryotic cells. The form of PP2A that controls mitosis is associated with a conserved regulatory subunit that is called B55 in vertebrates and Cdc55 in budding yeast. The activity of this form of PP2A can be inhibited by binding of conserved Igo/ENSA proteins. Although the mechanisms that activate Igo/ENSA to bind and inhibit PP2A are well understood, little is known about how Igo/Ensa are inactivated. Here, we have analyzed regulation of Igo/ENSA in the context of a checkpoint pathway that links mitotic entry to membrane growth in budding yeast. Protein kinase C (Pkc1) relays signals in the pathway by activating PP2ACdc55. We discovered that constitutively active Pkc1 can drive cells through a mitotic checkpoint arrest, which suggests that Pkc1-dependent activation of PP2ACdc55 plays a critical role in checkpoint signaling. We therefore used mass spectrometry to determine how Pkc1 modifies the PP2ACdc55 complex. This revealed that Pkc1 induces changes in the phosphorylation of multiple subunits of the complex, as well as dissociation of Igo/ENSA. Pkc1 directly phosphorylates Cdc55 and Igo/ENSA, and phosphorylation site mapping and mutagenesis indicate that phosphorylation of Cdc55 contributes to Igo/ENSA dissociation. Association of Igo2 with PP2ACdc55 is regulated during the cell cycle, yet mutation of Pkc1-dependent phosphorylation sites on Cdc55 and Igo2 did not cause defects in mitotic progression. Together, the data suggest that Pkc1 controls PP2ACdc55 by multiple overlapping mechanisms.
Keywords: cell cycle, cell growth, protein kinase C (PKC), protein phosphatase 2 (PP2A), Saccharomyces cerevisiae, B55, Cdc55, ENSA, Igo2, Pkc1
Introduction
Eukaryotic cells show enormous diversity in size and shape. The size and shape of a cell must ultimately be determined by mechanisms that control the extent of growth at specific locations. In budding yeast, dynamic changes in the pattern of growth illustrate how mechanisms that control growth at specific locations can influence cell size and shape (1–3). A newborn yeast cell undergoes isotropic growth over its entire surface during G1 phase. When sufficient isotropic growth has occurred, growth of the mother cell ceases, and polarized growth is initiated to form a bud. After a brief interval of polar growth, the daughter bud switches to isotropic growth, which continues during mitosis. The duration of each growth phase must be tightly controlled to ensure that yeast cells maintain their characteristic size and shape.
Cell size checkpoints delay key cell cycle transitions until sufficient growth has occurred (4, 5). Analysis of checkpoint signals in budding yeast led to the hypothesis that cell size and shape could be controlled by signals originating at sites of membrane growth (6). In this model, polar growth of the daughter cell membrane is driven by vesicles that carry a signaling molecule. As the vesicles arrive at the site of polar membrane growth, the signaling molecule becomes activated, thereby generating a signal that is proportional to growth. When the strength of the signal surpasses a threshold, a signaling pathway is initiated that activates mitotic Cdk1, which triggers the switch from polar to isotropic growth. The hypothesized growth-dependent signaling pathway would determine the extent of polar bud growth, which would influence both the size and shape of cells.
Proteins that could mediate growth-dependent signaling were identified by analyzing the signals triggered by an arrest of membrane growth. Plasma membrane growth is dependent upon vesicles from the secretory pathway, so membrane growth can be arrested by shifting temperature-sensitive sec mutants to the restrictive temperature (7). Mutants that block polarized growth of the daughter cell membrane cause a rapid cell cycle checkpoint arrest before mitosis (6). Checkpoint signaling is dependent upon the Rho1 GTPase, which is transported to the site of polar membrane growth on vesicles and becomes activated when vesicles fuse with the membrane (8, 9). Checkpoint signals are relayed by an atypical protein kinase C (Pkc1), which is activated by binding of GTP-bound Rho1 (10). Phosphorylation of Pkc1 is dependent upon membrane growth and appears to be proportional to the extent of membrane growth (6). Pkc1 binds and activates a protein phosphatase 2A (PP2A)3 complex that includes the Cdc55 regulatory subunit and redundant accessory proteins Zds1 and Zds2 (referred to as PP2ACdc55-Zds). PP2ACdc55-Zds regulates the budding yeast homologs of Wee1 and Cdc25, known as Swe1 and Mih1, which control entry into mitosis via inhibitory phosphorylation of Cdk1 (6, 11–17). A particularly important event appears to be dephosphorylation of Mih1 by PP2ACdc55-Zds, which is likely necessary for activation of Mih1 to initiate mitosis (11, 12). The Zds1/2 proteins target PP2ACdc55 to dephosphorylate Mih1 (12). Proteins in the pathway are connected by binding interactions and localize to the site of membrane growth, so they are well positioned to relay signals about membrane growth (8, 10, 12, 16, 18–23).
Growth-dependent signaling suggests a simple conceptual framework for coordinated control of cell growth, size, and shape. Linking checkpoint signals to the site of growth would provide a simple mechanism for controlling the extent of growth at specific locations, and it would provide a robust mechanism for cell size control that is adaptable to cells of diverse size and shape. Pkc1, a key checkpoint component, also links ribosome biogenesis to membrane growth (24, 25). Thus, it is conceivable that multiple aspects of cell growth are controlled via signals originating at sites of membrane growth. However, many key aspects of the model remain untested. Here, we have further investigated the role of Pkc1 in checkpoint signaling and how it activates PP2ACdc55-Zds. Previous work discovered that Pkc1 binds and activates PP2ACdc55-Zds and that checkpoint signaling is dependent upon Pkc1 (6). However, it was unclear whether signals from Pkc1 are sufficient to trigger entry into mitosis in checkpoint-arrested cells. In addition, the mechanism by which Pkc1 activates PP2ACdc55 to dephosphorylate Mih1 is unknown. Answering these questions is an essential step toward testing whether growth-dependent signaling controls cell size and shape.
Results
Pkc1 Can Over-ride a Checkpoint Arrest Caused by Blocking Membrane Growth
Previous work found that inactivation of PKC1 causes loss of checkpoint signaling, whereas overexpression of a constitutively active PKC1 (referred to as PKC1*) from the GAL1 promoter causes activation of PP2ACdc55 and dephosphorylation of Mih1 and Zds1 (6). Together, these experiments demonstrate that Pkc1 plays an important role in checkpoint signaling; however, they do not test whether Pkc1 is sufficient for checkpoint signaling. To better define the role of Pkc1 in checkpoint signaling, we tested whether expression of PKC1* at levels seen in vivo could drive cells through a checkpoint arrest. We used basal expression of PKC1* from the uninduced CUP1 promoter to produce Pkc1* protein at levels similar to endogenous Pkc1 (Fig. 1A). To induce a checkpoint arrest, cells carrying the conditional sec6-4 mutant were shifted to the restrictive temperature, which causes rapid cessation of polar membrane growth (6, 26). We released sec6-4, sec6-4 swe1Δ, and sec6-4 CUP1-PKC1* cells from a G1 arrest and then shifted cells to the restrictive temperature in late G1 to block polar membrane growth. Levels of the Clb2 mitotic cyclin were assayed to determine whether cells underwent a checkpoint arrest before mitosis. The sec6-4 cells underwent a checkpoint arrest and failed to produce significant amounts of Clb2. The sec6-4 swe1Δ cells failed to arrest, as observed previously (Fig. 1B). Expression of PKC1* eliminated the checkpoint arrest in a manner similar to swe1Δ.
FIGURE 1.
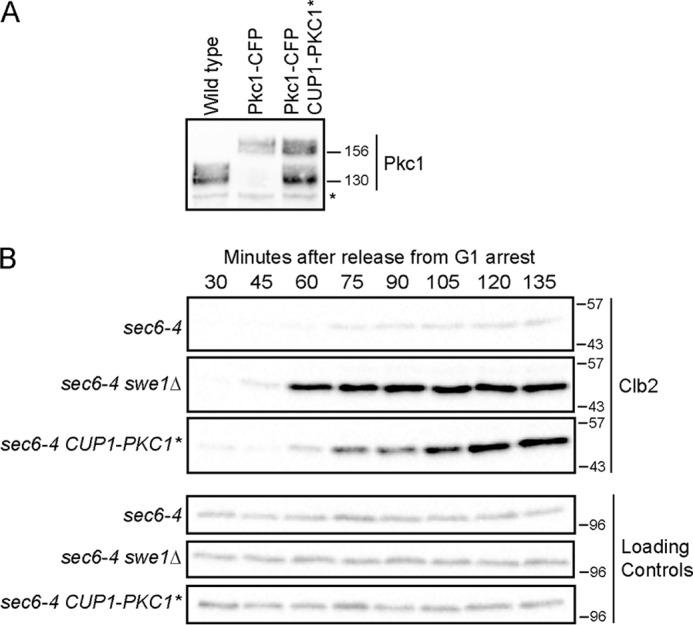
Expression of Pkc1* overrides a mitotic checkpoint arrest. A, to examine levels of Pkc1* protein expressed from CUP1-PKC1*, the expression construct was introduced into a strain in which endogenous PKC1 was tagged with cyan fluorescent protein (Pkc1-CFP). Pkc1-CFP migrated above the untagged Pkc1*. Extracts from log phase wild type, PKC1-CFP, and PKC1-CFP CUP1-PKC1* cells were probed by Western blotting with anti-Pkc1 antibody. The asterisk denotes a background band that serves as a loading control. Numbers shown next to Western blots indicate molecular mass in kilodaltons. B, cells were released from a G1 arrest and shifted to the restrictive temperature (34 °C) at 45 min after release. Levels of Clb2 were assayed by Western blotting.
Previous work found that overexpression of Zds1, which recruits Pkc1 to the PP2ACdc55 complex, also drives cells through a checkpoint arrest (6). Together, these data indicate that Pkc1 is both necessary and sufficient for checkpoint signaling. The mechanism by which Pkc1 relays checkpoint signals is therefore of central importance for understanding how mitosis is linked to membrane growth.
Pkc1 Controls the Composition and Phosphorylation of the PP2ACdc55-Zds Complex
Because Pkc1 associates with the PP2ACdc55-Zds complex, we reasoned that it plays a direct role in controlling PP2ACdc55 (6). We therefore used quantitative mass spectrometry to determine whether Pkc1 induces changes in the composition or modification of the PP2ACdc55-Zds complex. To do this, we used large scale immunoaffinity chromatography to isolate the PP2ACdc55-Zds complex from control cells and from cells in which PP2ACdc55 was strongly activated via expression of PKC1* from the GAL1 promoter. We then compared the complexes by quantitative mass spectrometry.
The purified complexes are shown in Fig. 2. Purification was performed in the presence of high salt and phosphatase inhibitors to remove weakly associated proteins and to preserve phosphorylation. The Zds1 protein purified from GAL1-PKC1* cells migrated with a faster mobility, consistent with previous work (6).
FIGURE 2.
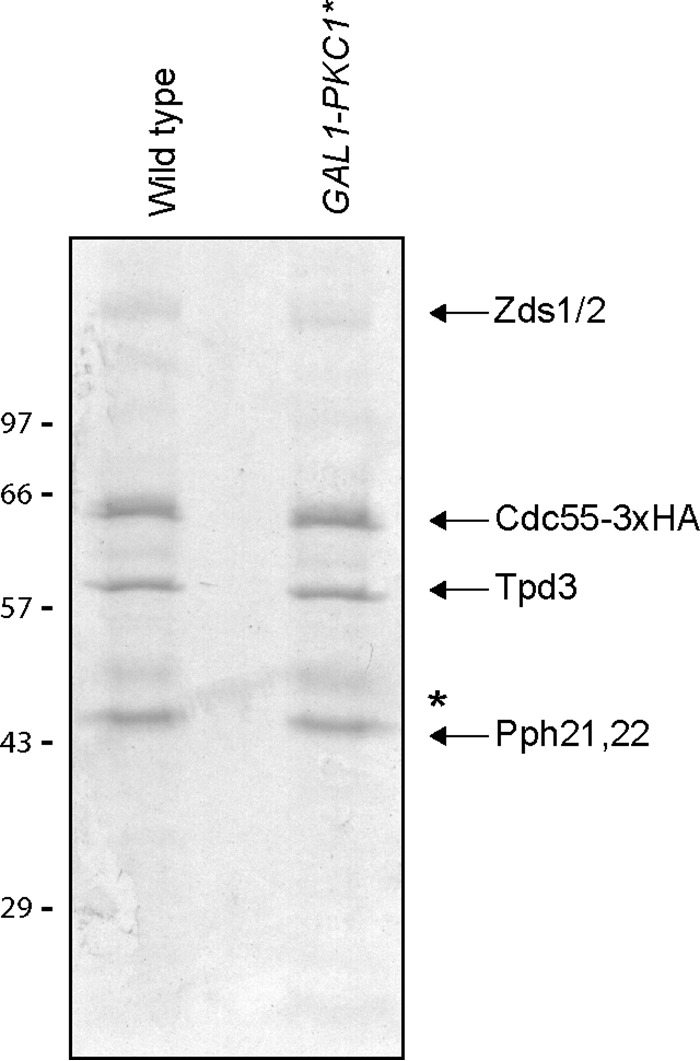
Pkc1* regulates the composition and phosphorylation state of the PP2ACdc55-Zds1 complex. Coomassie blue-stained gel showing PP2ACdc55–3XHA-Zds complexes purified from control cells and from cells expressing PKC1* from the GAL1 promoter. Complexes were purified by anti-HA immunoaffinity chromatography with competitive peptide elution. Numbers shown next to Western blots indicate molecular mass in kilodaltons. The asterisk indicates IgG heavy chains from the immunoaffinity column.
The purified complexes were digested with trypsin, and the resulting peptides were covalently modified by reductive dimethylation (27) to generate light (control) and heavy (GAL1-PKC1*) stable isotope-labeled pools. The samples were combined and analyzed by mass spectrometry to identify and measure changes in associated proteins and protein phosphorylation between the two samples. Individual peptide ratios (GAL1-PKC1*:WT) were log2-transformed, and the median of all measured peptides for each protein was used to assess relative protein abundances (Table 1 and supplemental Table S1). Relative levels of phosphorylation were derived in a similar fashion, using the median log2(GAL1-PKC1*:WT) for all measured peptides containing a given phosphorylation site (supplemental Table S2). Positive values indicate that protein abundance or phosphorylation increased in the presence of elevated PKC1 activity, whereas negative values indicate a decrease.
TABLE 1.
Quantitative analysis of the PP2ACdc55-Zds complex, relative protein abundances for core complex members in GAL-PKC1* and control cells
Ratios were normalized to the bait protein Cdc55. The complete list of proteins identified in the complexes appears in supplemental Table S1.
| ORF | Gene symbol | No. of peptides | Median log2 | S.D. | -Fold change |
|---|---|---|---|---|---|
| YMR273C | ZDS1 | 210 | −0.8 | 0.7 | −1.8 |
| YGL190C | CDC55 | 134 | 0.0 | 0.5 | 1.0 |
| YAL016W | TPD3 | 91 | −0.1 | 0.5 | −1.1 |
| YML109W | ZDS2 | 67 | −0.1 | 0.4 | −1.1 |
| YDL188C | PPH22 | 15 | 0.1 | 0.4 | 1.1 |
| YHR132W-A | IGO2 | 12 | −3.3 | 1.8 | −9.7 |
| YBL105C | PKC1 | 12 | 3.3 | 1.1 | 9.7 |
| YDL134C | PPH21 | 9 | 0.0 | 0.2 | 1.0 |
Table 1 lists proteins associated with the PP2ACdc55-Zds complex whose abundance could be quantified. Of these, Igo2 showed a large decrease in abundance within the complex upon overexpression of PKC1*. Pkc1 showed increased abundance; however, this was likely a result of the overexpression of PKC1*. Previous studies found that Pkc1 associates with the Zds1/2 proteins (6, 16, 22). The fact that Pkc1 could be detected in the PP2ACdc55-Zds complex after multiple high salt washes suggests that it interacts with high affinity.
Igo2 and its paralog Igo1 are the budding yeast homologs of vertebrate endosulfine proteins called ENSA and Arpp-19, which bind and inhibit vertebrate PP2ACdc55 during mitosis. The discovery that expression of PKC1* causes Igo2 to dissociate from PP2ACdc55 suggested a mechanism by which Pkc1 could promote activation of PP2ACdc55. A complete list of proteins and peptides identified from the PP2ACdc55 complex purifications appears in supplemental Table S1.
There are several technical challenges associated with identification of in vivo phosphorylation sites. In many cases, only a fraction of the endogenous protein is phosphorylated at any given time, so sites are not quantitatively phosphorylated. A unique challenge associated with analyzing phosphorylation sites on a phosphatase is that it is likely that some auto-dephosphorylation will occur during purification, even in the presence of competitive phosphatase inhibitors. Finally, in the samples that we used for comparison, the PP2ACdc55-Zds complex isolated from log phase cells served as the control sample. Because endogenous Pkc1 is present, a fraction of the PP2ACdc55-Zds complex should already be modified in the control sample, which will reduce the ratio of phosphorylated to unphosphorylated peptides when comparing phosphorylation sites between control cells and GAL1-PKC1* cells.
With these limitations in mind, we searched for phosphorylation sites that showed evidence of Pkc1-dependent regulation. We were able to identify and quantify sites on multiple proteins in the complex. The supplemental Table S2 lists all identified phosphopeptides in the core PP2ACdc55-Zds complex along with site localization assessments using the Ascore method (28) and peptide quantification.
Several sites showed evidence of Pkc1-dependent regulation (Table 2). In Cdc55, a site at Ser-410 and another at Ser-444 or Ser-453 appeared to show increased phosphorylation in cells that express PKC1*. An additional site, at Ser-479, showed evidence of 4–5-fold increased phosphorylation in GAL1-PKC1* cells; however, it was identified and quantified from only a single peptide so the evidence was not as strong as for the other sites. Two sites within Zds1, at Ser-444 or Ser-445 and Ser-691, showed evidence of decreased phosphorylation, consistent with our previous discovery that PKC1* induces dephosphorylation of Zds1 (6). The catalytic subunit of PP2A is encoded by nearly identical redundant genes called PPH21 and PPH22. We found evidence that Pph22 undergoes PKC1*-dependent regulation at Ser-38 or Thr-43.
TABLE 2.
Pkc1*-regulated phosphorylation sites identified in the PP2ACdc55-ZDS complex
psm is peptide spectral matches; quant is quantified.
| ORF | Gene | Site | Maximum Ascore | No. of psm | No. of quants | Median log2 | S.D. | -Fold change |
|---|---|---|---|---|---|---|---|---|
| YGL190C | CDC55 | Ser-410 | 1000 | 2 | 2 | 3.6 | 0.3 | 12.3 |
| YGL190C | CDC55 | Ser-444 or Ser-453 | 19/28 | 11 | 4 | 1.1 | 0.2 | 2.2 |
| YDL188C | PPH22 | Ser-38 or Thr-43 | 78 | 12 | 2 | 1.2 | 0.1 | 2.3 |
| YMR273C | ZDS1 | Ser-444 or Ser-445 | 0/13 | 9 | 5 | −2.3 | 0.3 | −4.9 |
| YMR273C | ZDS1 | Ser-691 | 21 | 8 | 4 | −2.0 | 0.3 | −4.0 |
| YML109W | ZDS2 | Ser-808 | 26 | 12 | 3 | 0.9 | 0.6 | 1.9 |
Rim15 Activates Binding of Igo2 to PP2ACdc55
We first focused on the roles of Igo1 and Igo2 in controlling PP2ACdc55. In vertebrates, endosulfine proteins are phosphorylated by the Greatwall kinase on a conserved serine, which activates them to bind and inhibit PP2AB55 (29, 30). The closest homolog of the Greatwall kinase in yeast is Rim15. Recent work found that Igo1 binds to PP2ACdc55 and that binding is dependent upon both Rim15 and the conserved serine (31, 32). However, Igo2 has not been characterized. Cells carrying deletions of both IGO1 and IGO2 are viable and show no phenotype in rapidly growing cells at low or moderate temperatures. At high temperatures (38 °C), igo1Δ igo2Δ cells grow slowly and show defects in mitosis (32). The mitotic defects and slow growth are rescued by swe1Δ, which indicates that they are due to misregulation of Cdk1 inhibitory phosphorylation.
The PP2ACdc55-Zds complex isolated from control cells included Igo2 phosphorylated on the conserved serine targeted by Rim15/Greatwall kinase (Ser-63) (supplemental Table S1). We used coimmunoprecipitation to further characterize the interaction between Igo2 and PP2ACdc55. To detect Igo2, we generated a polyclonal antibody against the full-length protein. Because of the high sequence similarity between Igo1 and Igo2, the antibody cross-reacted with Igo1. Because Igo1 and Igo2 differ slightly in molecular weight, the behavior of both proteins could be followed. Igo1 and Igo2 both migrated as multiple bands in log phase cells, suggesting that they undergo post-translational modification (Fig. 3A). In immunoprecipitation assays using log phase CDC55–3XHA cells, Igo2 interacted with PP2ACdc55 and the interaction was dependent upon Rim15 and S63 (Fig. 3B). The interaction of Igo1 with PP2ACdc55 appeared to be less robust. The slower migrating forms of Igo1/2 were still present in rim15Δ cells, which indicated that they were not due to phosphorylation of Ser-63 by Rim15 and that other kinases must be involved in phosphorylation of Igo1/2.
FIGURE 3.

Endosulfine interaction with PP2ACdc55 is conserved in budding yeast Igo2. A, extracts from wild type, igo1Δ, and igo2Δ cells were probed by Western blotting with an antibody raised against the Igo2 protein. B, anti-HA antibodies were used to immunoprecipitate PP2ACdc55–3XHA from extracts made from log phase wild type, rim15Δ, and igo2-S63A igo1Δ cells. Coprecipitation of Igo1/2 was assayed by Western blotting using anti-Igo antibody. Asterisks indicate the positions of background bands. Filled circle indicates the position of a background band that only appears in HIS3 cells and overlaps with a slower migrating form of Igo1. Numbers shown next to Western blots indicate molecular mass in kilodaltons.
Pkc1 Dissociates Igo2 from PP2A
We next used the coprecipitation assay to test whether Pkc1* dissociates Igo2 from PP2ACdc55, as suggested by the mass spectrometry. Igo2 bound to PP2ACdc55 in both control and GAL1-PKC1* cells prior to induction of PKC1* expression (Fig. 4A). Expression of PKC1* caused Igo2 to dissociate from PP2ACdc55.
FIGURE 4.

Pkc1 regulates interaction of Igo2 with PP2ACdc55. A, anti-HA antibodies were used to immunoprecipitate PP2ACdc55–3XHA from wild type and GAL1-PKC1* cells before and after induction of PKC1*. Coprecipitation of Igo2 was assayed by Western blotting using anti-Igo2 antibody. B, cells were grown to log phase in YEP media containing 2% glycerol and 2% ethanol. Galactose was added at t = 0. Igo1/2 phosphorylation was assayed by Western blotting. C, extracts from log phase wild type, cdc55Δ, and rts1Δ cells were probed by Western blotting with anti-Igo2 antibody. D, Coomassie Blue-stained gel and autoradiography of in vitro kinase reactions carried out with purified Igo2 and Pkc1*. A shift in the electrophoretic mobility of Igo2 upon phosphorylation by Pkc1 is not observed in the upper panel because a lower percentage of acrylamide and a shorter run time were used, which did not resolve the forms. E, in vitro kinase reactions carried out with purified Igo2 and Pkc1* were probed by Western blotting. F, cells were released from a G1 arrest into fresh media at 30 °C, and samples were collected every 15 min. Anti-HA antibodies were used to immunoprecipitate PP2ACdc55–3XHA. Samples of the crude extracts were taken pre- and post-binding and probed for Zds1, Cdc55–3XHA, and Igo2 to determine the extent of immunodepletion. The asterisk indicates a background band. G, Cdc55–3XHA immunoprecipitations from F were probed for Cdc55–3XHA, Zds1, and Igo2 by Western blotting. H, crude extracts used for immunoprecipitations in G were probed for Pkc1, Clb2, and Igo2 to assess the timing of cell cycle events. The asterisk marks a background band that also serves as a loading control. Numbers shown next to the Western blots indicate molecular mass in kilodaltons.
We noticed that expression of PKC1* caused Igo2 to shift to a slower migrating form (Fig. 4A). To examine this more carefully, we analyzed the electrophoretic mobility of Igo1 and Igo2 as a function of time during induction of PKC1* expression. Expression of PKC1* caused shifts in the mobility of both Igo1 and Igo2 (Fig. 4B).
Although Igo2 migrated as multiple bands, only the fastest migrating form associated with PP2ACdc55 (Fig. 4A). A model that could explain this observation is that PP2ACdc55 acts on associated Igo2 to keep it in the dephosphorylated state. In this model, phosphorylation of Igo2 could be a cause or a consequence of dissociation from PP2ACdc55. To examine this further, we tested whether phosphorylation of Igo1 or Igo2 is dependent upon PP2ACdc55 in vivo. To do this, we assayed Igo1 and Igo2 phosphorylation in wild type and cdc55Δ cells. We also assayed their phosphorylation in cells lacking Rts1, which is the other B-type regulatory subunit of PP2A in budding yeast. Both Igo1 and Igo2 were hyperphosphorylated in cdc55Δ cells compared with wild type and rts1Δ cells (Fig. 4C). This suggests that PP2ACdc55 dephosphorylates associated Igo2 or that PP2ACdc55 inhibits the kinase that phosphorylates Igo2. Xenopus ENSA also appears to be dephosphorylated by PP2AB55 (33). Note that GAL1-PKC1* caused full hyperphosphorylation of Igo2 and dissociation from PP2ACdc55. In contrast, cdc55Δ, which should also cause dissociation of Igo2 from PP2ACdc55 (29), caused only a fraction of Igo2 to undergo full hyperphosphorylation. Together, these observations suggest that dissociation of Igo2 from PP2ACdc55 is not sufficient to induce efficient phosphorylation of Igo2. Rather, signals from Pkc1 appear to contribute to phosphorylation of Igo2.
Although immunoprecipitation with anti-HA antibodies quantitatively depleted Cdc55–3XHA from extracts, it did not significantly deplete Igo1 or Igo2. This suggests that there is a large excess of Igo1/2 relative to PP2ACdc55 or that Igo1/2 interact with PP2ACdc55 with low affinity.
To test whether Pkc1 can directly phosphorylate Igo2, we performed in vitro kinase reactions using Pkc1*-3XHA purified from yeast and Igo2 purified from bacteria. Purified Pkc1* phosphorylated Igo2 in reactions containing [γ-32P]ATP (Fig. 4D). In addition, Pkc1* induced a quantitative electrophoretic mobility shift in Igo2 (Fig. 4E). The shift was less than the shift observed in vivo, which suggests that Pkc1 phosphorylates only a subset of the sites that cause the shift in vivo.
We next tested whether association of Igo2 with PP2ACdc55 is regulated during the cell cycle. CDC55–3XHA cells were released from a G1 arrest, and samples were taken at 15-min intervals as the cells progressed through the cell cycle. The PP2ACdc55–3XHA complex was immunoprecipitated and probed for associated Igo2 (Fig. 4, F and G). To assess the timing of Igo2 association relative to other cell cycle events, we also assayed levels of the Clb2 mitotic cyclin and phosphorylation of Pkc1 (Fig. 4H). Cdc55 and Zds1 were quantitatively depleted from the extracts in the immunoprecipitations, but Igo2 was not (Fig. 4F). Igo2 associated with PP2ACdc55 early in the cell cycle but was almost completely dissociated by 60 min, when cells were entering mitosis. Dissociation of Igo2 was correlated with maximal Pkc1 hyperphosphorylation, which occurred at 60 min (Fig. 4, G and H). Together, these observations are consistent with a model in which Pkc1 reaches maximal activity at entry into mitosis, which triggers dissociation of Igo2 from the PP2ACdc55-Zds complex. The weak signal for Igo1 and the presence of IgG background bands in the co-immunoprecipitations made it difficult to clearly discern whether Igo1 associated with PP2ACdc55 in a cell cycle-dependent manner.
In Vivo Analysis of Igo1/2 Function
To characterize the role of Igo1/2 in regulating cell cycle progression, we created a strain carrying deletions of both IGO1 and IGO2 (igo1/2Δ). We released wild type and igo1/2Δ cells from a G1 arrest and assayed levels of the mitotic cyclin Clb2. We also assayed phosphorylation of Swe1 and Mih1. We detected no significant difference in the timing of mitosis or in the regulation of Swe1 or Mih1 phosphorylation, consistent with a previous study (data not shown) (32). However, when cells were allowed to grow to saturation, we observed an increase in cell size in igo1/2Δ cells relative to wild type cells that was dependent upon SWE1 (Fig. 5A). In addition, there was a loss of slower migrating forms of Igo2 as cells approached saturation (Fig. 5B). Together, these results suggest that regulation of Igo1/2 plays a role in controlling mitotic Cdk1 inhibitory phosphorylation when growth is slowed due to limited nutrient availability. A loss of Igo function also causes an increase in cell size when nutrients become limiting in fission yeast, which suggests that this function of Igo proteins is conserved (34).
FIGURE 5.

Analysis of in vivo roles of Igo2. A, log phase wild type, igo1Δ igo2Δ and igo1Δ igo2Δ swe1Δ cells were inoculated into YPD medium at an optical density of 0.5 and allowed to grow to saturation at 30 °C. At 7.5 h, samples were taken, and the cells were photographed. At this time, the optical density was ∼3.5. Scale bar, 10 μm. B, log phase RIM15–3XHA cells were inoculated into YPD medium at an optical density of 0.5 and allowed to grow to saturation at 30 °C. Samples were taken at 1.5-h intervals and adjusted to contain equal numbers of cells. Igo1 and Igo2 were probed by Western blotting. The asterisk marks the position of a background band that only appears in HIS3 cells and overlaps with a slower migrating form of Igo1. C, sequence alignment of Igo2 and Xenopus ENSA. Identical residues are in white letters and in red boxes and conserved residues are in red letters and in white boxes. Arrows indicate location of the Greatwall/Rim15 phosphorylation site, potential Pkc1 phosphorylation sites determined by mass spectrometry, and a potential PKA phosphorylation site previously identified on Xenopus ENSA. The alignment was made using ESPript (47). D, extracts from log phase wild type, igo1Δ igo2Δ Igo2, igo1Δ igo2Δ igo2-ps1, and igo1Δ igo2Δ igo2-ps2 cells were probed by Western blotting with anti-Igo2 antibody. Filled circle indicates the position of a HIS3 marker-dependent background band that overlaps with the slower migrating form of Igo1 in strains carrying igo1Δ::HIS3. Numbers shown next to the Western blots indicate molecular mass in kilodaltons.
We next addressed the role of Igo2 phosphorylation. We considered the possibility that phosphorylation of Igo2 is necessary for dissociation from PP2ACdc55. In this model, a phosphorylation site mutant could have a dominant negative phenotype by causing a failure to dissociate from PP2ACdc55. To test this, we used quantitative mass spectrometry to map sites on Igo2 that are phosphorylated by Pkc1 in vitro. Because of the small size of Igo2, we obtained nearly complete sequence coverage and were able to quantify peptides harboring all 31 phosphorylatable serine, threonine, and tyrosine residues. The data revealed that Igo2 is phosphorylated by Pkc1 on several sites (Fig. 5C, Table 3, and supplemental Table 3). By monitoring the disappearance of unphosphorylated peptides and the appearance of phosphorylated forms, we were able to determine the fractional phosphorylation site occupancy (Table 4). The strongest site, Ser-119, was phosphorylated at >90% occupancy. Another site, at Ser-106, was phosphorylated at 48%, Finally, a third site, most likely at Ser-99 was phosphorylated at 46%, although the data could not rule out phosphorylation at Thr-91 or Ser-94.
TABLE 3.
Igo2 in vitro phosphorylation site identification
psm is peptide spectral matches.
| Site | No. of psm | Maximum Ascore |
|---|---|---|
| Ser-31 | 1 | 6 |
| Ser-63 | 1 | 27 |
| Thr-91 or Ser-94 or Ser-99 | 20 | |
| Thr-91 | 2 | 17 |
| Ser-94 | 3 | 46 |
| Ser-99 | 15 | 82 |
| Ser-106 or Ser-107 or Ser-108 or Ser-113 | 75 | |
| Ser-106 | 55 | 38 |
| Ser-107 | 17 | 10 |
| Ser-108 | 2 | 21 |
| Ser-113 | 1 | 31 |
| Ser-119 | 21 | 53 |
TABLE 4.
Inferred in vitro Igo2 phosphorylation site occupancy
| Site | No. of quantitative peptides | Median log2[+ATP/−ATP] | S.D. | -Fold change | Occupancy |
|---|---|---|---|---|---|
| % | |||||
| Ser-31 | 8 | 0.04 | 0.35 | 1.03 | 3 |
| Ser-63 | 8 | 0.04 | 0.35 | 0.97 | 0 |
| Thr-91 | 1 | −0.14 | 1.10 | 9 | |
| Thr-91 or Ser-94 | 20 | −0.17 | 0.40 | 1.13 | 11 |
| Thr-91 or Ser-94 or S99 | 3 | −0.89 | 0.15 | 1.85 | 46 |
| Ser-106a | 2 | −0.94 | 0.13 | 1.92 | 48 |
| Ser-119 | 2 | −3.61 | 0.03 | 12.21 | 92 |
a There are six serines on these peptides, and Ascore analysis indicates Ser-106 is the most likely regulated site.
To test the role of Igo2 phosphorylation in vivo, we made two phosphorylation site mutants. In the first, we mutated Ser-94, Ser-99, Ser-119, Ser-121, and Ser-122 to alanines. We refer to this mutant as igo2-ps1. In the second, we also mutated Ser-113, Ser-115, and the serine cluster at Ser-106 to Ser-109. We refer to this mutant as igo2-ps2. In both of these mutants, Ser-121/Ser-122, Ser-113/Ser-115, and Ser-107–109 were mutated due to their presence on phosphorylated peptides identified by mass spectrometry. By mutating these sites, we aimed to account for potential errors in site assignment and to reduce the chance that neighboring phosphorylation sites could be phosphorylated when preferred sites were eliminated. Several proteome-wide analyses of protein phosphorylation have detected potential phosphorylation sites at Ser-106, Ser-107, Ser-108, Ser-109, Ser-113, Ser-115, Ser-119, Ser-121, and Ser-122, which indicates that they are likely phosphorylated in vivo (see the BioGRID website).
Plasmids expressing the mutants from the IGO2 promoter were integrated in igo1Δ igo2Δ cells as the sole source of Igo proteins. Both mutants eliminated the slower migrating forms observed for wild type Igo2, indicating that relevant phosphorylation sites were mutated (Fig. 5D). However, we did not detect a phenotype for the mutants. They showed no temperature sensitivity or size defects in response to starvation, and regulation of Clb2, Swe1, Mih1, and Cdk1 inhibitory phosphorylation appeared to be normal. The single S119A mutant also showed no phenotype.
This analysis indicates that the function of Igo1/2 becomes particularly important under limiting nutrient conditions. In addition, the fact that association of Igo2 with PP2ACdc55 is regulated in a Pkc1-dependent manner during a normal cell cycle (Fig. 4, G and H) suggests that Igo1/2 may represent one of several overlapping mechanisms for control of PP2ACdc55, which would explain why igo1/2Δ cells do not have a phenotype under normal conditions. The observation that Pkc1 induces changes in the phosphorylation of multiple components of the PP2ACdc55-Zds complex is also consistent with the possibility that multiple overlapping mechanisms control the activity of PP2ACdc55.
Pkc1 Directly Phosphorylates Cdc55
We next focused on Pkc1-dependent regulation of Cdc55. Mass spectrometry identified two sites on Cdc55 that showed evidence of increased phosphorylation when Pkc1* was expressed (Table 2). Although the structure of yeast PP2ACdc55 has not been determined, we were able to align the amino acid sequence of Cdc55 with the sequence and structure of the homologous human PP2AB55 complex (Fig. 6A) (35). This revealed that the regulated sites are located within an insertion loop region of Cdc55. The phosphorylation sites are surface-exposed within the complex and are located far from the PP2A catalytic site and far from the interface with the other core components of the PP2A complex. The location of the sites suggests that they could regulate interaction of Cdc55 with other binding partners or induce a conformational change in Cdc55 that regulates the functions of PP2ACdc55.
FIGURE 6.

Pkc1 phosphorylates PP2ACdc55. A, alignment of Cdc55 phosphorylation sites with the structure of human B55 within the PP2AB55 complex (Protein Data Bank code 3DW8). The regulatory, catalytic, and scaffolding subunits are colored green, violet, and cyan, respectively. The insertion region containing Cdc55 phosphorylation sites is colored red on the structure of B55 and marked with a red bar in the sequence alignment (Cdc55 residues 400–465 correspond to B55 residues 370–392). White characters on red boxes denote identical residues, and red characters on white boxes denote conserved residues. Arrows indicate location of phosphorylation sites. Structures were generated using PyMOL (PyMOL Molecular Graphics System, Version 0.99, Schrödinger, LLC). The alignment was made using ESPript (47). B, Coomassie Blue-stained gel of immunoaffinity chromatography purified Pkc1*-3XHA and PP2ACdc55–3XHA. The asterisk marks antibody heavy chain from the immunoaffinity column. The arrows denote the components of the PP2A complex that co-purify with Cdc55. C, autoradiography film of in vitro kinase reactions carried out using purified PP2ACdc55–3XHA and Pkc1*-3XHA. Purified proteins were incubated alone or together in the presence of [γ-32P]ATP with and without okadaic acid. The PP2ACdc55 complex phosphorylated in the presence of okadaic acid was disrupted by the addition of SDS to a final concentration of 1% and incubated at 100 °C. The sample was then diluted into RIPA buffer and incubated with either anti-Cdc55 or control anti-GST antibodies. Reactions were loaded onto a 9% SDS-polyacrylamide gel. Numbers shown next to the Western blots or gels indicate molecular mass in kilodaltons.
To determine whether Pkc1 can directly phosphorylate Cdc55, we used immunoaffinity chromatography to purify PP2ACdc55–3XHA-Zds and Pkc1*-3XHA from yeast (Fig. 6B). We then tested whether Pkc1* could phosphorylate Cdc55 in vitro using [γ-32P]ATP. Because PP2ACdc55 is a phosphatase that could oppose phosphorylation by Pkc1, we performed kinase reactions in the presence or absence of the PP2A inhibitor okadaic acid. Pkc1* underwent autophosphorylation in the absence of PP2ACdc55 (Fig. 6C). There was no detectable phosphorylation of PP2ACdc55 in the absence of Pkc1*. Multiple proteins underwent phosphorylation in the presence of Pkc1*, and phosphorylation was strongly enhanced by okadaic acid. It also appeared that the band corresponding to Pkc1* autophosphorylation disappeared in the presence of PP2ACdc55, which could be due to dephosphorylation of Pkc1* by PP2ACdc55. Because Pkc1 binds to PP2ACdc55 via the Zds1/2 proteins, it would likely be a strong target for PP2ACdc55, even in the presence of an inhibitor like okadaic acid.
Several bands migrated near the molecular weight of Cdc55. To test whether one of these bands corresponded to Cdc55, we disrupted the phosphorylated PP2ACdc55 complex with 1% SDS. We then neutralized the SDS with an excess of non-ionic detergent. The reaction was split in two, and immunoprecipitation was carried out with either an anti-Cdc55 antibody or a control anti-GST antibody. This confirmed that Cdc55 was phosphorylated in the presence of Pkc1*. Together, these results show that Cdc55 is likely a direct substrate of Pkc1. Several other bands are prominently phosphorylated in the combined presence of Pkc1 and PP2ACdc55. Additional work will be necessary to characterize these bands.
Phosphorylation of Cdc55 Regulates Association with Igo2
To test for functions of Cdc55 phosphorylation, we generated a phosphorylation site mutant in which likely Pkc1-dependent phosphorylation sites were mutated to alanines. We mutated Ser-410 because it showed the clearest increase in phosphorylation in response to PKC1* expression. The data for Ser-410 was particularly clear because it was repeatedly detected on a phosphopeptide that included no other serines or threonines (supplemental Table S2). We also mutated Ser-401 and Ser-403 because they were present, along with Ser-410, on a longer phosphorylated peptide in which it was difficult to distinguish which site was phosphorylated or regulated in response to Pkc1 (supplemental Table S2). Ser-444 was mutated because it was a likely target of regulation, and Ser-443 was mutated due to its close proximity to Ser-444. We refer to the phosphorylation site mutant as cdc55-ps1.
The phosphorylation site mutant eliminated a slower migrating form of Cdc55–3XHA in vivo, which was previously found to be due to phosphorylation (36), indicating that relevant sites were mutated (Fig. 7A). Mutation of Ser-410 alone failed to eliminate the slower migrating form. The slower migrating form also disappeared in zds1/2Δ cells, consistent with a role for Zds1/2 in targeting phosphorylation of Cdc55 (Fig. 7A). Although the slower migrating form could be detected in untagged Cdc55, the two forms were better resolved using an HA-tagged version of Cdc55.
FIGURE 7.
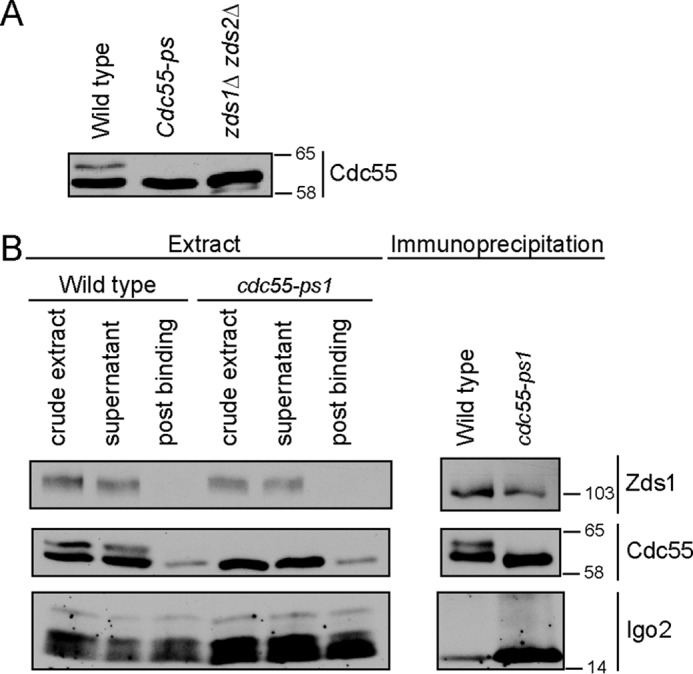
cdc55-ps1 mutant causes increased association with PP2ACdc55. A, extracts from log phase wild type, cdc55-ps1, and zds1Δ zds2Δ were probed by Western blotting using anti-Cdc55 antibody. Numbers shown next to Western blots indicate molecular mass in kilodaltons. B, anti-HA antibodies were used to immunoprecipitate PP2ACdc55–3XHA and PP2Acdc55-ps1–3XHA from wild type and cdc55-ps1 cells, respectively. Coprecipitation of Igo2 and Zds1 was assayed by Western blotting.
We used an immunoprecipitation assay to determine whether the cdc55-ps1 mutant affected the composition of the PP2ACdc55 complex. Association of Zds1 was unaffected, but association of Igo2 with PP2ACdc55 was increased in cdc55-ps1 cells (Fig. 7B). These results are consistent with the data showing that overexpression of Pkc1* leads to phosphorylation of Cdc55 and dissociation of Igo2 from PP2ACdc55. However, we did not detect any further phenotypes of the cdc55-ps1 mutant. It did not cause slow proliferation or cell size defects, and cells carrying the mutation entered mitosis with normal timing. Furthermore, the mutant did not cause defects in phosphorylation of Swe1 or Mih1.
Discussion
Activity of Pkc1 Is Necessary and Sufficient for Checkpoint Signaling
Previous studies pointed to Pkc1 as an important component of the checkpoint signaling pathway that links mitotic entry to membrane growth (6). Pkc1 is required for dephosphorylation of Mih1, which appears to be a critical step in the events that lead to mitotic entry in response to polar membrane growth. In addition, expression of constitutively active Pkc1 from the GAL1 promoter induces rapid quantitative dephosphorylation of Mih1. Expression of constitutively active Rho1 GTPase, which binds and activates Pkc1, also induces dephosphorylation of Mih1 that is dependent upon Pkc1. Finally, Pkc1 undergoes hyperphosphorylation during polar membrane growth that appears to be dependent upon and proportional to membrane growth.
Despite the clear importance of Pkc1, it was uncertain whether Pkc1 activity is sufficient for checkpoint signaling. Expression of constitutively active Pkc1 from the strong GAL1 promoter did not drive cells through a checkpoint arrest; however, a high level expression of constitutively active Pkc1 is toxic, so it could cause nonspecific effects. Here, we discovered that low level expression of constitutively active Pkc1 from the CUP1 promoter drives cells through a checkpoint arrest caused by blocking membrane growth. Together with the previous studies, this observation demonstrates that Pkc1 activity is both necessary and sufficient for checkpoint signaling. Thus, an understanding of how Pkc1 relays checkpoint signals is essential for understanding how mitotic entry is linked to membrane growth.
Pkc1 Induces Multiple Changes in the Composition and Phosphorylation of the PP2ACdc55-Zds Complex
We used mass spectrometry to investigate how Pkc1 relays checkpoint signals that activate PP2ACdc55, which revealed that Pkc1 induces multiple changes in the composition and phosphorylation of the PP2ACdc55-Zds1 complex. It causes dissociation of Igo2 from the complex, as well as increased phosphorylation of sites on Cdc55 and Pph21/22, and decreased phosphorylation of sites on Zds1. The mapping data do not provide comprehensive identification of phosphorylation events. Even under the best circumstances, it is difficult to detect peptides that span an entire protein sequence by mass spectrometry. For Cdc55 and Zds1, we obtained sequence coverage of 68 and 66%, respectively, and for Pph21/22 we obtained 45% coverage, so sites may have been missed. Phosphorylation sites could also have been missed because they were dephosphorylated during purification of the PP2ACdc55-Zds complex.
For further analysis, we focused on Igo2 and Cdc55. Igo2 was chosen because it is a conserved regulator of PP2ACdc55, and release of Igo2 from PP2ACdc55 suggested a mechanism by which Pkc1 could activate PP2ACdc55 (29–31). We focused on Cdc55 because it is a highly conserved core component of PP2ACdc55.
We made a phosphorylation site mutant of Zds1 that lacked the sites that underwent significant dephosphorylation upon expression of Pkc1* (sites Ser-444/445 and Ser-691/693/695). The mutations reduced, but did not eliminate, phosphorylation of Zds1, as detected via electrophoretic mobility shift, so it is likely that the mapping missed several sites (data not shown). The mutant did not cause an obvious phenotype when provided as the sole source of Zds proteins.
Evidence for Functional Differences between Igo1 and Igo2
In vertebrates, the Greatwall kinase activates ENSA to bind and inhibit PP2AB55 (29, 30). Inhibition of PP2AB55 by ENSA is thought to be essential for Cdk1 to phosphorylate its mitotic targets. In budding yeast, inactivation of Igo1 and Igo2 does not cause major mitotic defects under normal conditions, which indicates that they do not play an essential role in mitosis. Nevertheless, key aspects of the function and regulation of Igo1 and Igo2 are conserved in budding yeast. Previous work found that phosphorylation of Igo1 by Rim15, the budding yeast Greatwall homolog, activates it to bind and inhibit PP2ACdc55 (31, 32). Here, we found that Rim15 regulates Igo2 in a similar manner. However, we also observed several striking differences in the behavior of Igo1 and Igo2. First, Igo1 does not associate with PP2ACdc55 during an α-factor-induced G1 arrest and weakly associates throughout the cell cycle with a sharp peak during early mitosis (32). In contrast, we found that Igo2 associates with PP2ACdc55 during a G1 arrest and throughout the early cell cycle before dissociating as cells enter mitosis. Second, Igo2 undergoes dephosphorylation as cells deplete nutrients, but Igo1 does not. Third, Igo2 appears to be the predominant Igo protein that binds to PP2ACdc55 in rapidly growing cells.
Pkc1 Controls Binding of Igo2 to PP2ACdc55
Previous studies established that Rim15/Greatwall kinase promotes association of Igo/ENSA with PP2ACdc55, but nothing was known about the signals that control dissociation of Igo/ENSA. Here, we discovered several new mechanisms for control of Igo1/2 proteins. Pkc1 causes Igo2 to undergo hyperphosphorylation and dissociation from PP2ACdc55. Pkc1 can directly phosphorylate Cdc55, which appears to contribute to the mechanism by which Pkc1 dissociates Igo1/2 from PP2ACdc55. Hyperphosphorylation of Igo1/2 may also contribute to their dissociation from PP2ACdc55; however, at this point we cannot distinguish whether hyperphosphorylation of Igo1/2 is a cause or consequence of dissociation. A further discovery is that Igo1/2 are hyperphosphorylated in cdc55Δ cells, which suggests that they are dephosphorylated by PP2ACdc55. Thus, it is possible that PP2ACdc55 opposes release of the Igo1/2 proteins. Together, these observations provide new insight into mechanisms that control association of Igo1/2 proteins with PP2ACdc55.
What Are the Functions of Igo1 and Igo2?
Previous work found that igo1Δ igo2Δ cells show mitotic defects at high temperatures that are due to misregulation of Cdk1 inhibitory phosphorylation (32). We further discovered that igo1Δ igo2Δ show misregulation of cell size and Cdk1 inhibitory phosphorylation under nutrient-limiting conditions. Inactivation of the fission yeast homolog of Igo1/2 also causes misregulation of cell size and Cdk1 inhibitory phosphorylation under limiting nutrient conditions (34). Together, these observations suggest that Igo1/2 play a role in controlling mitotic inhibitory phosphorylation of Cdk1.
To characterize the in vivo functions of Pkc1-dependent phosphorylation of Cdc55 and Igo2, we mapped and mutated Pkc1-dependent phosphorylation sites. The cdc55 phosphorylation site mutants eliminated Cdc55 phosphorylation in vivo and caused increased association of Igo2 with PP2ACdc55. However, they did not cause an obvious phenotype. Similarly, igo2 phosphorylation site mutants eliminated Igo2 phosphorylation in vivo but did not cause a phenotype. Thus, the physiological functions of Pkc1-dependent phosphorylation of Cdc55 and Igo2 remain unknown. One explanation could be that regulation of Igo2 is one of several overlapping mechanisms by which Pkc1 controls PP2ACdc55, so when the mechanism is eliminated other mechanisms can compensate. An important observation that could support this model is our discovery that association of Igo2 with PP2ACdc55 is regulated during the cell cycle, which suggests that it plays a role in cell cycle-dependent regulation of PP2ACdc55. The observation that Pkc1 activity drives multiple changes in the composition and phosphorylation of the PP2ACdc55-Zds complex is consistent with the possibility that activation of PP2ACdc55 requires multiple Pkc1-dependent events.
The timing of dissociation of Igo2 from PP2ACdc55 during the cell cycle is interesting. In previous work, we found that Pkc1 undergoes gradual hyperphosphorylation during polar bud growth that peaks just prior to mitosis. The data further suggested that Pkc1 activates PP2ACdc55 to trigger dephosphorylation of Mih1 and entry into mitosis (6). Here, we discovered that dissociation of Igo2 from PP2ACdc55 occurs before entry into mitosis and is correlated with maximal hyperphosphorylation of Pkc1. Because Pkc1 triggers dissociation of Igo2 from PP2ACdc55, the data are consistent with a model in which gradually rising Pkc1 activity during polar membrane growth leads to dissociation of Igo2 from PP2ACdc55, which could contribute to activation of PP2ACdc55. Testing this model will require a fuller understanding of the mechanisms by which Pkc1 activates PP2ACdc55. A particularly important step will be in vitro reconstitution of PP2ACdc55 activation by Pkc1.
Experimental Procedures
Reproducibility
All experiments shown in the figure panels were carried out for a minimum of three biological replicates that yielded reproducible results.
Strain Construction
The genotypes of the strains used in this study are listed in Table 5. All strains are in the W303-1A background (leu2-3,112 ura3-1 can1-100 ade2-1 his3-11,15 trp1-1 bar1Δ GAL+). Cells were grown in YPD (yeast extract/peptone/dextrose) media supplemented with 40 mg/liter adenine, except where noted. Standard PCR-based approach was used for making gene deletions and epitope tagging at the endogenous locus (38, 39). To generate strains in which the endogenous CDC55 or IGO2 genes were replaced with phosphorylation site mutants, we first replaced the genes with the Candida albicans URA3 gene. The phosphorylation site mutants were then amplified by PCR and recombined at the endogenous locus, and 5-fluoroorotic acid was used to select against the URA3 marker. Expression of mutants was verified by Western blotting, and mutations were verified by sequencing of a PCR product amplified from genomic DNA. Strains DK2112 and DK2113 were generated by digesting plasmid pDK120 with NsiI to target integration at the URA3 locus in strains DK186 and DK2016, respectively. Strain DK2044 was generated by digesting plasmid pVT30 with ApaI to target integration at the URA3 locus in strain HT195. Strains DK2361, DK2369, and DK2371 were generated by digesting plasmid pVT31, pVT34, and pVT35, respectively, with NcoI to target integration at the URA3 locus in strain DK1976.
TABLE 5.
Plasmids used in this study
| Name | Selection markers | |
|---|---|---|
| pDK120 | CUP1-PKC1* | URA3 |
| pVT30 | GAL1-PKC1* | URA3 |
| pVT31 | YIplac211-IGO2 | URA3 |
| pVT33 | YIplac211-igo2 S63A | URA3 |
| pVT34 | YIplac211-igo2-ps1 | URA3 |
| pVT35 | YIplac211-igo2-ps2 | URA3 |
| pVT37 | YIplac211-igo2 S119A | URA3 |
| pVT38 | YIplac211-CDC55 | URA3 |
| pVT45 | YIplac211-cdc55-ps1 | URA3 |
| pVT41 | YIplac211-cdc55-ps2 | URA3 |
| pDN4 | GAL1-PKC1*–3XHA | URA3 |
| pCN01 | pDONR221-IGO2 | KAN |
| pCN03 | pDEST17-IGO2 | AMP |
Plasmid Construction
Plasmids used in this study are listed in Table 6. To create an integrating vector that expresses constitutively active PKC1 (PKC1*) from the CUP1 promoter, the PKC1* open reading frame (ORF) was amplified by PCR and cloned into the KpnI and SalI sites of pSB131 to create pDK120 (oligos 5′-GCGCGGTCGACATGAGTTTTTCACAATTGGAGCAGAACATTAAAAAAAAGA-3′ and 5′-ATAGGTACCTCATAAATCCAAATCATCTGGCATAAAGG-3′). Digestion of this plasmid with NsiI targets integration at URA3. An integrating plasmid that expresses PKC1* from the GAL1 promoter was created by amplifying the PKC1* ORF and cloning into the SalI and EagI sites of pDK20 to create pVT30 (oligos 5′-GCGCGGTCGACATGAGTTTTTCACAATTGGAGCAGAACATTAAAAAAAAGA-3′ and 5′-CGCCGGCCGTCATAAATCCAAATCATCTGGCAT-3′). Digestion of this plasmid with ApaI targets integration at URA3. To create an integrating vector that expresses Igo2 from its endogenous promoter, a DNA fragment containing the IGO2 ORF and sequences immediately upstream and downstream of the ORF was amplified and cloned into the EcoRI and BamHI sites of YIplac211 to create pVT31 (oligos 5′-GCGCGAATTCCTCTGGTTTGTTTATATAGTGTTAACTTGG-3′ and 5′-GCGCGGATCCGTTCTGTGGTTGTTCTTTGATGGCAC-3′). Digestion of this plasmid with NcoI targets integration at URA3. Mutations in IGO2 were introduced by site-directed mutagenesis or DNA synthesis (DNA2.0, Menlo Park, CA) and were verified by DNA sequencing. pVT33 and pVT37 contain S63A and S119A mutants of IGO2, respectively. pVT34 and pVT35 contain mutant constructs igo2-ps1 (S94A, S99A, S119A, S121A, and S122A) and igo2-ps2 (S94A, S99A, S119A, S121A, S122A, S106A/S109A, S113A, and S115A), respectively. An integrating vector that expresses CDC55 from its endogenous promoter was created by cloning a DNA fragment that contained the CDC55 ORF and sequences immediately upstream and downstream of the ORF into the KpnI and HindIII sites of YIplac211 to create pVT38 (oligos 5′-GCGCGGTACCGCGGAATGCTATCTGTATATATTGC-3′ and 5′-GCGCAAGCTTCCTGTTGTCAACAGGCACATCGA-3′). This plasmid can be cut with StuI to target integration at URA3. To generate a plasmid containing cdc55-ps1, serine residues (S401A, S403A, S410A, S443A, and S444A) were mutated by site-directed mutagenesis in pVT38 to generate pVT45.
TABLE 6.
Strains used in this study
| Name | MAT | Genotype | Ref. or source |
|---|---|---|---|
| DK186 | a | 48 | |
| DK209 | a/x | 49 | |
| DK2016 | a | PKC1-CFP::HIS5 | 6 |
| DK2112 | a | pDK120 [CUP1-PKC1*, URA3] | This study |
| DK2113 | a | PKC1-CFP::HIS5 pDK120 [CUP1-PKC1*, URA3] | This study |
| HT195 | a | CDC55–3XHA::His3MX6 | 6 |
| DK2044/45 | a | CDC55–3XHA::His3MX6 pVT30 [GAL1-PKC1*, URA3] | This study |
| DK2289 | a | CDC55–3XHA::His3MX6 igo1::kanMX6 igo2-S63A | This study |
| DK2116 | a | CDC55–3XHA::His3MX6 rim15::kanMX6 | This study |
| DK1945 | a | igo2::kanMX6 | This study |
| DK2305 | a | Igo1::kanMX6 | This study |
| DK1976 | a | igo1::His3MX6 igo2::kanMX6 | This study |
| DK1979 | a | igo1::His3MX6 igo2::kanMX6 swe1::URA3 | This study |
| DK2156 | a | cdc55::URA3 | This study |
| SH650 | a | cdc55::kanMX6 | 11 |
| DK2850 | a | cdc55::cdc55-ps1 (S401A, S403A, S410A, S443A, S444A) | This study |
| DK2846 | a | cdc55::cdc55-ps2 (S410A) | |
| DK2337 | a | cdc55-ps1–3XHA::KanMX6 | This study |
| HT129 | a | zds1:: LEU2 zds2::TRP1 | This study |
| DK2489 | a | cdc55::cdc55-ps1 zds1::His3MX6 zds2::kanMX6 | This study |
| DK2361 | a | igo1::His3MX6 igo2::kanMX6 pVT3 [IGO2, URA3] | This study |
| DK2369 | a | igo1::His3MX6 igo2::kanMX6 pVT34 [igo2-ps1, URA3] | This study |
| DK2371 | a | igo1::His3MX6 igo2::kanMX6 pVT35 [igo2-ps2, URA3] | This study |
| DK1790 | a | pDN4 [GAL1-PKC1*–3XHA, URA3] | 6 |
| DK354 | a | CDC55–3XHA::His3MX6 zds1::LEU2 zds2::TRP1 | 11 |
| DK647 | a | rts1::kanMX6 | 50 |
| DK2142 | a | RIM15–3XHA::His3MX6 | This study |
Immunoaffinity Purification of PP2ACdc55–3XHA Complexes for Mass Spectrometry
PP2ACdc55–3XHA was purified from control cells (HT195) and from cells that overexpress PKC1* from the GAL1 promoter (DK2045). Cells were grown overnight at 30 °C in YEP containing 2% glycerol and 2% ethanol to OD600 ∼0.8. PKC1* expression was induced for 45 min by addition of galactose to 2% final concentration. Cells were pelleted, resuspended in ∼50 ml of cold 50 mm Hepes-KOH, pH 7.6, and then repelleted. The supernatants were removed, and the cell pellets were flash-frozen in liquid nitrogen. To generate cell powder, frozen cell pellets were ground in a pre-chilled Krupps coffee mill in the presence of dry ice for 2 min, followed by grinding under liquid nitrogen with a mortar and pestle for an additional 20 min.
Cell powder, 15 g, was resuspended in 25 ml of extract buffer containing high concentrations of salt and phosphatase inhibitors (50 mm Hepes, pH 7.6, 700 mm NaCl, 150 mm NaF, 150 mm BGP, 0.25% Tween 20, and 1 mm PMSF). Crude extracts were stirred for 7–10 min at 4 °C, and the extracts were centrifuged at 40,000 rpm in a pre-chilled Ti-70 rotor for 1 h. Clarified extracts were incubated with 600 μg of purified polyclonal rabbit anti-HA antibody bound to 500 μl of Affi-Gel 10-protein A-agarose beads (Bio-Rad) for 3 h at 4 °C. After incubation with the extract, the beads were washed batchwise three times with 15 ml of cold extract buffer without PMSF, transferred to an empty 1.5-ml Biospin column (Bio-Rad), and washed 10 times with 1 ml of extract buffer without PMSF followed by five times with 1 ml of extract buffer without PMSF or Tween 20. To elute the complex from the column, 250 μl of elution buffer (50 mm Hepes-KOH, pH 7.6, 250 mm KCl, 1 mm MgCl2, 1 mm EGTA, 5% glycerol) containing HA dipeptide at 0.5 mg/ml was added to the column. The initial flow-through was discarded. After a 30-min incubation at room temperature, another aliquot was added, and the flow-through fraction was collected. This process was repeated four more times. The fourth elution used buffer without HA dipeptide. Fractions were combined and flash-frozen in liquid nitrogen. For mass spectrometry, 750 μl of the eluate was precipitated in the presence of 10% trichloroacetic acid (TCA) for 10 min on ice, followed by centrifugation for 10 min in a tabletop microcentrifuge at 13,000 rpm. The supernatant was aspirated off, and the pellet was washed two times with 1 ml of acetone and then dried.
Peptide Digestion and Reductive Dimethylation
TCA pellets from Cdc55–3XHA affinity purifications were resuspended in 150 μl of 1 m urea, 50 mm Tris-HCl, pH 8.8, with 20 ng/μl sequencing grade trypsin (Promega). Digestion proceeded overnight at 37 °C. Digested peptides were acidified by the addition of 20 μl of 10% trifluoroacetic acid, and the resultant precipitate was pelleted by centrifuging for 2 min at 21,000 × g. The supernatants were loaded onto pre-wet hand-packed C18 solid phase extraction STAGE tips (40). The columns were washed with 1% formic acid, and the peptides were reduced and alkylated by the sequential addition of 5 mm dithiothreitol (DTT), 15 mm iodoacetamide and then washed with phosphate/citrate buffer (227 mm Na2HPO4, 86 mm NaH2C6H5O7, pH 5.5). Control sample peptides were labeled by reductive dimethylation (27) with “light” reductive dimethylation reaction mix (0.8% formaldehyde (Sigma, catalog no. F1635), 120 mm NaCNBH3 (Sigma, catalog no. 296945), in phosphate/citrate buffer). In parallel, PKC1* sample peptides were labeled with “heavy” reductive dimethylation reaction mix (0.8% d2-formaldehyde (Isotec, catalog no. 492620), 120 mm NaCNBD3 (CDN, catalog no. D-1797), in phosphate/citrate buffer). The columns were washed with 1% formic acid, and the peptides were eluted with 70% acetonitrile, 1% formic acid, dried under vacuum, and stored at −20 °C. Equal amounts of control (light) and PKC1* (heavy) peptides were combined and dried under vacuum. TCA pellets from His6-Igo2 in vitro phosphorylation reactions were digested and labeled as described above for PP2ACdc55-Zds complexes, labeling the control (−ATP) reaction with light reagents and the experimental (+ATP) reaction with heavy reagents.
Mass Spectrometry
Peptides were resuspended in 5% formic acid and analyzed on an LTQ Orbitrap Discovery or LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Peptides were separated on a 100-μm inner diameter fused-silica column packed with 20 cm of Maccel C18AQ resin (3 μm, 200 Å, Nest Group, Southborough, MA) using a gradient of 5–26% acetonitrile in 0.125% formic acid over 150 min at a flow rate of ∼300 nl/min.
Peptides were detected in a hybrid dual-cell quadrupole linear ion trap-orbitrap mass spectrometer (LTQ Orbitrap Velos, Thermo Fisher Scientific) using a data-dependent Top20-MS2 method or on LTQ Orbitrap Discovery using a similar Top10-MS2 method. For each cycle, one full MS scan of m/z = 300–1500 was acquired in the Orbitrap with AGC target = 1 × 106. Each full scan was followed by the selection of the most intense ions, up to 20 (10 for Discovery), for collision-induced dissociation and MS2 analysis in the LTQ. An AGC target of 2.5 × 103 was used for MS2 scans. Ions selected for MS2 analysis were excluded from re-analysis for 90 s. Precursor ions with charge = 1+ or unassigned were excluded from selection for MS2 analysis. Lockmass, employing atmospheric polydimethylsiloxane (m/z = 371.1012) as an internal standard, was used in all runs to calibrate orbitrap MS precursor masses.
Peptide Identification and Filtering
MS2 spectra from PP2ACdc55-Zds complex runs were searched using SEQUEST version 28 (revision 13) against a composite database containing the translated sequences of all predicted open reading frames of Saccharomyces cerevisiae and its reversed complement, using the following parameters: a precursor mass tolerance of ±20 ppm; 1.0-Da product ion mass tolerance; lysC digestion; up to two missed cleavages; static modifications of carbamidomethylation on cysteine (+57.0214), dimethyl adducts (+28.0313) on lysine, and peptide N termini; and dynamic modifications of methionine oxidation (+15.9949), heavy dimethylation (+6.0377) on lysine and peptide N termini, and phosphate (+79.9663) on serine, threonine, and tyrosine.
Peptide spectral matches were filtered to 1% false discovery rate (FDR) using the target-decoy strategy (41) combined with linear discriminant analysis (42) using several different parameters, including Xcorr, ΔCn′, precursor mass error, missed cleavages, and observed ion charge state. Linear discriminant models were calculated for each LC-MS/MS run using peptide matches to forward and reversed protein sequences as positive and negative training data. Peptide spectral matches within each run were sorted in descending order by discriminant score and filtered to a 1% FDR as revealed by the number of decoy sequences remaining in the data set. The data were further filtered to control protein level FDRs. Peptides from all runs in each experiment were combined and assembled into proteins. Protein scores were derived from the product of all linear discriminant analysis peptide probabilities, sorted by rank, and filtered to 1% FDR as described for peptides. The FDR of the remaining peptides fell dramatically after protein filtering. Remaining peptide matches to the decoy database were removed from the final dataset.
Spectra from Igo2 in in vitro phosphorylation reactions were searched using SEQUEST against a custom database containing the His6-Igo2 sequence along with common contaminants and the reverse sequences. Search parameters were the same as for PP2ACdc55-Zds complexes except that no enzyme specificity was indicated. Peptides were filtered to 1% false-positives as described above allowing for a single non-tryptic peptide terminus.
Relative peptide quantification was calculated using the Vista program (43) to extract the areas of heavy and light peptide traces and determine their ratios for each peptide. For protein complex affinity purification experiments, a total signal-to-noise of heavy and light species of ≥10 was required. Protein ratios were calculated as described (44) using the median log2 ratio of all peptides for each protein. Protein ratios were normalized to the bait protein levels.
Phosphorylation site assignment was evaluated using the Ascore algorithm (28). The Ascore = −10 log(p), thus Ascore = 13 represents p = 0.05 and 95% probability of correct assignment. In many cases, the same peptide was chosen for MS/MS multiple times resulting in different site assignments with varying Ascores.
Phosphorylation site occupancy for in vitro Igo2 phosphorylation was calculated based on the depletion of unphosphorylated peptides in the presence of PKC1 kinase activity using the median ratio of peptides harboring residues identified as phosphorylated in the phosphopeptide analysis. The fractional phosphorylation occupancy = 1 − median(heavy/light). For these analyses we required signal-to-noise ≥100 for both heavy and light peptides.
Immunoaffinity Purification of Proteins for in Vitro Assays
PP2ACdc55–3XHA was purified using a slightly modified version of the protocol used to isolate complexes for mass spectrometry. Cell powder was lysed in a high salt extract buffer lacking phosphatase inhibitors (50 mm Hepes, 1 m NaCl, 0.25% Tween 20, and 1 mm PMSF). For Pkc1*-3XHA, DK1790 cells were grown overnight at 30 °C to A600 ∼0.7 in YEP with 2% glycerol and 2% ethanol. Induction of PKC1* was initiated by the addition of galactose to 2% final concentration. Protein expression was allowed to proceed for 3 h. Cells were collected, washed, and processed as described previously. Purification was carried out using the high salt buffer without phosphatase buffers.
Cell Cycle Time Courses
Cells were grown overnight at room temperature to an A600 ∼0.5 and synchronized in G1 by addition of α-factor to either 0.5 μg/ml (bar1Δ strains) or 15 μg/ml (BAR1 strains) for 3 h and 30 min. Cells were released from the arrest by washing three times with fresh YPD medium pre-warmed to the specified temperature. For time courses involving a temperature shift, cultures were incubated at room temperature prior to the shift and were transferred to a shaking water bath to shift the temperature.
At each time point, 1.6-ml samples were collected in screw-cap tubes. Cells were pelleted; the supernatant was removed, and 250 μl of glass beads was added before freezing in liquid nitrogen. To lyse cells, 140 μl of sample buffer (65 mm Tris-HCl, pH 6.8, 3% SDS, 10% glycerol, 5% β-mercaptoethanol, 50 mm NaF, and 100 mm β-glycerophosphate) was added. PMSF was added to the sample buffer to a final concentration of 1 mm immediately before use. To lyse cells, tubes were placed in a disruptor (Multibeater-8; BioSpec) at top speed for 2 min. The tubes were immediately removed, centrifuged for 15 s in a microcentrifuge, and then placed in a boiling water bath for 6 min. After boiling, the tubes were centrifuged again for 5 min. Extracts made by this protocol typically have a protein concentration of 1 μg/μl. For Western blotting, each gel lane was loaded with 5 μl (for Zds1, Clb2, Cdc28 phospho-Tyr-19, and Cdc55), 15 μl (for Igo2 and Swe1), or 20 μl (for Mih1).
Western Blotting
SDS-PAGE and Western blotting were performed as described previously (17, 45). Polyacrylamide gels with dimensions of 145 × 65–70 mm (resolving gel) were run with a power source set to limit current to 20 mA per gel and a maximum of 165 V. For purified proteins, 9% gels were run until the dye front ran off the bottom of the gel. For Pkc1 Western blottings, electrophoresis was performed on a 9% polyacrylamide gel until a 57.6-kDa marker ran off the bottom of the gel. For Igo2 Western blottings, electrophoresis was performed on a 15% polyacrylamide gel until a 14-kDa marker reached the bottom of the gel. For Cdc55 Western blottings, electrophoresis was performed on a 9% polyacrylamide gel until a 40-kDa marker reached the bottom of the gel. Western blots were transferred for 75 min at 800 mA at 4 °C in a transfer tank (Hoeffer) in a buffer containing 20 mm Tris base, 150 mm glycine, and 20% methanol or in a TransBlot Turbo semi-dry transfer system (Bio-Rad). Blots were probed overnight at 4 °C with affinity-purified rabbit polyclonal antibodies raised against the specified protein. Blots were probed with an HRP-conjugated donkey anti-rabbit secondary antibody (GE Healthcare). HRP was detected with the Advansta ECL reagents.
Coimmunoprecipitation
Coimmunoprecipitation of Igo2 with PP2ACdc55–3XHA was assayed as described previously with the following modifications (46). Strains DK186 (untagged control), HT195 (CDC55–3XHA), DK2116 (CDC55–3XHA rim15Δ), and DK2289 (CDC55–3XHA igo1Δ igo2-S63A) were grown overnight to an OD600 ∼0.7 in 50 ml of YPD medium at room temperature. The cells were pelleted, resuspended in 1 ml of YPD, pelleted again in a 2-ml tube, and the supernatant was removed and the pellet was frozen in liquid nitrogen. For experiments requiring galactose induction of PKC1*, strains HT195 (CDC55–3XHA) and DK2044 (CDC55–3XHA GAL1-PKC1*) were grown overnight to OD600 ∼0.7 in 100 ml of YEP supplemented with 2% glycerol and 2% ethanol at 30 °C. 50 ml of culture was pelleted, resuspended in 1 ml of the same media, pelleted again in a 2-ml tube, and frozen in liquid nitrogen. Galactose was added to the remaining culture to a final concentration of 2%, and PKC1* expression was allowed to proceed for 45 min at 30 °C.
Immunoaffinity beads were made by binding 15 μg of purified polyclonal rabbit HA antibody to 15-μl protein A beads in the presence of phosphate-buffered saline containing 500 mm NaCl and 0.1% Tween 20. Cell extracts were made by adding 300 μl of acid-washed glass beads to frozen cell pellets followed by 300 μl of lysis buffer (50 mm Hepes-KOH, pH 7.6, 75 mm B glycerol phosphate, 50 mm NaF, 1 mm MgCl2, 1 mm EGTA, 5% glycerol, 0.25% Tween 20, and 1 mm PMSF). The tubes were immediately placed into a bead-beater (Biospec Multibeater-8) and shaken at top speed for 25 s. The tubes were briefly spun at 14,000 rpm in a microcentrifuge and then placed in an ice-water bath for 5 min. 250 μl of the supernatant was transferred to a new 1.5-ml tube and replaced with 250 μl of lysis buffer. The tubes were beaten again for 25 s. 250 μl of the supernatant was removed, pooled with the first supernatant, and centrifuged at 14,000 rpm in a microcentrifuge for 10 min at 4 °C. The supernatant was added to immunoaffinity beads equilibrated in lysis buffer. The tubes were rotated gently end over end at 4 °C for 1 h and 45 min. The immunoprecipitation was monitored by collecting 20-μl samples of the extract before the microcentrifuge spin, before treatment with antibody, and after antibody binding and pelleting of the beads. The samples were frozen in liquid nitrogen for analysis by Western blotting. The beads were washed three times with 400 μof lysis buffer without PMSF. Cdc55–3XHA and associated proteins were eluted from the beads by the addition of 150 μl of elution buffer (50 mm Hepes-KOH, pH 7.6, 1 m NaCl, 1 mm MgCl2, 1 mm EGTA, 5% glycerol, and 0.5 mg/ml HA dipeptide) at room temperature. The beads were incubated for 15 min and gently agitated every few minutes to allow for mixing of the beads. The beads were pelleted in a microcentrifuge, and 125 μl of the supernatant was removed, taking care to avoid the antibody-containing beads. This process was repeated once more with the exception that 150 μl of the supernatant was removed. The supernatants were pooled and precipitated by the addition of TCA to 10%. The resulting pellet was resuspended in 25 μl of 1× sample buffer; 12 and 1 μl were loaded onto a 15 and 9% polyacrylamide gel and used for Western blottings to probe for Igo2 and Cdc55–3XHA, respectively. For Western blotting of the crude extracts, 40 μl of 1× sample buffer was added to 20 μl of crude extract; the samples were boiled, and 10–20 μl was loaded onto an SDS-polyacrylamide gel.
Antibodies
Antibodies that recognize Igo1 and Igo2 were generated by immunizing a rabbit with a full-length His6-Igo2 fusion. To make a plasmid that expresses a His6-Igo2 fusion, the IGO2 open reading frame was amplified in a two-step process and recombined into the vector pDONR221 (Gateway; Life Technologies, Inc.) to create pCN01. First, a tobacco etch virus cleavage site was placed upstream of IGO2 using oligos 5′-GAAAACCTGTACTTCCAGTCCATGTCAGAGGATCTTTCACCTAC-3′ and 5′-GTACAAGAAAGCTGGGTCTCATTTATTTGGAGATCTTGGTGG-3′. Second, attB1 and attB2 recombination sites were added using oligos 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCGAAAACCTGTACTTCCAGTCC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTC-3′. The IGO2 fragment was then recombined into pDEST17 to create pCN03, which expresses Igo2 as a His6 fusion. The His6-Igo2 fusion was purified and used to immunize a rabbit using standard protocols. The serum was run over a His6-Igo2 column to purify anti-Igo2 antibodies. To test the specificity of the antibody, we probed extracts from wild type, igo1Δ, and igo2Δ cells. Because of their high sequence similarity, anti-Igo2 was found to cross-react with Igo1. Igo1 and Igo2 differ enough in molecular weight that their corresponding bands can be resolved by SDS-PAGE (Fig. 3A).
Anti-Clb2 was generated as described previously and verified with a clb2Δ strain (37). Anti-Pkc1 was generated, as described previously (6), and verified with a Pkc1-CFP fusion strain, which shifts Pkc1 to a higher molecular weight (see Fig. 1). Anti-Zds1 was generated, as described previously (12), and verified with a zds1Δ strain. Anti-Cdc55 was a gift from Adam Rudner and was verified with a cdc55Δ strain.
Kinase Assays
For protein kinase assays, purified proteins were combined, with substrate in excess, in kinase assay buffer (50 mm Hepes-KOH, pH 7.6, 10% glycerol, 1 mm DTT, 1 mm ATP, 2 mm MgCl2, 0.02% Tween 20). KCl was added to reactions to obtain a final concentration of 90 mm. Where indicated, the protein phosphatase inhibitor okadaic acid was added to a final concentration of 50 μm. To demonstrate that Pkc1* can phosphorylate Igo2, 50-μl radiolabeled reactions were carried out containing 6 μg of His6-Igo2 and 12 μl of Pkc1*-3XHA in kinase assay buffer. To show that Pkc1* can induce an electrophoretic shift in Igo2, 30-μl reactions were carried out containing 100–200 ng of Igo2 and 6–12 μl of Pkc1*-3XHA. The His6 tag was cleaved off the Igo2 fusion protein using GST-tobacco etch virus prior to incubating with Pkc1*-3XHA. To identify Pkc1 phosphorylation sites on Igo2 by mass spectrometry, identical 50-μl reactions containing 3 μg of His6-Igo2 and 15 μl of Pkc1*-3XHA were set up. ATP was added to one reaction, whereas the other reaction was retained as a control. To demonstrate that Pkc1* can phosphorylate PP2ACdc55, 100-μl radiolabeled reactions containing 25 μl of PP2ACdc55–3XHA and 8 μl of Pkc1*-3XHA were carried out. Cold kinase reactions were initiated by addition of ATP to a final concentration of 1 mm. For 32P labeling, assays contained 10–15 μCi of [γ-32P]ATP and 100 μm cold ATP. Reactions were incubated at 30 °C for 30 min with gentle agitation every 10 min to ensure sample mixing. Reactions were quenched by addition of 4× sample buffer and frozen in liquid nitrogen. To demonstrate that one of the phosphorylated bands corresponded to Cdc55, we doubled the Pkc1*-3XHA/Cdc55–3XHA reaction. At the end of the reaction, SDS was added to 1%, and the sample was boiled for 2–4 min to disrupt the PP2ACdc55 complex. RIPA buffer (150 mm NaCl, 1.0% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0) was then added to the reaction to bring the SDS concentration down to 0.1%, and the supernatant was split equally between two tubes, one containing anti-Cdc55 beads and the other containing anti-GST control beads. These tubes were rotated end over end at room temperature for 2 h and washed three times in RIPA buffer. After the third wash, 25 μl of 2× protein sample buffer was added to the beads. The beads were boiled for 6 min, and 20 μl was loaded onto a 9% SDS-polyacrylamide gel.
Microscopy
Photographs of yeast cells were taken using an Axioskop microscope fitted with a ×100 Plan-Neofluar 1.3 NA objective and an AxioCam HRm camera (Carl Zeiss). Images were acquired using AxioVision software.
Author Contributions
V. T. and D. R. K. designed the study, interpreted the results, and wrote the manuscript. V. T. carried out the experiments shown in Figs. 1–7, with assistance from A. W., J. F., and R. L. N. D. and S. P. G. carried out and interpreted the mass spectrometry analysis.
Supplementary Material
Acknowledgments
We thank members of the laboratory for helpful advice and critical reading of the manuscript. We also thank Kate Schuber, Christian Nelson, and Tyler Liban for assistance with experiments. Finally, we thank Adam Rudner for providing anti-Cdc55 antibody.
This work was supported by the University of California Cancer Research Coordinating Committee and National Institutes of Health Grant GM069602. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1–S3.
- PP2A
- protein phosphatase 2A
- FDR
- false discovery rate
- oligo
- oligonucleotide.
References
- 1. Hartwell L. H., and Unger M. W. (1977) Unequal division in Saccharomyces cerevisiae and its implications for the control of cell division. J. Cell Biol. 75, 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnston G. C., Pringle J. R., and Hartwell L. H. (1977) Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp. Cell Res. 105, 79–98 [DOI] [PubMed] [Google Scholar]
- 3. Lew D. J., and Reed S. I. (1993) Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J. Cell Biol. 120, 1305–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jorgensen P., and Tyers M. (2004) How cells coordinate growth and division. Curr. Biol. 14, R1014–R1027 [DOI] [PubMed] [Google Scholar]
- 5. Turner J. J., Ewald J. C., and Skotheim J. M. (2012) Cell size control in yeast. Curr. Biol. 22, R350–R359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anastasia S. D., Nguyen D. L., Thai V., Meloy M., MacDonough T., and Kellogg D. R. (2012) A link between mitotic entry and membrane growth suggests a novel model for cell size control. J. Cell Biol. 197, 89–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Novick P., and Schekman R. (1979) Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 76, 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamochi W., Tanaka K., Nonaka H., Maeda A., Musha T., and Takai Y. (1994) Growth site localization of Rho1 small GTP-binding protein and its involvement in bud formation in Saccharomyces cerevisiae. J. Cell Biol. 125, 1077–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abe M., Qadota H., Hirata A., and Ohya Y. (2003) Lack of GTP-bound Rho1p in secretory vesicles of Saccharomyces cerevisiae. J. Cell Biol. 162, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamada Y., Qadota H., Python C. P., Anraku Y., Ohya Y., and Levin D. E. (1996) Activation of yeast protein kinase C by Rho1 GTPase. J. Biol. Chem. 271, 9193–9196 [DOI] [PubMed] [Google Scholar]
- 11. Pal G., Paraz M. T., and Kellogg D. R. (2008) Regulation of Mih1/Cdc25 by protein phosphatase 2A and casein kinase 1. J. Cell Biol. 180, 931–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wicky S., Tjandra H., Schieltz D., Yates J. 3rd, Kellogg D. R. (2011) The Zds proteins control entry into mitosis and target protein phosphatase 2A to the Cdc25 phosphatase. Mol. Biol. Cell 22, 20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin F. C., and Arndt K. T. (1995) The role of Saccharomyces cerevisiae type 2A phosphatase in the actin cytoskeleton and in entry into mitosis. EMBO J. 14, 2745–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minshull J., Straight A., Rudner A. D., Dernburg A. F., Belmont A., and Murray A. W. (1996) Protein phosphatase 2A regulates MPF activity and sister chromatid cohesion in budding yeast. Curr. Biol. 6, 1609–1620 [DOI] [PubMed] [Google Scholar]
- 15. Yang H., Jiang W., Gentry M., and Hallberg R. L. (2000) Loss of a protein phosphatase 2A regulatory subunit (Cdc55p) elicits improper regulation of Swe1p degradation. Mol. Cell. Biol. 20, 8143–8156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yasutis K., Vignali M., Ryder M., Tameire F., Dighe S. A., Fields S., and Kozminski K. G. (2010) Zds2p regulates Swe1p-dependent polarized cell growth in Saccharomyces cerevisiae via a novel Cdc55p interaction domain. Mol. Biol. Cell 21, 4373–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harvey S. L., Enciso G., Dephoure N., Gygi S. P., Gunawardena J., and Kellogg D. R. (2011) A phosphatase threshold sets the level of Cdk1 activity in early mitosis. Mol. Biol. Cell 22, 3595–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rossio V., and Yoshida S. (2011) Spatial regulation of Cdc55-PP2A by Zds1/Zds2 controls mitotic entry and mitotic exit in budding yeast. J. Cell Biol. 193, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andrews P. D., and Stark M. J. (2000) Dynamic, Rho1p-dependent localization of Pkc1p to sites of polarized growth. J. Cell Sci. 113, 2685–2693 [DOI] [PubMed] [Google Scholar]
- 20. Gentry M. S., and Hallberg R. L. (2002) Localization of Saccharomyces cerevisiae protein phosphatase 2A subunits throughout mitotic cell cycle. Mol. Biol. Cell 13, 3477–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Qureshi-Emili A., Li Y., Godwin B., Conover D., Kalbfleisch T., et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627 [DOI] [PubMed] [Google Scholar]
- 22. Drees B. L., Sundin B., Brazeau E., Caviston J. P., Chen G. C., Guo W., Kozminski K. G., Lau M. W., Moskow J. J., Tong A., Schenkman L. R., McKenzie A. 3rd., Brennwald P., Longtine M., Bi E., et al. (2001) A protein interaction map for cell polarity development. J. Cell Biol. 154, 549–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Queralt E., and Uhlmann F. (2008) Separase cooperates with Zds1 and Zds2 to activate Cdc14 phosphatase in early anaphase. J. Cell Biol. 182, 873–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y., Moir R. D., Sethy-Coraci I. K., Warner J. R., and Willis I. M. (2000) Repression of ribosome and tRNA synthesis in secretion-defective cells is signaled by a novel branch of the cell integrity pathway. Mol. Cell. Biol. 20, 3843–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nanduri J., and Tartakoff A. M. (2001) The arrest of secretion response in yeast: signaling from the secretory path to the nucleus via Wsc proteins and Pkc1p. Mol. Cell 8, 281–289 [DOI] [PubMed] [Google Scholar]
- 26. TerBush D. R., Maurice T., Roth D., and Novick P. (1996) The exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 15, 6483–6494 [PMC free article] [PubMed] [Google Scholar]
- 27. Boersema P. J., Raijmakers R., Lemeer S., Mohammed S., and Heck A. J. (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 [DOI] [PubMed] [Google Scholar]
- 28. Beausoleil S. A., Villén J., Gerber S. A., Rush J., and Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 29. Mochida S., Maslen S. L., Skehel M., and Hunt T. (2010) Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330, 1670–1673 [DOI] [PubMed] [Google Scholar]
- 30. Gharbi-Ayachi A., Labbé J. C., Burgess A., Vigneron S., Strub J. M., Brioudes E., Van-Dorsselaer A., Castro A., and Lorca T. (2010) The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330, 1673–1677 [DOI] [PubMed] [Google Scholar]
- 31. Bontron S., Jaquenoud M., Vaga S., Talarek N., Bodenmiller B., Aebersold R., and De Virgilio C. (2013) Yeast endosulfines control entry into quiescence and chronological life span by inhibiting protein phosphatase 2A. Cell Rep. 3, 16–22 [DOI] [PubMed] [Google Scholar]
- 32. Juanes M. A., Khoueiry R., Kupka T., Castro A., Mudrak I., Ogris E., Lorca T., and Piatti S. (2013) Budding yeast greatwall and endosulfines control activity and spatial regulation of PP2A(Cdc55) for timely mitotic progression. PLoS Genet. 9, e1003575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams B. C., Filter J. J., Blake-Hodek K. A., Wadzinski B. E., Fuda N. J., Shalloway D., and Goldberg M. L. (2014) Greatwall-phosphorylated endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. eLife 3, e01695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chica N., Rozalén A. E., Pérez-Hidalgo L., Rubio A., Novak B., and Moreno S. (2016) Nutritional control of cell size by the Greatwall-Endosulfine-PP2A.B55 pathway. Curr. Biol. 26, 319–330 [DOI] [PubMed] [Google Scholar]
- 35. Xu Y., Chen Y., Zhang P., Jeffrey P. D., and Shi Y. (2008) Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31, 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hombauer H., Weismann D., Mudrak I., Stanzel C., Fellner T., Lackner D. H., and Ogris E. (2007) Generation of active protein phosphatase 2A is coupled to holoenzyme assembly. PLoS Biol. 5, e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kellogg D. R., and Murray A. W. (1995) NAP1 acts with Clb2 to perform mitotic functions and suppress polar bud growth in budding yeast. J. Cell Biol. 130, 675–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Longtine M. S., McKenzie A. 3rd., Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 39. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 40. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 41. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 42. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bakalarski C. E., Elias J. E., Villén J., Haas W., Gerber S. A., Everley P. A., and Gygi S. P. (2008) The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. J. Proteome Res. 7, 4756–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torres E. M., Dephoure N., Panneerselvam A., Tucker C. M., Whittaker C. A., Gygi S. P., Dunham M. J., and Amon A. (2010) Identification of aneuploidy-tolerating mutations. Cell 143, 71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson C. W., Baum P. R., and Gesteland R. F. (1973) Processing of adenovirus 2-induced proteins. J. Virol. 12, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mortensen E. M., McDonald H., Yates J. 3rd, and Kellogg D. R. (2002) Cell cycle-dependent assembly of a Gin4-septin complex. Mol. Biol. Cell 13, 2091–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gouet P., Courcelle E., Stuart D. I., and Metoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
- 48. Altman R., and Kellogg D. (1997) Control of mitotic events by Nap1 and the Gin4 kinase. J. Cell Biol. 138, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zimmerman Z. A., and Kellogg D. R. (2001) The Sda1 protein is required for passage through Start. Mol. Biol. Cell 12, 201–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Artiles K., Anastasia S., McCusker D., and Kellogg D. R. (2009) The Rts1 regulatory subunit of protein phosphatase 2A is required for control of G1 cyclin transcription and nutrient modulation of cell size. PLoS Genet. 5, e1000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.