

Abstract
The highly mutagenic A:8-oxoguanine (oxoG) base pair is generated mainly by misreplication of the C:oxoG base pair, the oxidation product of the C:G base pair. The A:oxoG base pair is particularly insidious because neither base in it carries faithful information to direct the repair of the other. The bacterial MutY (MUTYH in humans) adenine DNA glycosylase is able to initiate the repair of A:oxoG by selectively cleaving the A base from the A:oxoG base pair. The difference between faithful repair and wreaking mutagenic havoc on the genome lies in the accurate discrimination between two structurally similar base pairs: A:oxoG and A:T. Here we present two crystal structures of the MutY N-terminal domain in complex with either undamaged DNA or DNA containing an intrahelical lesion. These structures have captured for the first time a DNA glycosylase scanning the genome for a damaged base in the very first stage of lesion recognition and the base extrusion pathway. The mode of interaction observed here has suggested a common lesion-scanning mechanism across the entire helix-hairpin-helix superfamily to which MutY belongs. In addition, small angle X-ray scattering studies together with accompanying biochemical assays have suggested a possible role played by the C-terminal oxoG-recognition domain of MutY in lesion scanning.
Keywords: 8-oxoguanine (8-oxoG), base excision repair (BER), DNA, small angle X-ray scattering (SAXS), structural biology, disulfide trapping, lesion scanning
Introduction
Cells are under constant threat from a variety of chemical and biological agents that damage DNA. One of the most abundant forms of genotoxic damage is 8-oxoguanine (oxoG),2 which arises from the oxidation of guanine by the attack of the reactive oxygen species (1–3). oxoG only differs from its normal counterpart guanine by two atoms, one each at positions 7 (N7–H versus N7, respectively) and 8 (C8=O versus C8–H, respectively) (Fig. 1A). Such changes do not alter the Watson-Crick face of the nucleobase, leaving its ability to base pair with cytosine unaffected. However, during DNA replication, the replicative DNA polymerase prefers to base pair oxoG with A, because of the stable Hoogsteen base pair formed between the two (Fig. 1B) (4, 5). oxoG:A poses an enormous challenge to the cellular DNA repair system, because neither base in this pair provides correct information with which to repair the other: oxoG is constitutionally aberrant, whereas A is a normal base but is misincorporated at the position supposed to be occupied by C. Failing to repair oxoG:A lesions results in deleterious G:C to T:A transversion mutations. To avoid the genotoxic effects caused by oxoG:A, cells utilize the evolutionarily conserved MutY (MUTYH in humans) adenine DNA glycosylase to selectively cleave the adenine base from oxoG:A (4, 6). The resulting abasic site product undergoes short patch DNA repair synthesis to generate primarily oxoG:C, which is in turn is repaired by MutM (hOGG1 in humans), and a lesser amount of oxoG:A, which is recycled into the MutY repair pathway (1, 3, 7).
FIGURE 1.
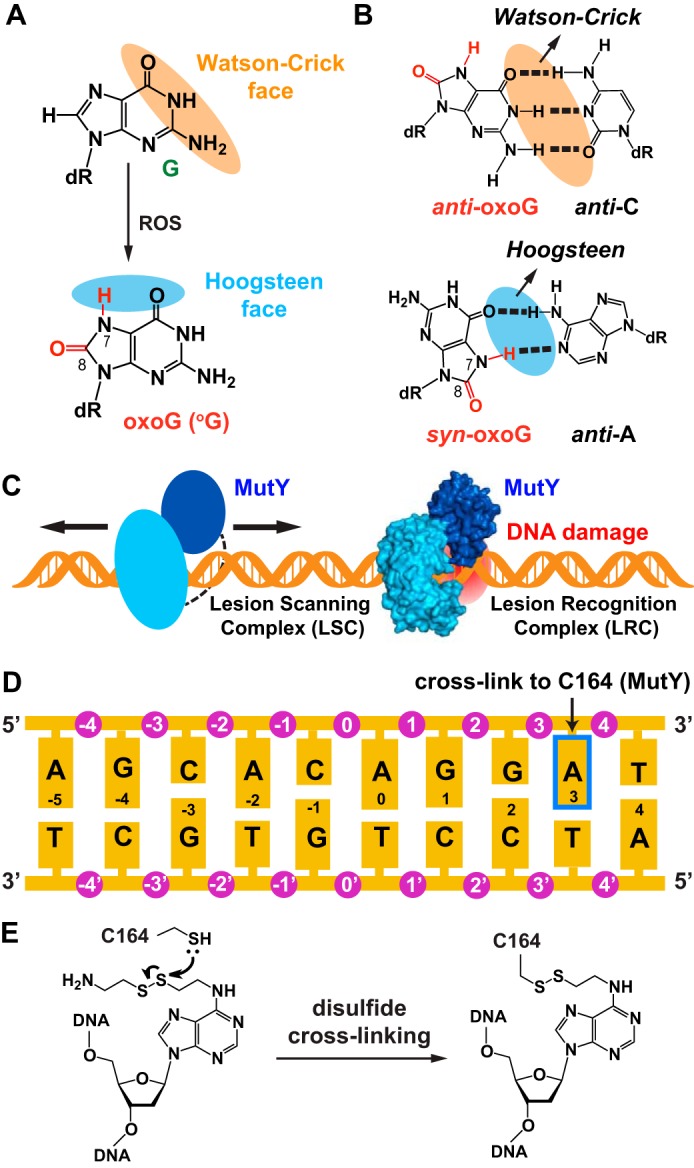
oxoG lesion in DNA. A, the primary oxoG lesion is formed by attack of reactive oxygen species (ROS) on a guanine residue. The oxidation leaves the Watson-Crick face of guanine unchanged. B, oxoG base exists in two alternative conformations in DNA. oxoG (anti-) forms Watson-Crick base pair with cytosine, whereas oxoG (syn-) forms a Hoogsteen base pair with adenine. C, schematic diagram of MutY scanning along the DNA for the lesion. MutY in the LRC is shown in surface representation, with the N-terminal domain colored cyan and the C-terminal domain colored blue. D, duplex DNA sequence used in this work. The arrow indicates the adenine that is modified (as shown in E) to cross-link to an engineered Cys residue in MutY. E, structure of the modified adenine used in disulfide cross-linking and of the covalent linkage it forms with MutY.
The pivotal role MutY/MUTYH plays in oxoG:A repair is underscored by observations that inactivation of MUTYH are directly correlated with a high rate of G:C to T:A transversion mutation in colorectal cancers (4, 8, 9). Defective repair of oxoG:A has also been shown to lead to mutations in other genes, such as the tumor suppressor APC (4, 10, 11) and the protooncogene KRAS (7, 12, 13). The oxoG:A mispair can also be generated by direct incorporation of doxoGTP by the replicative polymerase. Two recent studies have shown that the accumulation of doxoGTP led by the inhibition of the hMTH1 protein (which sanitizes the nucleotide pool by hydrolyzing the doxoGTP to doxoGMP) is toxic to human cancer cells (8, 9, 14, 15), further highlighting the importance of oxoG repair. As more and more evidence points to the importance of MUTYH in protecting the genome, it is of crucial importance that we understand the structural basis of its mechanism of action.
MutY is a member of the HhH-GPD superfamily of DNA glycosylases (10, 11, 16, 17). Its N-terminal domain (NTD) has a signature helix-hairpin-helix motif followed by a Gly/Pro-rich loop and a key catalytic aspartic acid residue. MutY bears an active site pocket that is responsible for the catalytic removal of the adenine base from the DNA (12, 13). The NTD alone has been shown to carry out catalytic removal of adenine from oxoG:A in vitro, although with much lower efficiency than the full-length MutY (14, 15, 18). The C-terminal domain (CTD) of MutY structurally resembles MutT, an 8-oxo-dGTPase, and in MutY is responsible for recognition of the oxoG nucleobase, providing the means by which MutY discriminates oxoG from T (16, 17, 19). Our lab has previously determined the structure of the MutY lesion recognition complex (LRC), where MutY (having a catalytically inactivating mutation D144N) is bound to duplex DNA bearing an oxoG:A lesion (12, 13, 19–21). The LRC structure shed light on the details of specific recognition MutY makes for the oxoG:A substrate. In the LRC, the adenine is extruded from the DNA duplex and resides in an extrahelical active-site pocket on the enzyme, poised to be excised by the protein. The oxoG, however, remains intrahelical and is stabilized by hydrogen bonding interactions with well conserved residues from both NTD and CTD. In a related LRC structure having a wild-type active site but uncleavable synthetic nucleobase, 2′-fluoro-2′-deoxyadenosine (fluorinated lesion-recognition complex (FLRC)), the substrate base is plunged more deeply into the active pocket than in the LRC and forms more extensive interactions with active site residues, suggesting that adenine excision proceeds via general acid catalysis (13, 18, 22). The FLRC structure is therefore considered to most accurately represent MutY poised to catalyze base-excision. We have also determined the structure of the MutY anti-substrate complex (ASC), in which the wild-type MutY is bound to DNA containing an oxoG:C base pair (19, 20). The ASC structure shows that MutY possesses an exo-site that acts as a decoy for cytosine, a mechanism that MutY utilizes to avoid cleaving C from the oxoG:C base pair. Although the molecular mechanisms of substrate recognition and anti-substrate recognition are well established both structurally and biochemically (12, 13, 19–21), the mechanism by which MutY scans along normal DNA remains largely unknown. Questions that remain to be addressed include how MutY achieves both fast and accurate DNA scanning, whether scanning requires both N-terminal and C-terminal domains, what features of the oxoG:A base pair allows discrimination from normal base pairs, and what conformational changes MutY undergoes upon encountering a lesion.
In this work, we determined the crystal structures of the N-terminal domain of MutY bound to either undamaged DNA or DNA bearing an intrahelical oxoG:A base pair, which are named the N-terminal lesion scanning complex (N-LSC) and the N-terminal intrahelical lesion recognition complex (N-ILRC), respectively. (Table 1 includes a summary of the acronyms of the MutY-DNA complexes mentioned in this work.) We also used small angle X-ray scattering (SAXS) to generate the envelope for the lesion-scanning complex (LSC), which has full-length MutY bound to normal DNA. Based on structural information obtained from the two X-ray methods, we conclude that both NTD and CTD are involved in lesion-scanning (the process where MutY scans the genome, which is mainly composed of undamaged DNA, for a lesion), and that the CTD changes its conformation going from the scanning mode to the lesion recognition mode. In addition, the N-ILRC structure suggests a possible mechanism for how MutY first detects an oxoG:A base pair while it is fully intrahelical.
TABLE 1.
List of the MutY-DNA complexes used in crystallography and SAXS experiments
| MutY-DNA complex | Protein component | DNA component |
|---|---|---|
| Lesion recognition complex (LRC) | Full-length MutY D144N, P164C | 11-mer double-stranded DNA containing an oxoG:A base pair |
| Fluorinated lesion recognition complex (FLRC) | Full-length MutY P164C | 11-mer double-stranded DNA containing an oxoG:FA base pair |
| Lesion scanning complex (LSC) | Full-length MutY D144N, P164C | 10-mer double-strand DNA containing a T:A base pair |
| N-terminal lesion scanning complex (N-LSC) | MutY-NTD D144N, P164C | 10-mer double-strand DNA containing a T:A base pair |
| N-terminal intrahelical lesion recognition complex (N-ILRC) | MutY-NTD D144N, V51Y, P164C | 10-mer double-strand DNA containing an oxoG:A base pair |
Results
Experimental System
Previous work has shown that the LRC does not readily crystallize by conventional crystallography methods. Crystallization was therefore facilitated by introducing an intermolecular disulfide cross-linker at a site that is 4 base pairs away from the target base pair (12, 13, 22). The relatively low affinity MutY exhibits toward undamaged DNA (20, 23), and the anticipated fleeting nature of such binding led us to introduce a similar cross-linking strategy to obtain a stable and homogenous protein-DNA complex suitable for crystallization. We first set out to crystallize the full-length MutY with undamaged DNA using the same cross-linking site that was used in the LRC (13, 23). Although we screened various DNA lengths and sequences, we consistently obtained crystals that diffracted poorly. Although we were able to place the NTD in the electron density maps obtained from the crystals using molecular replacement, the position of the CTD could not be assigned because of its poor electron density, which suggested that the CTD either exists in multiple conformations or is completely disordered in the LSC. Therefore, we truncated the C from MutY and crystallized the NTD (residues 1–229) bound to undamaged DNA. After screening several DNA sequences and using the cross-linking site P164C, which is well validated by the LRC and ASC structures, we obtained a stable MutY NTD-DNA complex by cross-linking the lesion recognition-competent but catalytically inactive D144N mutant version of the MutY NTD to a 10-mer undamaged DNA containing a thiol attached to the A3 nucleobase in the major groove (Fig. 1, D and E). The complex formed high quality crystals that enabled structure determination by molecular replacement and model building with refinement to 1.8 Å resolution (Table 2). To obtain the crystal structure of the N-ILRC, it was necessary to block the entry of adenine into the active site pocket either mechanistically or sterically. Hence, we introduced the V51Y mutation in close proximity to the extruded adenine in the LRC structure, in the hope that a more space-filling bulky residue would make the adenine remain intrahelical. Tainer and co-workers (12) have shown that the catalytic activity of Escherichia coli MutY-NTD is completely abolished when Val-45 (which is equivalent to Val-51 in Bacillus stearothermophilus) is mutated to tyrosine (15), probably because of hindered adenine nucleotide extrusion from the DNA duplex, which would be beneficial in our case. The same cross-linking strategy was applied to crystallize the N-ILRC, which contains the MutY-NTD (V51Y/D144N/P164C) bound to the same duplex DNA used for the N-LSC except for having an oxoG:A instead of a T:A base pair at position 0 (Fig. 1D). The crystal structure of this complex refined to 1.9 Å resolution revealed, as expected that the target oxoG:A base pair is completely intact and intrahelical. Finally, the full-length LSC prepared for SAXS studies has the same cross-linking site and DNA as in the N-LSC structure.
TABLE 2.
Crystallography data collection and refinement statistics
| N-LSC | N-ILRC | |
|---|---|---|
| PDB code | 5KN8 | 5KN9 |
| Data collection statistics | ||
| Resolution range (Å)b | 81.31–1.81 (1.85–1.81) | 97.86–1.93 (1.85–1.81) |
| Space group | P21 | P21 |
| Unit cell | ||
| a (Å) | 49.2 | 49.9 |
| b (Å) | 38.6 | 38.8 |
| c (Å) | 83.8 | 83.0 |
| α (°) | 90 | 90 |
| β (°) | 104 | 106 |
| γ (°) | 90 | 90 |
| Total reflectionsa | 102,066 (5851) | 84,628 (5745) |
| Unique reflectionsa | 28,148 (1646) | 23,193 (1552) |
| Completeness (%)a | 99.7 (99.9) | 99.5 (99.9) |
| Multiplicitya | 3.6 (3.6) | 3.6 (3.7) |
| Mean I/σ(I) a | 11.6 (1.3) | 10.9 (1.6) |
| Rmergea,b | 0.112 (1.044) | 0.110 (0.923) |
| CC½ | 0.996 (0.567) | 0.996 (0.567) |
| Refinement statistics | ||
| Rwork (%)a,b | 16.65 (27.13) | 16.87 (22.98) |
| Rfree (%)a,b | 21.96 (29.42) | 22.87 (30.53) |
| Number of non-hydrogen atoms | 2430 | 2395 |
| Macromolecules | 2239 | 2220 |
| Ligands | 9 | 10 |
| Water | 182 | 165 |
| Protein residues | 243 | 243 |
| RMSD from ideality | ||
| Bond length (Å) | 0.009 | 0.08 |
| Bond angles (°) | 1.474 | 1.316 |
| Ramachandran plot (%)c | ||
| Favored | 99.6 | 98.2 |
| Allowed | 0.4 | 1.8 |
| Outliers | 0 | 0 |
| Average B-factor | 30.30 | 34.80 |
| Macromolecules | 29.60 | 34.60 |
| Ligands | 20.00 | 25.40 |
| Solvent | 39.20 | 37.70 |
a The values in parentheses refer to the highest resolution shell.
b Rmerge = Σ|I − 〈I〉 |/Σ <I>, where I is the observed intensity. Rwork = Σ|Fo − Fc|/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree was calculated based on 5% data randomly selected and omitted throughout structure refinement (42).
c The values were calculated using MolProbity (43).
Structure of the N-terminal MutY Scanning Undamaged DNA
The overall architecture of the NTD in the N-LSC is very similar to that in the FLRC (heavy atom RMSD = 0.751; Fig. 2, A and B). The ways in which MutY interacts with the DNA, however, are very different in the two structures. In the FLRC, the NTD invades the DNA duplex from the minor groove side, flipping the FA into the active pocket and causing the DNA to bend severely at the target oxoG:A base pair. oxoG stays intrahelical, making hydrogen bonding interactions with amino acid residues from both the NTD and the CTD. Conversely, in the N-LSC, MutY associates with DNA loosely, and the DNA is almost canonical B-form (Fig. 2C); all base pairs remain completely intrahelical, as shown in the electron density map (Fig. 2D). We numbered the phosphate group that is 5′ to the target FA in the FLRC structure p0. (The same numbering applies to the N-LSC structure.) In the FLRC, MutY-NTD makes extensive phosphate contacts to both the 3′ (p1, p2, and p3) and the 5′ (p0 and p−1) side of the lesion on the target strand (Fig. 2, E and F). As a result, the target strand is severely bent as it exits from the active pocket. In the N-LSC, however, interactions with the DNA backbone are localized to the 3′ (p1, p2, and p3) side of the target base, from where the target strand diverges from MutY. In both structures, several amino acid residues from the HhH motif, namely Gly124, Tyr126, and Thr127, contact p2 and p3 using their backbone amide groups, although Thr127 also hydrogen bonds with p2 using its side chain (Fig. 2, E, F, I, and J). A calcium ion is also found in both structures to be coordinated by Ser118 and Val123 and contacts p3, further strengthening the protein interactions with the A strand. Gly145, which follows right after the HhH-GPD motif, contacts p1 with its main chain, providing the protein an additional point of contact on the DNA backbone. The fact that the HhH motif accounts for most of the phosphate contacts in the N-LSC suggests that MutY uses it to first engage the DNA in a nonspecific fashion without causing DNA bending or base extrusion. Crystal structure of other HhH-GPD superfamily DNA glycosylases such as AlkA (23, 24) and hOGG1,3 in complex with undamaged DNA also showed a similar mode of interaction, where the HhH motif of the glycosylase interacts with the two phosphate groups that are 3′ to the target base on the lesion-containing strand.
FIGURE 2.

Structures of the N-LSC and the 2′-FLRC. A, structure of the FLRC (PDB code 3G0Q) showing 2′-flouroadenosine (FA) in the active site. The NTD of MutY is colored in cyan, the C-terminal domain is in blue, and the iron-sulfur cluster is in orange (iron) and yellow (sulfur) sticks. The DNA backbone is colored green, and the oxoG and FA are in red (oxoG) and orange (2′-flouroadenosine) spheres. B, structure of the N-LSC (present work) showing the NTD scanning along undamaged DNA. The color scheme is the same as in A except that DNA backbone is colored gold. C, global changes in DNA structure. The MutY NTD in the N-LSC and the FLRC are superimposed. The protein is not displayed, but the DNA that is bound to it is. D, structure of the DNA in the N-LSC. The blue mesh is σA-weighted 2mFo − DFc map contoured at 1σ. E and I, schematic diagrams showing the protein-DNA interactions in the FLRC and the N-LSC, respectively. The phosphate groups that the protein contacts in both structures are colored brown. F and J, views of the target strand in the FLRC and N-LSC. The dotted lines indicate the contacts between MutY and the DNA backbone. G and K, close-up views of the DNA minor groove interactions by MutY in FLRC and N-LSC. The two short loops that intercalate the DNA in the FLRC are shown in magenta. H and L, FLRC and the N-LSC structures rotated 90° from panels G and K views, respectively.
In addition to the phosphate contacts, significant differences exist on the minor groove side of the target base pair. In the FLRC, two short loops (residues 47–49 and 87–89) from the six-helix barrel domain of the NTD penetrate the DNA duplex, extruding the FA nucleoside from the DNA and filling the resulting gap by hydrogen bonding to the oxoG base. Specifically, Tyr-88 inserts 5′ of oxoG to break the stacking between oxoG and the base 5′ to it, and Gln-48 plunges its side chain in between the two DNA strands, making contacts to both p1 of the A strand and the Watson-Crick face of the oxoG base (Fig. 2, E–G). Two residues from the same loop, Thr-49 and Leu-86, also form hydrogen bonds with oxoG (Fig. 2H). In the N-LSC structure; however, the protein backbone of both loops retract from the DNA (Fig. 2, K and L). Also, Gln-48 reorients its side chain by rotating it 180° to face away from the DNA, and instead of stacking with oxoG as in the FLRC, Tyr-88 assumes a different rotamer and points its hydroxyl group toward the guanine that is 5′ to the target T:A base pair. It is also apparent that the MutY footprint on the complementary strand shifts by around 1.5 bases toward the 5′ direction in transitioning from the lesion-scanning mode (N-LSC) to the lesion recognition mode (FLRC) (Fig. 2, C, E and I). This marked difference in complementary strand DNA conformation, as well as the protein retraction from undamaged DNA, has again been observed in the cases of AlkA (12, 13, 23) and hOGG1,3 suggesting that the HhH-GPD superfamily DNA glycosylases employ a common mode of interaction when scanning undamaged DNA. Such loop retraction and relatively loose association with the DNA backbone observed in the N-LSC explain the lower binding affinity of MutY to undamaged DNA and also provide a structural basis for MutY fast scanning (25–27).
Structure of the N-terminal MutY Interacting with an Intrahelical oxoG:A Base Pair
The N-ILRC structure highly resembles the N-LSC structure (heavy atom RMSD = 0.196), with the protein forming limited backbone contacts to the target strand and the DNA being almost canonical B-form. The only notable difference between the two structures is localized in the region of the target base pair. In the N-ILRC, the target oxoG:A exists as a Hoogsteen base pair, in which oxoG adopts a syn-glycosidic conformation, and adenine exists in its normal anti-glycosidic conformation (Fig. 3, A and F). The 2mFo − DFc map of the intrahelical adenine and the lack of positive electron density for an extruded base in the active site indicate that the N-ILRC has captured a state in which MutY makes its initial contact to DNA when the target base pair is fully intrahelical. The fact that there lacks any pronounced structural differences between the N-ILRC and the N-LSC suggests either of the following two possibilities: 1) NTD is able to initiate the base extrusion on its own, and the discrimination between oxoG:A and T:A takes place in a later stage of the extrusion process; :In this case, the CTD is not responsible for detecting the lesion in an early stage but may contribute to the stabilization of the extrahelical state where oxoG exists as an anti-conformer; or 2) the NTD itself is not sufficient for efficient scanning, and the CTD makes some crucial DNA contacts to detect the oxoG:A base pair when it is fully intrahelical. Apparently without the positional information of the CTD, the N-ILRC structure itself does not provide clues as to whether or not CTD participates in lesion scanning. However, comparison of the N-ILRC to the N-LSC or the FLRC seems to suggest a possible mechanism by which the CTD could play a role in intrahelical lesion recognition.
FIGURE 3.

Structures of the N-ILRC and the FLRC. A, the intrahelical oxoG:A base pair in the N-ILRC. The blue mesh is σA-weighted 2mFo − DFc map contoured at 1σ. The hydrogen bonding interactions are denoted by dotted lines. B, the lesion base pair in the FLRC where FA is extruded into the active pocket. C, structure of the intrahelical oxoG:A base pair and the T:A base pair. The N-ILRC is overlaid to the N-LSC, and only the target base pairs are shown. D, structure of the FLRC overlaid to the N-ILRC. The NTD of the two structures are superimposed. Only the protein portion of FLRC and the DNA of the N-ILRC are shown. E, overlay of the wild-type MutY active site pocket to MutY with the V51Y mutation. F, overlay of the FLRC DNA to the N-ILRC DNA. The N-ILRC DNA is shown in gray, oxoG is in red, and A is in orange. Color coding scheme is the same as Fig. 2 except as specifically noted.
The oxoG:A base pair shares marked structural similarity to the T:A base pair. As shown in Fig. 3C, the two base pairs look almost identical on the minor groove side, because the 8-carbonyl of oxoG overlays almost perfectly with the 2-carbonyl of thymine. There is, however, a pronounced difference between the two base pairs on the major groove side. The entire Watson-Crick face of oxoG, which contains three hydrogen bond donor/acceptors, faces the major groove, whereas the thymine protrudes its 5-methyl and 4-carbonyl. The difference in major groove hydrogen bonding capacities between oxoG:A and T:A base pairs led us to suspect that MutY might take it as one of the first clues that hints at the existence of oxoG:A. In fact, when we superimpose the FLRC on the N-ILRC, we found that a short loop in the CTD (residues 306–310) that is responsible for oxoG (anti-) recognition in the FLRC is positioned in a way that allows it to interact with oxoG:A from the major groove side (Fig. 3D). For example, His-309 could be a potential candidate for oxoG (syn-) recognition either by hydrogen bonding to the 2′ amine group of the oxoG base or simply by acting as a “stopper” residue that walks along the major groove and gets stalled at anything that is more bulky than a normal base.
Role of CTD in Lesion Scanning
Studies have shown that NTD alone is sufficient for catalysis (although at a slower rate than the full-length MutY) (13, 15, 28–30), but whether the NTD alone is sufficient for lesion scanning is still in question. Because of the lack of a crystal structure of full-length MutY bound to undamaged DNA, we turned to size exclusion chromatography (SEC)-SAXS to generate a conformational model of the LSC. Previous structures of hOGG1 in complex with undamaged DNA (23, 24)3 have shown that cross-linking the protein to the DNA at particular positions facilitates the extrusion of an undamaged base. To ensure that the target A in the T:A base pair remains fully intrahelical during the course of sample preparation and data collection, we turned to in vitro cleavage assays on disulfide cross-linked MutY complexes having either an oxoG or a T that base pairs with the A at the target site (Fig. 4). The reactions were initiated by adding the same cross-linker containing double-stranded DNA as used in the N-LSC or the N-ILRC to the full-length catalytically active MutY (bearing a P164C mutation). The mixture was incubated at room temperature. Free adenine, which inhibits MutY enzymatic activity by binding to its active site pocket and precluding the binding of any naturally occurring base from the DNA (12, 13, 19), was added to further ensure the blockage of base extrusion from the undamaged DNA. After 40 h of incubation, oxoG:A underwent nearly complete cleavage, whereas T:A remained intact, suggesting that T:A was strongly discriminated against by MutY, further implying that the target A in the T:A base pair remained intrahelical during the course of the experiment. To collect the SAXS data, the samples were passed through an SEC column, and the eluate flow was directed to the SAXS flow cell. By selecting exposures that correspond to the desired monomeric protein-DNA complex, we ensured that the results were free of concentration-dependent effects, which is often a problem in conventional SAXS experiments.
FIGURE 4.
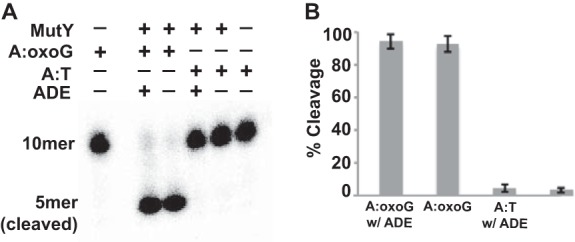
Glycosylase assay of the cross-linked complexes. To test the glycosylase activity of the cross-linked complex used in SAXS, we labeled the target strand (A strand), which contains the disulfide cross-linker with 32P and annealed it to the complimentary strand containing either an oxoG or a T that pairs with the A. The annealed DNA was added to P164C MutY with or without the presence of adenine. The reaction proceeded for 40 h at room temperature before it was quenched by NaOH (see “Experimental Procedures” for detailed procedures). A, a representative urea gel showing cleavage of A opposite oxoG by MutY and lack of cleavage of A opposite T under the same conditions. B, fraction of DNA cleaved in the reactions in A shown in bar diagram. Each data point shows an average of three parallel experiments, and the error bars show the standard deviation from the average.
The SAXS data for the LRC was first measured as a well established control complex for comparison between crystallographic structures and SAXS data. The calculated scattered intensity curve (the I(q) curve; see red line in Fig. 5A) from the LRC crystal structure (PDB code 1RRQ) was in good agreement with the experimental data, with a χ2 of 1.6 (see “Experimental Procedures” for detailed protocols). Furthermore, fitting of the crystal structure into the SAXS envelope reveals a satisfactory fit, with the NTD, the DNA, and the CTD all well accommodated within the envelope. (Fig. 5D). We next made a chimeric complex by superimposing the N-LSC to the LRC, then taking the CTD portion of the LRC, and merging it to the N-LSC. As shown in Fig. 5B, the predicted SAXS curve for this chimera displays a poor fit to the measured curve, with the χ2 of the fit being 3.8. Superposition of the chimera to the SAXS envelope of the LSC reveals that part of the CTD protrudes out of the envelope (Fig. 5E). Also, a short loop in the CTD that is responsible for oxoG recognition in the LRC clashes with the DNA in the chimera (see the inset of Fig. 5E), further suggesting that the CTD must have undergone domain movement from its position in the LRC. We then performed rigid body docking of the CTD to achieve a better fit to the envelope and arrived at a model LSC structure. At the given data resolution, it is hard to resolve the orientation of the CTD, and it is possible that the CTD exists as a mixture of multiple rotation-related conformers. However, the fact that the model LSC is better accommodated in the envelope than the chimera and that the χ2 is improved from 3.8 to 3.2 suggests that the CTD is unlikely to interact with the undamaged DNA as closely as it does with damage-containing DNA. The fact that CTD still remains in close proximity to the DNA as suggested by the LSC SAXS envelope implies that it is probably forming some DNA interactions that contribute to the scanning process. In agreement with this hypothesis is the observation that the experimentally determined Rg and Dmax values of the LSC are both slightly bigger that those of the LRC (Table 3 and Fig. 5G), providing further evidence for CTD movement.
FIGURE 5.

SAXS curves and low resolution envelopes of MutY-DNA complexes. A, experimental SAXS data (I(q) verses q) of the LRC shown as red dots. The theoretical scattering curve of the LRC (PDB code 1RRQ) is shown in red line. The inset shows the Guinier plot for LRC. B, experimental SAXS data of the LSC shown as black dots. The black line is the theoretical scattering curve (generated by CRYSOL; see “Experimental Procedures” for method details) of the chimera shown in E. The inset shows the Guinier plot for LSC. C, experimental SAXS data of MutY shown as blue dots. The inset shows the Guinier plot. D, superposition of the LRC crystal structure to the ab initio envelope (see “Experimental Procedures” for detailed protocols in SAXS data analysis). E, superposition of the chimera structure to the ab initio envelope of the LSC. The chimera was made by superimposing the LRC sthat of the N-LSC and fusing the CTD of LRC to the N-LSC. The inset shows the steric clash of the CTD with the DNA in N-LSC. F, superposition of the model LSC to the ab initio envelope of LSC. G, the p(r) function of the SAXS data of the two complexes.
TABLE 3.
SAXS-derived parameters for MutY-DNA complexes
|
Rg |
Dmax |
||||
|---|---|---|---|---|---|
| Guinier | p(r) | EOM | p(r) | EOM | |
| Å | Å | ||||
| LRC | 25.00 ± 2.06 | 25.6 | 24.8 | 85.0 | 82.5 |
| LSC | 27.29 ± 1.63 | 27.8 | 27.3 | 92.0 | 91.0 |
| MutYa | 28.24 ± 0.83 | ||||
a Because in the MutY sample, multiple conformations with different Rg and Dmax values exist (see Fig. 6, C and F), the Rg and Dmax values derived from the p(r) curve and the EOM analysis are not shown in this table.
Having established the position of CTD in the LSC, we wanted to explore its potential mobility in either the DNA-bound or the apo form by applying the ensemble optimization method (EOM) (25, 31). The NTD and CTD of MutY were treated as individual rigid bodies connected by a flexible linker. In the cases of the LRC and the LSC, the DNA is attached to the NTD and together treated as one rigid body. A pool of 10000 models was generated containing a random sampling of domain positions to cover the conformational space without creating steric clashes. A genetic algorithm was then applied to select an ensemble of models whose combined theoretical SAXS curve best fit the experimental data. The distribution of Rg and Dmax values are viewed as indicators of the conformational heterogeneity of the samples. Comparing the Rg and Dmax distribution of the LRC, we found that the selected ensemble shows a much narrower distribution than the random pool, with a peak Rg of 24.8 Å and a peak Dmax of 82.5 Å (Fig. 6A and D), which are in good agreement with the numbers determined from the experimental SAXS curve (Table 3). Similarly, the LSC displayed a narrow Rg and Dmax distribution, with the respective peak values being 27.3 and 91.0 Å (Fig. 6, B and E), which also agree well with those calculated from the scattering curve, assuming that the complex has one predominant conformation. We noticed that there exists a small portion of particles that are larger than the major species in both the LRC and the LSC, with Dmax values of 94.4 and 98.0 Å, respectively (Fig. 6, D and E). We believe that this shows that in both complexes, there is a small portion of molecules that exist in a more extended conformation that should not substantially affect the results of the envelope construction, because the scattering curve is mainly contributed by one dominating species. In contrast to the DNA-protein complexes, the apo protein displays a clear multimodel distribution. The Rg distribution has a major peak at 25.8 Å with a shoulder at 25.0 Å and a minor peak at 28.9 Å (Fig. 6C). Correspondingly, the Dmax values show a major peak at 85.2 Å, with a split at 82.0 Å and a minor peak at 89.8 Å (Fig. 6F). We hypothesize that the multiple peaks and shoulders observed here represent MutY in an equilibrium of multiple states in solution. However, upon binding to DNA, both the NTD and the CTD interact with DNA, and the whole complex adopts a relatively compact structure.
FIGURE 6.

EOM analysis of MutY and MutY-DNA complexes. A–C, EOM generated frequency distribution of the conformers as a function of the Rg of LRC, LSC, and MutY respectively. D–F, EOM generated frequency distribution of the conformers as a function of the Dmax of LRC, LSC, and MutY, respectively. The selected pool distribution is shown in red lines, and the random pool is in black.
Taken together, the SAXS analysis strongly suggest that both the NTD and CTD of MutY contact DNA during lesion scanning and hence that both domains contribute to the early stages of recognition of oxoG:A lesions, whereas they are intact and intrahelical. Nevertheless, both domains and their associated DNA contacts undergo substantial reorganization during the transition from an intrahelical oxoG:A lesion encounter complex to an extrahelical lesion recognition and repair complex.
Discussion
In this work, we have reported high resolution crystal structures of MutY-NTD in complex with DNA containing either an intrahelical T:A base pair (N-LSC) or an intrahelical oxoG:A base pair (N-ILRC). In both structures, MutY interacts with the DNA in a completely noninvasive manner, with only limited contacts localized on the 3′ side of the target base and with the two short loops responsible for base extrusion being fully retracted from the DNA. These observations led us to propose that the N-LSC and the N-ILRC have captured MutY interacting with DNA at the earliest stage of the base extrusion pathway, an earlier stage than previously observed experimentally for any DNA glycosylase. Previous structural studies of the HhH family DNA glycosylases have captured these proteins in complex with DNA at a late stage of the base extrusion pathway, where these proteins, despite their different substrate specificity, share a common base extrusion mechanism that involves the protein invading the DNA from the minor groove side, bending the DNA by interacting with phosphate groups on both sides of the lesion, and extruding the damaged base via insertion of a short loop (13, 28–30). Less is known, however, about how these proteins interact with DNA in the early stages of base extrusion. The two structures reported in this work show that MutY uses the signature HhH DNA-binding motif common to all HhH superfamily proteins to initiate DNA interactions, indicating that this could be a mechanism employed by all proteins of this superfamily. Such nonspecific DNA contacts by the HhH motif has also been observed in the AlkA undamaged DNA structure (23, 33), further suggesting the existence of a common lesion-scanning mechanism for the HhH DNA glycosylases. It is therefore reasonable to assume that the structurally related HhH proteins share a similar mode of interaction all along the base extrusion pathway.
We have also determined the position of the CTD in the LSC using SAXS, which has indicated that it is highly likely that the CTD also plays a role in lesion scanning. Unlike most other DNA glycosylases that recognize the damaged base, MutY recognizes the oxoG:A base pair, which requires extensive contacts with both strands of DNA. Therefore, it makes sense that the CTD, which is responsible for oxoG recognition, also participates in initial lesion detection.
Fig. 7 shows a step by step model of MutY lesion scanning and recognition based on this work and structural studies published previously by our lab (13, 18). MutY performs fast scanning on undamaged DNA by forming contacts to the phosphate backbone, mainly using the aforementioned HhH motif in the NTD. The CTD stays in proximity to DNA during scanning, probably interacting with the complimentary strand. As described previously, the short loop in CTD (residues 307–310) is a candidate for initial detection of syn-oxoG. Upon recognition of the intrahelical oxoG:A base pair, MutY initiates the extrusion of A and the flipping of oxoG (from the syn-conformation to the anti-conformation). In the extrahelical lesion recognition stage, the NTD makes extensive contacts to the target strand DNA backbone, and the CTD contacts the complimentary strand, both locking the DNA in the extrahelical conformation. Also, amino acid residues from several short loops in either the NTD or the CTD hydrogen bond with the anti-oxoG, filling the gap left by A upon its extrusion, and a few amino acid residues in the active pocket form specific interactions with A, positioning it correctly for catalysis (see Fig. 2 for details).
FIGURE 7.
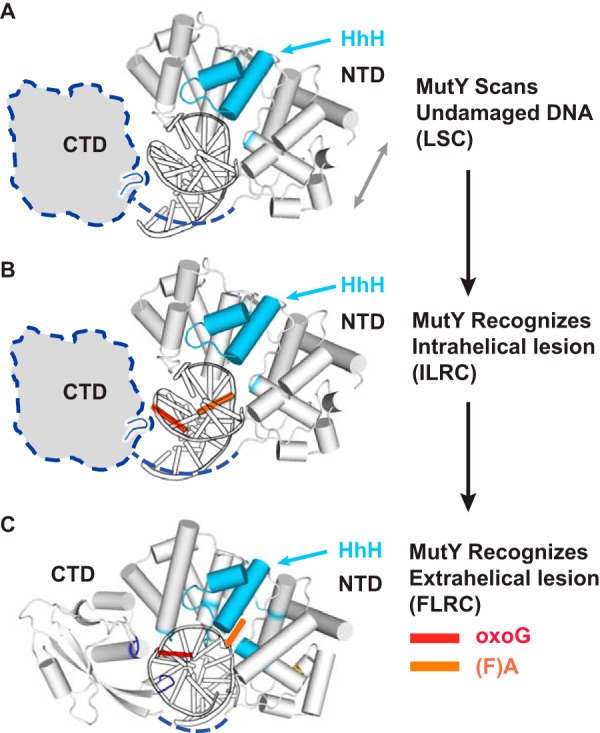
Model for MutY lesion scanning and recognition. A, model for MutY lesion scanning. B, model for intrahelical lesion recognition. The NTD and the DNA are shown in cartoon representation, and the CTD is shown as a shape encircled by dotted lines, with the short loop (residues 307–309) shown in its hypothetical position. C, MutY interacts with the extrahelical lesion (FLRC). Protein is shown in gray, with the residues and motifs interacting with DNA colored cyan (NTD) or blue (CTD). oxoG is colored in red, and (F)A is in orange.
Experimental Procedures
B. stearothermophilus MutY Preparation
To prepare the NTD MutY, point mutations (P164C, D144N, and K230Stop) were introduced into the B. stearothermophilus MutY gene by QuikChange mutagenesis using a parent construct wherein the gene was cloned into the pET28 (Novagen) expression vector, resulting in an ORF with an N-terminal His tag and thrombin cleavage site. Both the NTD and full-length MutY were overexpressed and purified using similar protocols as described previously (19).
Synthesis of Cross-linking DNA
DNA oligomers 5′-AGCACAGGAT-3′ and 5′-ATCC(oxoG)GTGCT-3′ (where italicized A indicates modified A with a disulfide tether for cross-linking; O6-Ph-dI and oxoG phosphoramidite were purchased from Glen Research) was synthesized on an ABI 392 DNA synthesizer using standard reagents. The tether-containing DNA was prepared using reported procedures (19).
Glycosylase Assay of the Cross-linked Complex
To test the cleavage activity of the cross-linked complex, we labeled the 5′ end of the target strand (the A strand, which has a disulfide cross-linker) with 32P. The resulting oligonucleotide was annealed to the complimentary strand (containing either an oxoG or a T base at the target site) and diluted 100 nm with buffer containing 10 mm Tris, pH 8. 1 μl of 400 nm full-length MutY (bearing P164C mutation for cross-linking) and 1 μl of the 100 nm duplex DNA solution were added to 8 μl of reaction buffer containing 50 mm Na2HPO4, pH 6.6, 100 mm NaCl, resulting in a 10-μl reaction. For groups that contain adenine as an inhibitor, 1 mm adenine was added to the reaction buffer prior to the addition of protein and DNA. The reaction mixture was incubated at room temperature for 40 h. 20 mm DTT was added to each reaction to reduce the disulfide cross-link, before 1.2 μl of 2 m NaOH was added to quench the reaction. The mixture was heated up to 90 °C for 15 min to facilitate strand scission.
Protein-DNA Cross-linking Reaction and SAXS Sample Preparation
The MutY-DNA complex was formed using reported procedures (19). The complex was purified by MonoQ ion exchange chromatography and dialyzed against buffer containing 10 mm Tris, pH 7.4, 90 mm NaCl, 10 μm β-ME at 4 °C overnight. (It has been shown in previous studies that 10 μm β-ME does not disrupt the disulfide cross-link (19).) The LSC sample also contained 1 mm adenine in the cross-linking reaction and all FPLC buffers. The purified complex was concentrated to ∼300 μm and flash frozen into liquid nitrogen for data collection.
SAXS Data Collection and Analysis
Prior to SAXS analysis, the MutY protein was purified by SEC using a Superdex 75 column (GE Healthcare), and the MutY-DNA complexes were purified using a MonoQ ion exchange column. SAXS data were collected at the 18ID Beamline at Argonne National Laboratory. The X-ray wavelength was 1.0 Å, and the data q range was 0.0037–0.3937 Å−1 (q = 4π sin θ/λ, 2θ is the scattering angle, and λ is the X-ray wavelength). Samples were first injected into a pre-equilibrated Superdex 200 increase column (GE Healthcare) before being subjected to X-ray. As samples eluted from the column, 1-s exposures were recorded every 2 s. All experiments were performed at room temperature. Positions of the sample peaks were determined by examining the scattered intensity. A few sample frames corresponding to the elution peak of the chromatogram were averaged to maximize the signal to noise ratio. Several frames immediately proximal to the sample peak were chosen as buffer frames, which were averaged and subtracted from the sample scattering to obtain the final SAXS curve. The data were reduced to I(q) versus q plots using a beamline-specific pipeline and further processed using the ATSAS program suite (31, 35). The radius of gyration was obtained from Guinier approximation (36), and GNOM (25, 33) was used to determine the pair distance distribution function, p(r), and the maximum macromolecule dimension Dmax. Theoretical SAXS curve of the crystal structures were calculated with CRYSOL (34, 37), and ab initio modeling was performed with DAMMIF (35, 38). The envelopes presented were generated by converting the ab initio models to an electron density map using Situs 2.7.2 (36, 39). EOM (25, 40) was used to perform the conformational heterogeneity analysis as described under “Results.”
Crystallization, Data Collection, and Structure Refinement
The purified MutY-DNA complex was concentrated to 225 μm and crystallized by the hanging drop vapor diffusion method at room temperature. The N-LSC was crystallized in 100 mm Tris, pH 8.5, 200 mm Ca(OAc)2, 18% (w/v) PEG 4000, and the N-ILRC was crystallized in 300 mm Ca(OAc)2, 14% (w/v) PEG 4000. Crystals appeared after several days and were allowed to grow for a couple of weeks. The crystals were transferred to cryoprotectant solution containing the original reservoir solution plus either 22% (N-LSC crystals) or 26% (N-ILRC crystals) glycerol and frozen into liquid nitrogen for data collection. Data were collected on Beamline 24-ID at NE-CAT at the Advanced Photon Source at the Argonne National Laboratory and processed using XDS (37, 41). Crystal of the N-LSC belongs to the space group P21, with unit cell dimensions of a = 49.2 Å, b = 38.6 Å, and c = 83.8 Å. The crystal of the N-ILRC also belongs to the space group P21, with unit cell dimensions of a = 49.9 Å, b = 38.8 Å, and c = 83.0 Å. The data collection statistics are summarized in Table 2. The coordinates of the N-terminal domain of the protein from the structure of P164C/D144N B. stearothermophilus MutY bound to oxoG:A-containing DNA (PDB code 1RRQ) were used as the initial search model. Election density for double stranded DNA was clearly evident in the σA-weighted mFo – DFc map right after molecular replacement. The model was improved by rigid body fit and subsequent simulated annealing, TLS and individual B-factor refinement using Phenix (38, 42). In both the N-LSC and the N-ILRC structures, the electron density of the target base pair was clearly evident in their respective positions in the σA-weighted mFo − DFc map after the adjacent nucleotides were put into the model. Model building was performed in Coot (39, 43) while constantly monitoring Rfree (40). Water molecules were added to the model using both automated methods in Phenix and manual inspection of difference maps. Refinement and model building statistics were shown in Table 2. The figures were prepared using PyMOL version 1.7 (41).
Author Contributions
L. W. and G. L. V. conceived the idea for the project. L. W. conducted most of the experiments and analyzed most of the results. S. C. and L. W. collected and analyzed SAXS data. L. W. and G. L. V. wrote the paper.
Acknowledgments
We thank the entire staff of Beamlines 24-ID of the Advance Photon Source at Argonne National Laboratory for expert assistance with X-ray data collection and processing. We thank Dr. Seung-Joo Lee, Dr. Danaya Pakotiprapha, and Dr. Gerard Hilinski for valuable feedback on the manuscript, as well as Alan Yang for assistance in protein purification.
Note Added in Proof
There was some overlap in text between the version of this article that was published as a Paper in Press on January 27, 2017 and Wang, L., Lee, S.-J., and Verdine, G. L. (2015) Structural basis for avoidance of promutagenic DNA repair by MutY adenine DNA glycosylase. J. Biol. Chem. 290, 17096–17105. The text overlap has now been removed.
This work was supported by National Institutes of Health Grants CA100742, P41 GM103622 (to BioCAT), and 1S10OD018090-01. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5KN8 and 5KN9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
U. Shigdel and G. L. Verdine, unpublished data.
- oxoG
- 8-oxoguanine
- ASC
- anti-substrate complex
- LRC
- lesion recognition complex
- FLRC
- fluorinated lesion recognition complex
- LSC
- lesion scanning complex
- N-LSC
- N-terminal LSC
- N-ILRC
- N-terminal intrahelical lesion recognition complex
- RMSD
- root mean square deviation
- SAXS
- small angle X-ray scattering
- PDB
- Protein Data Bank
- EOM
- ensemble optimization method
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- SEC
- size exclusion chromatography
- β-ME
- β-ME.
References
- 1. Michaels M. L., Cruz C., Grollman A. P., and Miller J. H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. U.S.A. 89, 7022–7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 [DOI] [PubMed] [Google Scholar]
- 3. Michaels M. L., and Miller J. H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol. 174, 6321–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al-Tassan N., Chmiel N. H., Maynard J., Fleming N., Livingston A. L., Williams G. T., Hodges A. K., Davies D. R., David S. S., Sampson J. R., and Cheadle J. P. (2002) Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232 [DOI] [PubMed] [Google Scholar]
- 5. Shibutani S., Takeshita M., and Grollman A. P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 349, 431–434 [DOI] [PubMed] [Google Scholar]
- 6. David S. S., O'Shea V. L., and Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lipton L., Halford S. E., Johnson V., Novelli M. R., Jones A., Cummings C., Barclay E., Sieber O., Sadat A., Bisgaard M.-L., Hodgson S. V., Aaltonen L. A., Thomas H. J., and Tomlinson I. P. (2003) Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 63, 7595–7599 [PubMed] [Google Scholar]
- 8. Gad H., Koolmeister T., Jemth A.-S., Eshtad S., Jacques S. A., Ström C. E., Svensson L. M., Schultz N., Lundbäck T., Einarsdottir B. O., Saleh A., Göktürk C., Baranczewski P., Svensson R., Berntsson R. P., et al. (2014) MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 508, 215–221 [DOI] [PubMed] [Google Scholar]
- 9. Huber K. V., Salah E., Radic B., Gridling M., Elkins J. M., Stukalov A., Jemth A.-S., Göktürk C., Sanjiv K., Strömberg K., Pham T., Berglund U. W., Colinge J., Bennett K. L., Loizou J. I., et al. (2014) Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 508, 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thayer M. M., Ahern H., Xing D., Cunningham R. P., and Tainer J. A. (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 14, 4108–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nash H. M., Bruner S. D., Schärer O. D., Kawate T., Addona T. A., Spooner E., Lane W. S., and Verdine G. L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol. 6, 968–980 [DOI] [PubMed] [Google Scholar]
- 12. Guan Y., Manuel R. C., Arvai A. S., Parikh S. S., Mol C. D., Miller J. H., Lloyd S., and Tainer J. A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat. Struct. Biol. 5, 1058–1064 [DOI] [PubMed] [Google Scholar]
- 13. Fromme J. C., Banerjee A., Huang S. J., and Verdine G. L. (2004) Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 427, 652–656 [DOI] [PubMed] [Google Scholar]
- 14. Manuel R. C., and Lloyd R. S. (1997) Cloning, overexpression, and biochemical characterization of the catalytic domain of MutY. Biochemistry 36, 11140–11152 [DOI] [PubMed] [Google Scholar]
- 15. Li X., Wright P. M., and Lu A. L. (2000) The C-terminal domain of MutY glycosylase determines the 7,8-dihydro-8-oxo-guanine specificity and is crucial for mutation avoidance. J. Biol. Chem. 275, 8448–8455 [DOI] [PubMed] [Google Scholar]
- 16. Noll D. M., Gogos A., Granek J. A., and Clarke N. D. (1999) The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity for 8-oxoguanine·adenine mispairs and may have evolved from MutT, an 8-oxo-dGTPase. Biochemistry 38, 6374–6379 [DOI] [PubMed] [Google Scholar]
- 17. Volk D. E., House P. G., Thiviyanathan V., Luxon B. A., Zhang S., Lloyd R. S., and Gorenstein D. G. (2000) Structural similarities between MutT and the C-terminal domain of MutY. Biochemistry 39, 7331–7336 [DOI] [PubMed] [Google Scholar]
- 18. Lee S., and Verdine G. L. (2009) Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc. Natl. Acad. Sci. 106, 18497–18502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang L., Lee S.-J., and Verdine G. L. (2015) Structural basis for avoidance of promutagenic DNA repair by MutY adenine DNA glycosylase. J. Biol. Chem. 290, 17096–17105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu A. L., Tsai-Wu J. J., and Cillo J. (1995) DNA determinants and substrate specificities of Escherichia coli MutY. J. Biol. Chem. 270, 23582–23588 [DOI] [PubMed] [Google Scholar]
- 21. Porello S. L., Leyes A. E., and David S. S. (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry 37, 14756–14764 [DOI] [PubMed] [Google Scholar]
- 22. Verdine G. L., and Norman D. P. (2003) Covalent trapping of protein-DNA complexes. Annu. Rev. Biochem. 72, 337–366 [DOI] [PubMed] [Google Scholar]
- 23. Bowman B. R., Lee S., Wang S., and Verdine G. L. (2010) Structure of Escherichia coli AlkA in complex with undamaged DNA. J. Biol. Chem. 285, 35783–35791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Banerjee A., Yang W., Karplus M., and Verdine G. L. (2005) Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 434, 612–618 [DOI] [PubMed] [Google Scholar]
- 25. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., and Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 26. Blainey P. C., van Oijen A. M., Banerjee A., Verdine G. L., and Xie X. S. (2006) A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 103, 5752–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blainey P. C., Luo G., Kou S. C., Mangel W. F., Verdine G. L., Bagchi B., and Xie X. S. (2009) Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 16, 1224–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bruner S. D., Norman D. P., and Verdine G. L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403, 859–866 [DOI] [PubMed] [Google Scholar]
- 29. Fromme J. C., and Verdine G. L. (2003) Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J. 22, 3461–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hollis T., Ichikawa Y., and Ellenberger T. (2000) DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 19, 758–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deleted in proof
- 33. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 34. Svergun D., Barberato C., and Koch M. (1995) CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 35. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wriggers W. (2010) Using Situs for the integration of multi-resolution structures. Biophys Rev. 2, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 41. Delano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 42. Brünger A. T. (1993) Assessment of phase accuracy by cross validation: the free R value: methods and applications. Acta Crystallogr. D Biol. Crystallogr. 49, 24–36 [DOI] [PubMed] [Google Scholar]
- 43. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]