

Abstract
Overexpression of the pro-angiogenic chemokine IL-8 (CXCL8) is associated with a poor prognosis in several solid tumors, including epithelial ovarian cancer (EOC). Even though histone deacetylase (HDAC) inhibition has shown remarkable antitumor activity in hematological malignancies, it has been less effective in solid tumors, including EOC. Here we report results that may explain the decreased efficiency of HDAC inhibition in EOC, based on our data demonstrating that HDAC inhibition specifically induces expression of IL-8/CXCL8 in SKOV3, CAOV3, and OVCAR3 cells. Suppression or neutralization of vorinostat-induced IL-8/CXCL8 potentiates the vorinostat inhibitory effect on cell viability and proliferation. The IL-8/CXCL8 expression induced by vorinostat in EOC cells is dependent on IκB kinase (IKK) activity and associated with a gene-specific recruitment of IKKβ and IKK-dependent recruitment of p65 NFκB to the IL-8/CXCL8 promoter. In addition, HDAC inhibition induces acetylation of p65 and histone H3 and their IL-8/CXCL8 promoter occupancy. In vivo results demonstrate that combining vorinostat and the IKK inhibitor Bay 117085 significantly reduces tumor growth in nude mice compared with control untreated mice or either drug alone. Mice in the combination group had the lowest IL-8/CXCL8 tumor levels and the lowest tumor expression of the murine neutrophil [7/4] antigen, indicating reduced neutrophil infiltration. Together, our results demonstrate that HDAC inhibition specifically induces IL-8/CXCL8 expression in EOC cells and that the mechanism involves IKK, suggesting that using IKK inhibitors may increase the effectiveness of HDAC inhibitors when treating ovarian cancer and other solid tumors characterized by increased IL-8/CXCL8 expression.
Keywords: chemokine, histone deacetylase (HDAC), histone deacetylase inhibitor (HDI), interleukin, ovarian cancer, CXCL8, IκB kinase, interleukin-8
Introduction
The proinflammatory and proangiogenic chemokine IL-8 (CXCL-8) contributes to the progression of epithelial ovarian cancer (EOC)2 through its induction of tumor cell survival, proliferation, angiogenesis, and metastasis (1, 2). At the transcriptional level, IL-8/CXCL8 expression is regulated by the transcription factor NFκB, which is constitutively activated in ovarian cancer (OC) cells and conveys a poor outcome (3–5). Activation of NFκB is mediated by the enzymes of the IκB kinase (IKK) complex, which, in addition to phosphorylating IκBα, leading to its proteasomal degradation, can phosphorylate NFκB subunits and other transcription factors and cofactors (6, 7). Although the proteasomal degradation of IκBα, resulting in the nuclear accumulation of NFκB subunits, represents a general step in NFκB activation, the specificity and duration of NFκB-regulated responses are mediated by the protein composition of NFκB complexes and their posttranslational modifications (8–11). Recent studies from our laboratory have demonstrated that IKK is specifically recruited to the NFκB-binding site in the IL-8/CXCL8 promoter, resulting in increased IL-8/CXCL8 transcription in EOC cells (12–14).
Histone deacetylases (HDACs), a family of enzymes that, by removing acetyl groups from histone and non-histone proteins, play an important role in the epigenetic regulation of gene expression and are frequently dysregulated in cancer cells (15). Expression of class I HDACs is increased in EOC cells and has been associated with a poor prognosis and development of chemoresistance (16–18). Inhibition of HDACs with small molecules has led to the development of HDAC inhibitors (HDIs), which have been used in the treatment of hematological malignancies because they exhibit excellent differential action on normal and cancer cells at therapeutic dosages (19, 20). Vorinostat (suberoylanilide hydroxamic acid) and romidepsin (depsipeptide, FK228) are Food and Drug Administration-approved HDIs that have been very effective in treating cutaneous T cell lymphoma and other hematological malignancies (21–23). In contrast, clinical trials with HDIs as single agents in the treatment of solid tumors, including EOC, have produced poor results, but the reasons are largely unknown.
Here we show that HDAC inhibition specifically induces IL-8/CXCL8 expression in ovarian cancer cells, resulting in increased cell viability and proliferation and decreased apoptosis. Our in vitro results indicate that the mechanism involves increased nuclear accumulation of p65 NFκB and its gene-specific, IKK-dependent recruitment to the IL-8/CXCL8 promoter, where it is acetylated. Our in vivo results demonstrate that combination of vorinostat and the IKK inhibitor Bay 117085 significantly reduces tumor growth in ovarian cancer xenografts compared with either drug alone. Together, our results suggest that using IKK inhibitors may increase the effectiveness of HDAC inhibitors in ovarian cancer.
Results
HDAC Inhibition Induces IL-8/CXCL8 Expression in Ovarian Cancer Cells
To determine whether HDAC inhibition regulates expression of NFκB-dependent proinflammatory genes in ovarian cancer cells, we first analyzed IL-8/CXCL8, TNFα, and IL-6 expression in SKOV3 cells incubated for 48 h with vorinostat. Interestingly, although vorinostat significantly increased IL-8 mRNA levels (Fig. 1A) and cytokine release (Fig. 1B), it did not have a substantial effect on TNFα or IL-6 mRNA and cytokine release levels. The increased IL-8 mRNA expression (Fig. 1C) and cytokine release (Fig. 1D) in SKOV3 cells incubated with 1.5 μm vorinostat, which approximately corresponds to the clinically used concentrations (24), were time-dependent. The IL-8 expression in SKOV3 cells was also significantly induced by romidepsin; 10 nm romidepsin, which approximately corresponds to the clinically used concentrations, increased IL-8 mRNA expression ∼30-fold (Fig. 1E). Fig. 1F illustrates a dose response of IL-8 expression in SKOV3 cells on vorinostat, romidepsin, and apicidin, another class I HDAC inhibitor, on an equimolar basis.
FIGURE 1.
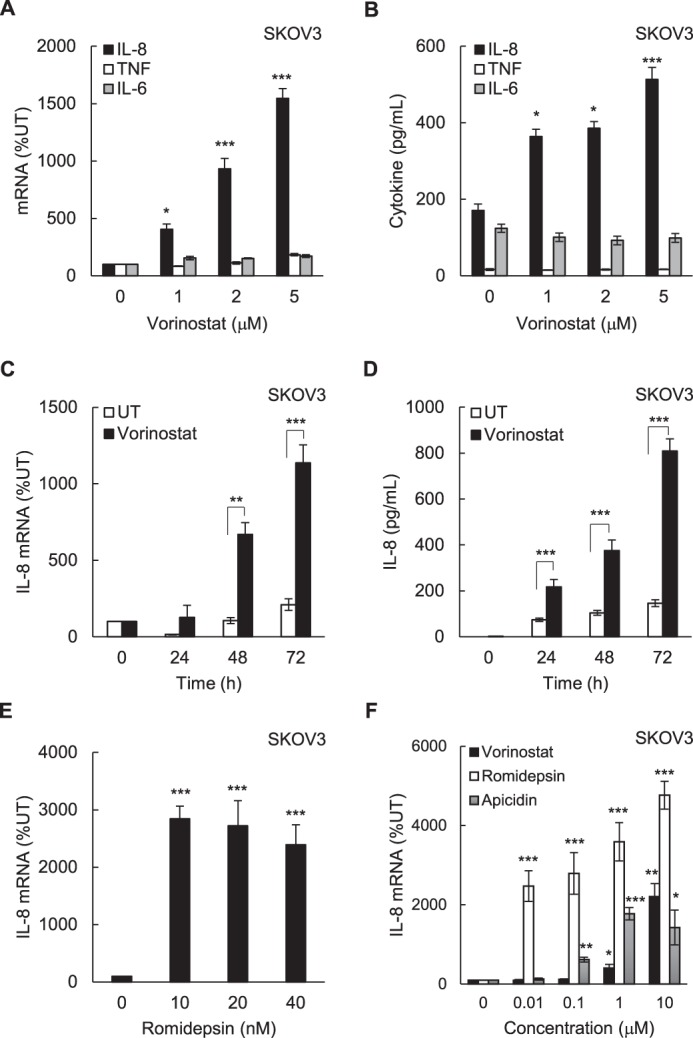
HDAC inhibition induces IL-8/CXCL8 expression in ovarian cancer SKOV3 cells. A and B, real-time RT-PCR analysis of mRNA levels (A) and ELISA of cytokine release of IL-8, TNFα, and IL-6 (B) in SKOV3 cells incubated with vorinostat for 48 h. C and D, real-time RT-PCR analysis (C) of IL-8 mRNA levels and ELISA of IL-8 cytokine release (D) in SKOV3 cells incubated with 1.5 μm vorinostat or control DMSO for up to 72 h. E, RT-PCR analysis of IL-8 mRNA in SKOV3 cells incubated with romidepsin for 48 h. F, RT-PCR of IL-8 mRNA in SKOV3 cells incubated for 48 h with equal concentrations of vorinostat, romidepsin, or apicidin. The values represent the mean ± S.E. of four experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with control UT cells.
To investigate whether HDAC inhibition increases the expression of other NFκB-dependent genes in ovarian cancer cells, we analyzed the mRNA levels of the NFκB-dependent chemokine CXCL5 (epithelial-derived neutrophil-activating peptide 78, ENA-78), which has the same NFκB binding promoter sequence as IL-8/CXCL8 (12); TGFβ1, which is also regulated by NFκB (the NFκB binding site is located +94 from the transcription start site (25); the NFκB-dependent anti-apoptotic genes cIAP-1 and Bcl2; the NFκB family proteins p65, p50, and IκBα; and the IL-8/CXCL8 receptors CXCR1 (IL-8RA) and CXCR2 (IL-8RB), which are expressed in ovarian cancer cells and are also regulated by NFκB (26–29). Interestingly, IL-8 was the only gene that was significantly induced in SKOV3 cells incubated for 48 h with 1.5 μm vorinostat compared with cells incubated for 48 h with control DMSO (Fig. 2A). IL-8 was also the only NFκB-dependent gene that was significantly induced by 1.5 μm vorinostat (48 h) in ovarian cancer CAOV3 (Fig. 2, B and C) and OVCAR3 cells (Fig. 2, D and E).
FIGURE 2.
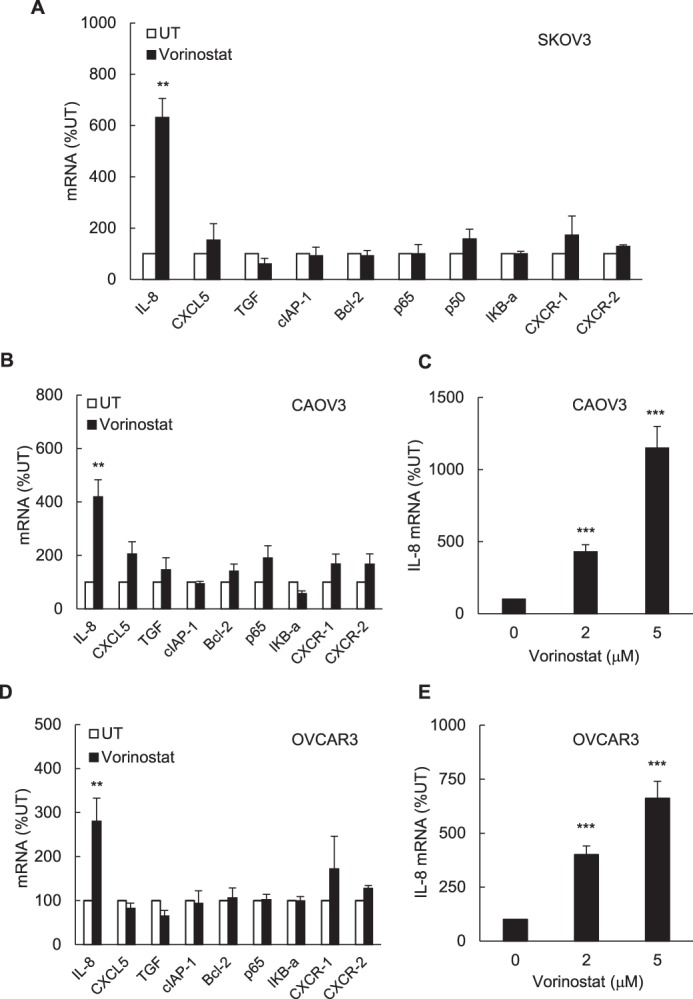
Regulation of NFκB-dependent genes by vorinostat in ovarian cancer cells. A and B, real-time RT-PCR analysis of mRNA levels of the NFκB-regulated genes IL-8/CXCL8, CXCL5, TGFβ1, cIAP-1, Bcl-2, p65, p50, IκBα, CXCR1, and CXCR2 in SKOV3 (A) and CAOV3 (B) cells incubated with 1.5 μm vorinostat or control DMSO for 48 h. C, RT-PCR of IL-8 mRNA in CAOV3 cells incubated with increasing vorinostat concentrations for 48 h. D, RT-PCR of the above NFκB-regulated genes in OVCAR3 cells incubated with 1.5 μm vorinostat or DMSO for 48 h. E, RT-PCR of IL-8 mRNA in OVCAR3 cells incubated with increasing vorinostat concentrations for 48 h. The values represent the mean ± S.E. of four experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with control UT cells.
Suppression of Vorinostat-induced IL-8/CXCL8 expression Augments Vorinostat Proapoptotic and Antiproliferative Effects in Ovarian Cancer Cells
Because IL-8/CXCL8 induces survival and proliferation of cancer cells (30–33), we wanted to determine whether inhibition of vorinostat-induced IL-8 expression might enhance the vorinostat proapoptotic and antiproliferative effects in OC cells. SKOV3 cells were transfected with IL-8-specific or control siRNA and incubated for 48 h with increasing vorinostat concentrations. As expected, vorinostat increased IL-8 mRNA levels (Fig. 3A) and cytokine release (Fig. 3B) in cells transfected with control siRNA. IL-8 mRNA levels (Fig. 3A) and cytokine release (Fig. 3B) were significantly decreased in cells transfected with two different IL-8-specific siRNAs: IL-8 siRNA-1 (sc-39631) and IL-8 siRNA-2 (sc-156051), which is a pool of three different siRNA duplexes. Consistent with previous studies demonstrating that HDAC inhibition induces apoptosis in OC cells (34–36), vorinostat decreased cell viability, as measured by trypan blue exclusion (Fig. 3C), and increased apoptosis, as measured by nucleosome release into the cytoplasm (13), in cells transfected with control siRNA (Fig. 3D). Importantly, IL-8 suppression by IL-8-specific siRNA-1 or siRNA-2 significantly potentiated vorinostat-decreased cell viability (Fig. 3C). Suppression of IL-8 by si-RNA-1 also significantly increased apoptosis (Fig. 3D) and decreased the colony formation rate (Fig. 3E) and cell proliferation (Fig. 3F) in vorinostat-treated SKOV3 cells. These results indicated that suppression of vorinostat-induced IL-8 signaling might augment the vorinostat proapoptotic and antiproliferative effects in OC cells.
FIGURE 3.
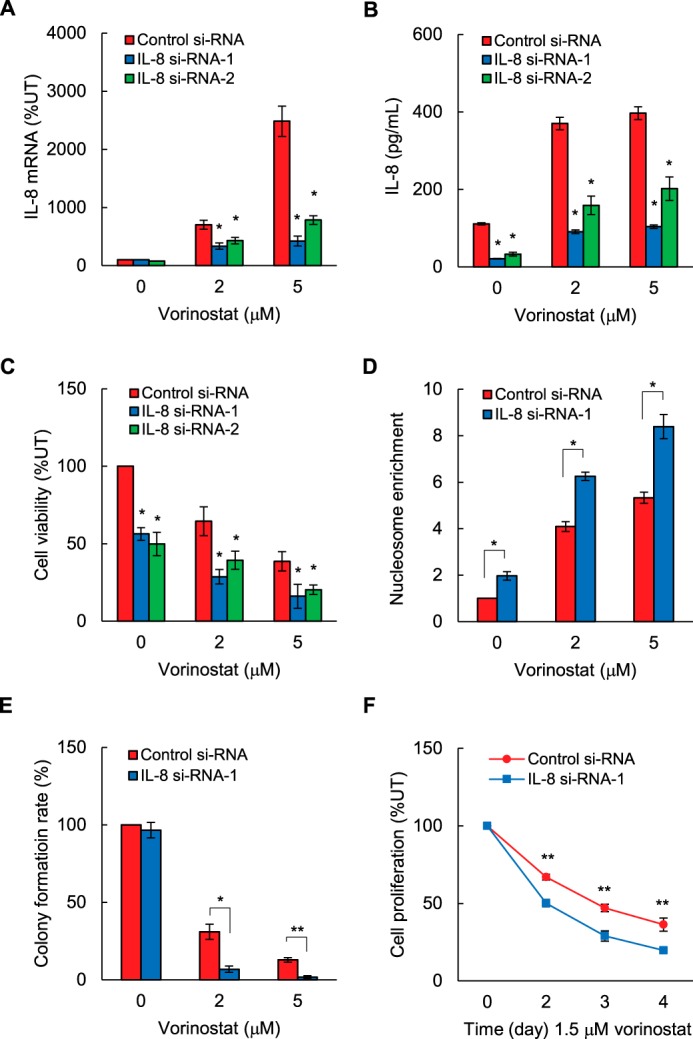
Suppression of vorinostat-induced IL-8/CXCL8 expression enhances vorinostat proapoptotic and antiproliferative effects in OC cells. A and B, SKOV3 cells were transfected with control siRNA or IL-8-specific siRNA-1 or siRNA-2, treated for 48 h with 0, 2, and 5 μm vorinostat, and analyzed for IL-8 mRNA expression by real-time RT-PCR (A) and for IL-8 release by ELISA (B). C, cell viability was measured by trypan blue exclusion in cells transfected with control, IL-8 siRNA-1, or IL-8 siRNA-2 and incubated with vorinostat for 48 h. D and E, apoptosis was analyzed by cytoplasmic nucleosome enrichment assay (D), and the colony formation rate was evaluated as the percentage of plated cells that formed colonies (E) in cells transfected with control or IL-8 siRNA-1 and incubated with 0, 2, and 5 μm vorinostat for 48 h. F, cell proliferation was measured by CellTiter 96 One Solution cell proliferation assay in cells transfected with control or IL-8 siRNA-1 and incubated with 1.5 μm vorinostat or control DMSO. The values represent the mean ± S.E. of four experiments. *, p < 0.05; **, p < 0.01 compared with cells transfected with the corresponding control siRNA.
To validate the above results and determine whether they can be duplicated by neutralization of the produced IL-8, we analyzed the viability and proliferation of OC cells incubated for 48 h with vorinostat in the presence of IL-8-neutralizing antibody. As shown in Fig. 4A, compared with control mouse IgG1, incubation with mouse anti-IL-8 IgG1 significantly decreased the viability of untreated and vorinostat-treated SKOV3 and CAOV3 cells. IL-8 neutralization also significantly reduced the proliferation of SKOV3 and CAOV3 cells (Fig. 4B), indicating that inhibition of IL-8/CXCL8 signaling enhances the vorinostat antiproliferative effect in OC cells.
FIGURE 4.
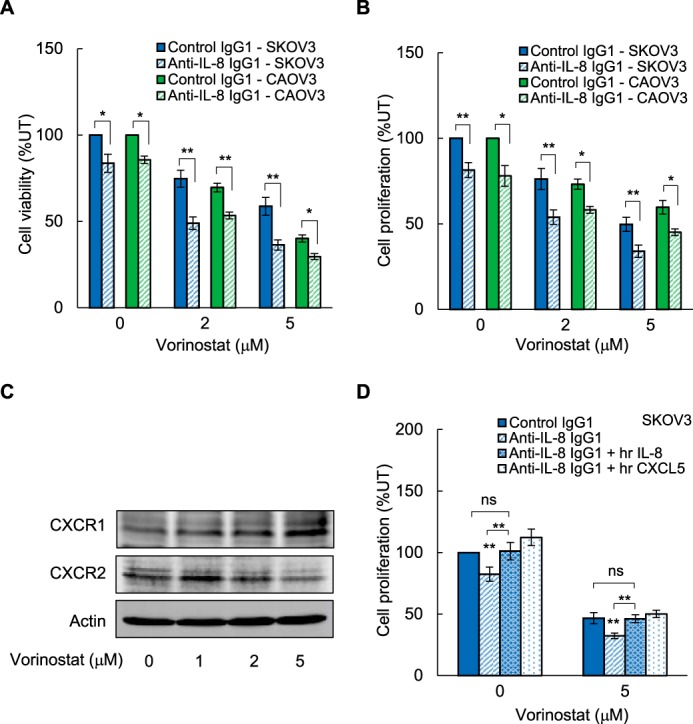
Neutralization of vorinostat-induced IL-8/CXCL8 potentiates the vorinostat inhibitory effect on OC cell viability and proliferation. A and B, SKOV3 and CAOV3 cells were incubated for 48 h with 0, 2, and 5 μm vorinostat in the presence of 2 μg/ml of control mouse IgG1 or 2 μg/ml of mouse anti-human IL-8 IgG1, and cell viability (A) and proliferation (B) were measured as described above. C, Western blotting analysis of CXCR1 and CXCR2 in WCEs of SKOV3 cells incubated with 0, 2, and 5 μm vorinostat for 48 h. Each lane corresponds to ∼5 × 104 cells. D, proliferation of SKOV3 cells incubated for 48 h with 0 and 5 μm vorinostat in the presence of 2 μg/ml of control mouse IgG1 or anti-IL-8 IgG1 with and without 0.5 μg/ml of exogenously added recombinant human IL-8/CXCL8 or CXCL5 proteins. The values represent the mean ± S.E. of three experiments. *, p < 0.05; **, p < 0.01 compared with cells incubated with control mouse IgG1.
IL-8 mediates its functions through binding to its receptors, CXCR1 and CXCR2, which are expressed in OC cells and are also regulated by NFκB (26–29, 37–39). To determine whether HDAC inhibition might regulate the expression of CXCR1 and CXCR2 in OC cells, we analyzed the protein levels of both receptors in SKOV3 cells incubated for 48 h with increasing vorinostat concentrations. As shown in Fig. 4C, vorinostat did not have a marked effect on the protein levels of CXCR1 and CXCR2; this is consistent with the mRNA data in Fig. 2 showing that vorinostat does not have a significant effect on CXCR1 and CXCR2 mRNA levels in EOC cells.
Although the chemokine receptor CXCR1 predominantly binds IL-8/CXCL8, the receptor CXCR2 binds several CXC chemokines, including CXCL5/ENA-78 (29–33). To test whether recombinant human IL-8/CXCL8 or CXCL5 proteins could reverse the decreased proliferation of cells incubated with anti-IL-8 antibody, we measured the proliferation of SKOV3 cells incubated in the absence and presence of vorinostat (5 μm, 48 h) with anti-IL-8 antibody and exogenously added recombinant IL-8/CXCL8 or CXCL5 proteins. As shown in Fig. 4D, addition of both IL-8/CXCL8 and CXCL5 proteins was able to reverse the reduced cell proliferation in the presence of anti-IL-8-neutralizing antibody, indicating that the CXCR1/2 receptors are functional.
Vorinostat-induced IL-8/CXCL8 Expression Is Dependent on IKK Activity and Associated with IKKβ Recruitment to the IL-8/CXCL8 Promoter
To investigate the transcriptional mechanisms responsible for the vorinostat-induced IL-8 expression in OC cells, we first analyzed the cytoplasmic and nuclear levels of NFκB proteins in vorinostat-treated SKOV3 cells. As we have shown previously (12), both p65 and p50 are localized predominantly in the nucleus in OC cells (Fig. 5A). Vorinostat further increased the nuclear levels of p65 and Lys-314/315-acetylated p65, whereas it did not have a marked effect on the nuclear levels of p50 NFκB. IκBα is localized both in the cytoplasm and in the nucleus in OC cells (12); 2 and 5 μm vorinostat decreased IκBα levels in both compartments (Fig. 5, A and B). In addition, vorinostat substantially increased Lys-9/14 acetylation of histone H3 (Fig. 5, A and B).
FIGURE 5.

Vorinostat-induced IL-8/CXCL8 expression is dependent on IKK activity and associated with IKKβ recruitment to the IL-8 promoter in ovarian cancer cells. A, Western blotting of CEs and NEs prepared from SKOV3 cells incubated with vorinostat for 48 h and analyzed by using p65, Lys-314/315 ac-p65, p50, IκBα, histone H3, Lys-9/14 ac-histone H3, and actin antibodies. Each lane corresponds to ∼5 × 104 cells. B, densitometric evaluation of IκBα levels in CEs and NEs (top panel) and of p65, Lys-314/315 ac-p65, p50, histone H3, and Lys-9/14 ac-histone H3 in NEs (bottom panel) of SKOV3 cells shown in A. The protein densities were normalized to actin. The values in untreated cells were arbitrarily set to 1, and the other values are presented relative to these values. The data represent the means of three experiments ± S.E. *, p < 0.05 compared with untreated cells. C and D, real-time RT-PCR analysis of IL-8 mRNA (C) and ELISA of the released IL-8 (D) in SKOV3 cells preincubated for 12 h with 5 μm Bay 117085, 5 mm aspirin, 10 μm SB 203580, or control DMSO and then treated for 48 h with 1.5 μm vorinostat. E, ChIP analysis of IKKα, IKKβ, and IKKϵ occupancy at the IL-8 promoter quantified by real-time PCR in SKOV3 cells incubated for 48 h with increasing concentrations of vorinostat. F, ChIP of IKKβ occupancy at the IL-8, TNFα, and IL-6 promoters in SKOV3 cells incubated for 48 h with vorinostat. The results in E and F are presented as -fold difference in occupancy over the human IGX1A (SA Biosciences) sequence control and represent the mean ± S.E. of three experiments. G, real-time RT-PCR analysis of IKKα, IKKβ, and IKKϵ mRNA levels in SKOV3 cells incubated for 48 h with increasing vorinostat. H, Western blotting analysis of IKKβ and control actin levels in WCEs of SKOV3 cells incubated for 48 h with increasing vorinostat (top panel). Each lane corresponds to ∼5 × 104 cells. Bottom panel, densitometric evaluation of IKKβ (shown in the top panel) normalized to actin. The values are presented relative to the value in untreated cells, which was set to 1. The data represent the means of three experiments ±S.E.
Because we have shown previously that the IL-8 transcription in OC cells is mediated by gene-specific recruitment of IKKβ to the IL-8 promoter (12), we wanted to determine whether vorinostat-induced IL-8 expression is dependent on IKK activity and associated with IKKβ promoter recruitment. SKOV3 cells were preincubated for 12 h with IKK inhibitors, 5 μm Bay 117085 (4) and 5 mm aspirin (41), as well as with a p38 inhibitor, 10 μm SB 203580 (42), followed by 48-h incubation with 1.5 μm vorinostat. Although p38 inhibition did not have a significant effect on IL-8 expression, IKK inhibition significantly decreased the vorinostat-induced IL-8 mRNA levels (Fig. 5C) and cytokine release (Fig. 5D).
To determine whether HDAC inhibition might affect IKK recruitment to the IL-8 promoter, we measured the recruitment of IKKα, IKKβ, and IKKϵ to the IL-8 promoter in vorinostat-treated SKOV3 cells by ChIP. Interestingly, although vorinostat markedly increased IKKβ recruitment to the IL-8 promoter, it did not have a substantial effect on the recruitment of IKKα or IKKϵ (Fig. 5E). In addition, vorinostat-induced IKKβ recruitment to the IL-8 promoter was gene-specific because IKKβ recruitment to the TNFα and IL-6 promoters (Fig. 5F) and CXCL5, TGFβ1, cIAP-1, and Bcl-2 promoters (data not shown) was much lower. The vorinostat-induced IKKβ recruitment to the IL-8 promoter suggested that HDAC inhibition might increase the IKKβ expression in OC cells. To test this hypothesis, we analyzed IKKα, β, and ϵ expression in SKOV3 cells incubated for 48 h with vorinostat. However, as shown in Fig. 5G, vorinostat did not have any significant effect on the IKK mRNA levels. Vorinostat also did not have any significant effect on the protein levels of IKKβ (Fig. 5H), indicating that HDAC inhibition specifically increases recruitment of the already synthesized nuclear IKKβ to the IL-8 promoter.
Vorinostat Induces p65, Lys-314/315-acetylated p65, and Lys-9/14-acetylated Histone H3 Occupancy at the IL-8/CXCL8 Promoter
Because vorinostat increased the nuclear levels of p65, Lys-314/315 ac-p65, and Lys-9/14 ac-histone H3 (Fig. 5, A and B), we wanted to determine whether they are recruited to the IL-8 promoter. Consistent with the gene-specific induction of IL-8 expression in vorinostat-treated EOC cells (Figs. 1 and 2), vorinostat significantly increased p65 recruitment to the IL-8 promoter, whereas it did not increase p65 recruitment to the TNFα or IL-6 promoters (Fig. 6A) or CXCL5, TGFβ1, cIAP-1, or Bcl2 promoters (data not shown). Importantly, p65 recruitment to the IL-8 promoter was IKK-dependent because preincubation with Bay 117085 significantly decreased vorinostat-induced p65 promoter recruitment (Fig. 6B).
FIGURE 6.
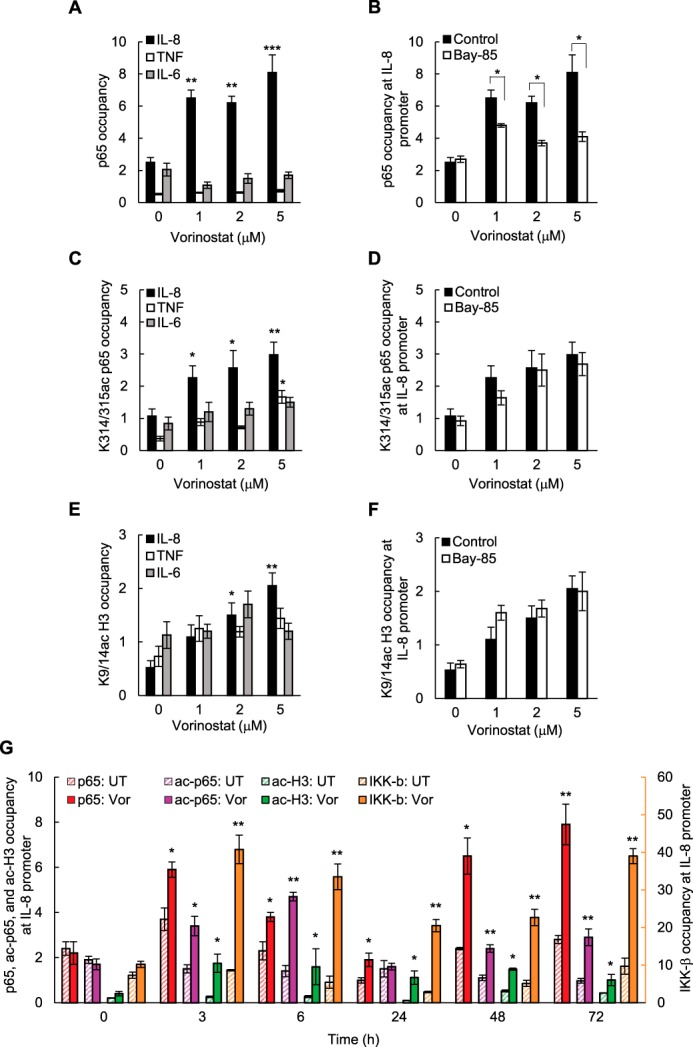
Vorinostat induces p65, Lys-314/315-acetylated p65, and Lys-9/14-acetylated histone H3 recruitment to the IL-8/CXCL8 promoter in ovarian cancer cells. A, ChIP analysis of p65 occupancy at the IL-8, TNFα, and IL-6 promoters quantified by real-time PCR in SKOV3 cells incubated for 48 h with vorinostat. B, ChIP of p65 occupancy at the IL-8 promoter in SKOV3 cells preincubated 12 h with 5 μm Bay 117085 or control DMSO and treated for 48 h with increasing concentrations of vorinostat. C, ChIP of Lys-314/315 ac-p65 occupancy at the IL-8, TNFα, and IL-6 promoters in SKOV3 cells incubated for 48 h with vorinostat. D, ChIP of Lys-314/315 ac-p65 occupancy at the IL-8 promoter in SKOV3 cells preincubated for 12 h with 5 μm Bay 117085 or control DMSO and treated for 48 h with vorinostat. E, ChIP of Lys-9/14 ac-histone H3 occupancy at the IL-8, TNFα, and IL-6 promoters in SKOV3 cells incubated for 48 h with vorinostat. F, ChIP of Lys-9/14 ac-histone H3 occupancy at the IL-8 promoter in SKOV3 cells preincubated for 12 h with 5 μm Bay 117085 or control DMSO and treated for 48 h with vorinostat. G, time course of p65, Lys-314/315 ac-p65, Lys-9/14 ac-histone H3, and IKKβ occupancy at the IL-8/CXCL8 promoter in SKOV3 cells incubated with 1.5 μm vorinostat (Vor) or control DMSO and analyzed by ChIP. The data in A–G are presented as -fold difference in occupancy of the particular protein at the particular locus in comparison with the human IGX1A (SA Biosciences) locus and represent the mean ± S.E. of three experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with cells treated with DMSO.
Seven acetylated lysines have been identified within p65 NFκB, including Lys-122, Lys-123, Lys-218, Lys-221, Lys-310, Lys-314, and Lys-315 (9). We found that vorinostat significantly increased the occupancy of Lys-314/315 ac-p65 at the IL-8 promoter but not at the TNF and IL-6 promoters (Fig. 6C). We did not detect recruitment of p65 acetylated at Lys-122/123, Lys-218, Lys-221, and Lys-310 to the IL-8, TNF, or IL-6 promoters (data not shown). Interestingly, acetylation of p65 at Lys-314/315 has been associated previously with a differential regulation of NFκB target genes (43–45). In contrast to p65, however, Lys-314/315 ac-p65 occupancy at the IL-8 promoter was not dependent on IKK activity (Fig. 6D). Vorinostat also increased Lys-9/14 ac-histone H3 occupancy at the IL-8 promoter (Fig. 6E) independent of IKK activity (Fig. 6F). To investigate the kinetics of the recruitment, we measured a time course of p65, Lys-314/315 ac-p65 (ac-p65), IKKβ, and Lys-9/14 ac-histone H3 (ac-H3) occupancy at the IL-8/CXCL8 promoter in vorinostat-treated SKOV3 cells. Interestingly, p65, ac-p65, and IKKβ were recruited to the IL-8 promoter in two waves; the first occurring between 3 and 6 h and the second wave occurring 48–72 h after HDAC inhibition. During the first wave, p65 and IKKβ occupancy at the IL-8 promoter increased first, reaching a maximum 3 h after HDAC inhibition, followed by ac-p65 occupancy, which reached a maximum 6 h after vorinostat treatment (Fig. 6G). Together, these in vitro results indicate that HDAC inhibition induces histone acetylation and IKK-dependent p65 recruitment to the IL-8 promoter; the promoter-associated p65 is then acetylated, resulting in increased IL-8/CXCL8 transcription.
Combination of Vorinostat and IKK Inhibitor Enhances Vorinostat Effectiveness in Reducing Tumor Growth in Vivo
To investigate whether IKK inhibition enhances vorinostat effectiveness in ovarian cancer in vivo, we examined the effect of vorinostat and Bay 117085 alone and in combination on ovarian tumor growth in nude mice. Female athymic nude mice were implanted subcutaneously with SKOV3 cells. After tumors (∼70 mm3) developed, the mice were randomly divided into four groups (n = 7) and injected i.p. for 28 days with the following: vehicle control (PBS), Bay 117085 (5 mg/kg) every other day, vorinostat (50 mg/kg) every other day, and a combination of Bay 117085 (5 mg/kg) and vorinostat (50 mg/kg). As evaluated by body weight, the treatment regimen was well tolerated because the mice did not lose any weight (Fig. 7A).
FIGURE 7.
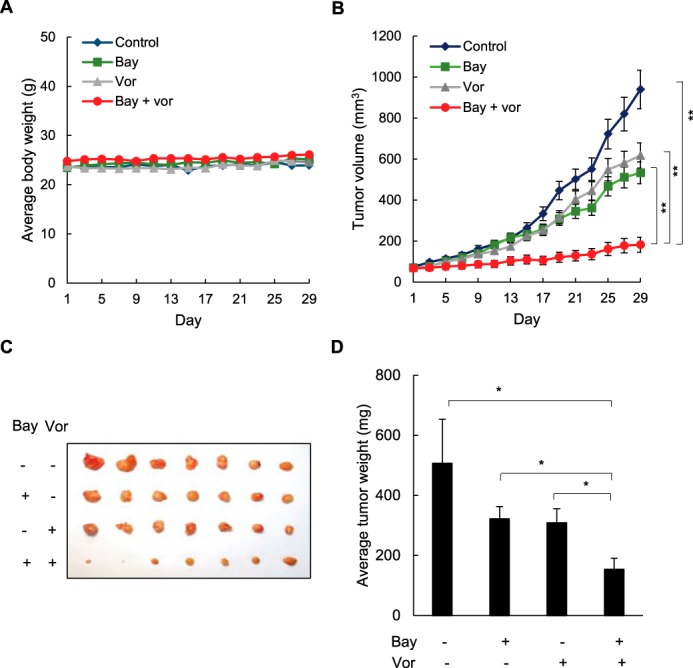
Combination of vorinostat and Bay 117085 enhances vorinostat effectiveness in reducing tumor growth in nude mice implanted with ovarian cancer xenografts. A, average body weight of mice in four treatment groups (n = 7) (control, Bay 117085, vorinostat (Vor), and Bay 117085/vorinostat combination) over the course of 4 weeks. B, average tumor volumes in the four treatment groups (n = 7) over the course of 4 weeks. C, excised SKOV3 tumors implanted subcutaneously in mice (n = 7) after 4 weeks of treatment. D, average weight of the excised tumors (n = 7) at the end of the 4-week treatment period. The values represent the mean ± S.E. *, p < 0.05; **, p < 0.01.
After 28 days of therapy, Bay 117085 reduced the average tumor volume by about 40% compared with the control group, whereas vorinostat reduced the average tumor volume by about 30% (Fig. 7B). Remarkably, by the final day of treatment, the combination of Bay 117085 and vorinostat decreased the tumor volume by about 80% compared with control animals and by about 70% compared with vorinostat alone. The reduced tumor volume in mice treated with the combination Bay 117085/vorinostat therapy corresponded to tumor appearance (Fig. 7C), and to average tumor weight at the end of the experiment (Fig. 7D). The average tumor weight in the combination group was reduced by about 70% compared with control mice and by about 50% compared with vorinostat-treated mice (Fig. 7D).
Combination of Vorinostat and IKK Inhibitor Is Associated with Reduced IL-8/CXCL8 Levels in Mouse Tumor Xenografts
Because our results showed that IL-8 promotes cell survival and proliferation in vorinostat-treated OC cells in vitro (Figs. 3 and 4) and because the vorinostat/Bay 117085 combination therapy resulted in the slowest tumor growth in vivo (Fig. 7), we reasoned that combination therapy might be associated with reduced IL-8 expression in implanted ovarian tumor xenografts. To analyze IL-8 expression in vivo, we measured IL-8 mRNA and protein levels in tumor tissues and IL-8 cytokine release in plasma samples obtained from mice at the end of the treatment. Because mice do not have a homolog of the CXCL8/IL-8 gene, which is present in other species, including humans (46), all IL-8 detected in mouse tumors and plasma samples was derived from the implanted SKOV3 cell xenografts. Indeed, mice in the combination group had the lowest levels of IL-8 tumor mRNA (Fig. 8A), IL-8 plasma concentrations (Fig. 8B), as well as IL-8 protein tumor expression (Fig. 8, C and D). Compared with the control untreated group, IL-8 plasma concentration in the combination group was about 80% lower (Fig. 8B), and the IL-8 protein tumor levels were about 60% lower (Figs. 8, C and D). As a control, we also analyzed the tumor and plasma levels of TGFβ1. In contrast to IL-8, however, the tumor levels of human TGFβ1 mRNA in the combination group did not decrease and were essentially unchanged in all groups (Fig. 8A). Similarly, TGFβ1 plasma concentrations (Fig. 8B) and tumor protein levels (Fig. 8C) were comparable in all treatment groups. Even though additional mechanisms likely contribute to the lowest IL-8 levels in the combination group, these results indicate that vorinostat/Bay 117085 therapy inhibits IL-8 expression in vivo.
FIGURE 8.
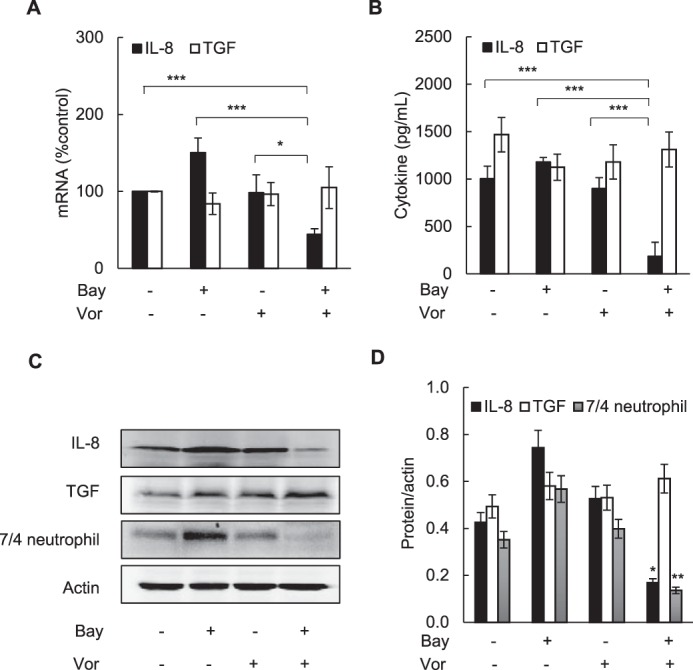
Combination of vorinostat and Bay 117085 decreases IL-8/CXCL8 levels in vivo. A, IL-8 and TGFβ1 mRNA levels analyzed by real-time RT-PCR in excised tumors from the four treatment groups (n = 7). Vor, vorinostat. B, IL-8 and TGFβ1 release measured by ELISA in mouse plasma samples in the four treatment groups (n = 7) at the end of the experiment. C, representative Western blotting of tumor WCEs analyzed using antibodies against IL-8/CXCL8, TGFβ1, murine neutrophil [7/4] antigen, and actin. D, densitometry evaluation of IL-8/CXCL8, TGFβ1, and the murine neutrophil [7/4] antigen in tumor samples; the band intensities were normalized to actin (n = 5). The values represent the mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001).
Interestingly, although mice do not possess a homolog of the human IL-8/CXCL8 gene, murine neutrophils express the IL-8/CXCL8 receptors CXCR1 and CXCR2 and can respond to human IL-8 (47–49). To investigate whether the lowest tumor IL-8 levels in the combination group were associated with reduced neutrophil recruitment, we analyzed the tumor protein levels of the murine neutrophil [7/4] antigen. Consistent with the tumor IL-8 levels, the combination group had the lowest tumor neutrophil [7/4] antigen levels (Figs. 8C), which were about 60% lower in the combination group than in the control untreated group (Fig. 8D), indicating reduced neutrophil recruitment.
Discussion
Even though HDAC expression is increased in ovarian cancer cells, clinical trials targeting the HDAC activity in EOC as well as in other solid tumors have been disappointing (50–52). In this study, we present evidence that may explain the decreased efficacy of HDAC inhibitors in EOC based on our data demonstrating that HDAC inhibition specifically induces expression of IL-8/CXCL8, which induces cell proliferation and survival in EOC cells. The induced IL-8 expression is dependent on IKK activity and associated with gene-specific, IKK-dependent recruitment of p65 NFκB to the IL-8 promoter, followed by increased Lys-314/315 ac-p65 promoter occupancy (Fig. 9). Importantly, our in vivo results demonstrate that combination of HDIs and IKK inhibitors significantly reduces tumor growth in ovarian cancer xenografts compared with either drug alone. These data provide the first evidence demonstrating that HDAC inhibition specifically induces IL-8/CXCL8 expression in EOC cells and suggest that using IKK inhibitors may increase the effectiveness of HDIs in ovarian cancer.
FIGURE 9.
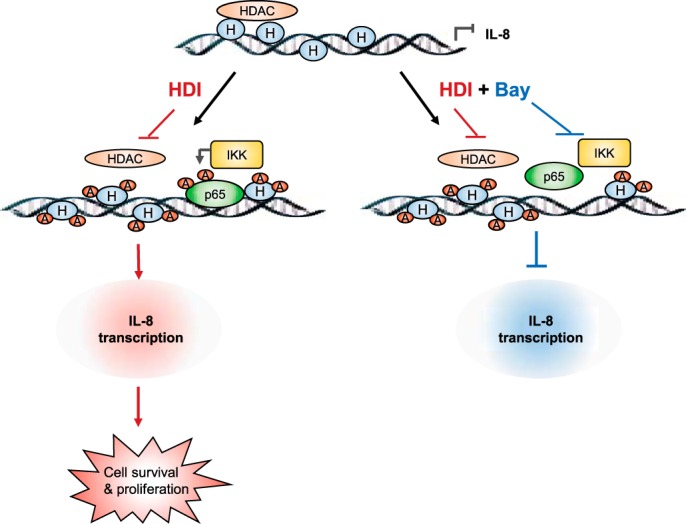
Proposed model of IL-8 regulation by HDAC and IKK in ovarian cancer cells. In ovarian cancer cells, the IL-8 promoter is occupied by an HDAC, which limits IL-8 transcription. Inhibition of HDAC activity increases IKKβ recruitment to the IL-8 promoter, facilitating recruitment of p65 NFκB and its acetylation, resulting in increased IL-8 transcription, cell survival, and proliferation. Inhibition of IKK activity decreases p65 recruitment to the IL-8 promoter, suppressing HDI-induced IL-8 expression.
The rationale for developing HDIs as anti-cancer agents was based on their ability to induce hyperacetylation of histones and non-histone proteins, resulting in increased differentiation, apoptosis, and cell cycle arrest of cancer cells (19, 20). However, accumulating evidence indicates that HDIs also activate the NFκB prosurvival pathway, which limits their lethality and contributes to cell resistance (53–56). Previous studies have shown that the mechanism by which HDAC inhibition increases NFκB-dependent transcription involves increased IκBα degradation and nuclear accumulation of p65 and increased acetylation of p65, resulting in transcriptional activation of NFκB (57–59). Our results are consistent with these studies and demonstrate that HDAC inhibition increases IL-8/CXCL8 mRNA and protein levels in EOC cells, and this is associated with decreased IκBα levels and increased nuclear levels of p65, Lys-314/315 ac-p65, and acetylated histone H3. However, because IL-8/CXCL8 protein release is observed 24 h after HDAC inhibition, whereas IL-8 mRNA is substantially induced 48 h after vorinostat treatment (Fig. 1), it seems likely that additional mechanisms regulate the HDI-induced IL-8 protein expression and release in EOC cells, especially at the beginning of IL-8 induction.
Interestingly, among the tested NFκB-regulated genes (IL-8/CXCL8, TNFα, IL-6, CXCL5, TGFβ1, cIAP-1, Bcl-2, p65, p50, IκBα, CXCR-1, and CXCR2), only IL-8/CXCL8 was significantly induced by vorinostat in EOC cells (Figs. 1 and 2). In addition, vorinostat induced p65 recruitment only to the IL-8 promoter but not to other NFκB-dependent promoters. Vorinostat-induced p65 recruitment to the IL-8 promoter was dependent on IKK activity and correlated with IKKβ recruitment and IL-8 transcription in EOC cells. In addition, HDAC inhibition increased the occupancy of Lys-314/315 ac-p65 and Lys-9/14 ac-histone H3 at the IL-8 promoter in EOC cells. However, in contrast to p65 promoter occupancy, which was IKK-dependent, the occupancy of Lys-314/315 ac-p65 was IKK-independent. Thus, it seems likely that HDAC inhibition first increases histone acetylation at the IL-8 promoter, thus facilitating IKK-dependent recruitment of p65 and its subsequent acetylation, resulting in increased IL-8 transcription. This model is also supported by the time course of p65, Lys-314/315 ac-p65, Lys-9/14 ac-histone H3, and IKKβ occupancy at the IL-8 promoter, indicating that Lys-9/14 ac-histone H3, p65, and IKKβ are recruited first, followed by increased occupancy of ac-p65 (Fig. 6G). However, because appreciable levels of IL-8/CXCL8 mRNA are induced 48 h after HDAC inhibition (Fig. 1C), these data suggest that histone acetylation or p65/IKKβ recruitment alone are not sufficient to induce the IL-8 transcription and that Lys-314/315 ac-p65 promoter occupancy is required for maximum IL-8/CXCL8 expression in EOC cells. The recruited IKKβ may phosphorylate p65 or other transcription factors or regulators, thus facilitating IL-8 transcription. But why is p65 NFκB recruited only to the IL-8 promoter in vorinostat-treated EOC cells? One possible scenario is that, in EOC cells, the IL-8 promoter is occupied by an HDAC, which prevents or limits IL-8 transcription. Inhibition of HDAC activity would then facilitate histone acetylation, followed by p65 recruitment and its acetylation (Fig. 9). However, because the expression of CXCL5, which has the same NFκB binding promoter sequence as IL-8/CXCL8 (12), is not increased by vorinostat in EOC cells (Fig. 2), it seems likely that the NFκB binding promoter sequence alone is not sufficient to determine the specificity of HDAC and p65 recruitment.
Importantly, because suppression of vorinostat-induced IL-8 expression increases the vorinostat proapoptotic and antiproliferative effects in EOC cells (Fig. 3) and because mice in the combination group had the smallest tumors and lowest IL-8 levels (Figs. 7 and 8), our data suggest that induced IL-8 expression may represent one of the mechanisms responsible for the limited effectiveness of HDAC inhibitors in ovarian cancer. This is supported by previous studies demonstrating that IL-8 mediates tumor progression and angiogenesis in ovarian cancer (1, 2) as well as by our data demonstrating that neutralization of the vorinostat-induced IL-8 potentiates the vorinostat inhibitory effect on OC cell viability and proliferation (Fig. 4). In addition, Sonnemann et al. (60) have shown that HDIs and aspirin synergistically induce cell death in OC cells in vitro independent of cyclooxygenase. Because aspirin and other nonsteroidal anti-inflammatory drugs, in addition to inhibiting cyclooxygenase activity, inhibit IKK activity (41, 61), it is possible that the observed HDI/aspirin synergistic effect was mediated by IKK inhibition and suppression of the IL-8 expression induced by HDAC inhibition.
Targeting IKK activity and NFκB-dependent transcription of prosurvival genes induced by HDAC inhibition has been investigated in the treatment of multiple myeloma and other hematological malignancies (55). In ovarian cancer and other solid tumors, combination of IKK and HDAC inhibitors has never been considered, perhaps because of the limited effectiveness of these drugs as single agents. Our in vivo data show that IKK inhibition significantly enhances the vorinostat effectiveness in inhibiting OC tumor growth in SKOV3 mouse xenografts. Compared with vorinostat alone, the combination of vorinostat/IKK inhibitor reduced tumor volume and tumor weight by about 70% and 50%, respectively (Fig. 7). The smallest tumor size in the combination group was associated with the lowest IL-8 tumor levels and plasma concentrations and with the lowest tumor expression of the murine neutrophil [7/4] antigen, indicating reduced tumor infiltration with mouse neutrophils in the combination group (Fig. 8).
Collectively, our data demonstrate that HDAC inhibition specifically induces IL-8/CXCL8 expression in ovarian cancer cells, resulting in their increased viability and proliferation. The responsible mechanism involves gene-specific and IKK-dependent p65 recruitment to the IL-8 promoter. Inhibition of IKK activity reduces vorinostat-induced IL-8 expression and release in OC cells in vitro, and combining vorinostat with the IKK inhibitor significantly reduces tumor growth of OC xenografts in nude mice. Our results suggest that, by suppressing IL-8 expression, IKK inhibitors may increase the effectiveness of HDIs in ovarian cancer and perhaps other solid tumors characterized by increased IL-8/CXCL8 expression.
Experimental Procedures
Antibodies and Reagents
The antibodies against human p65 NFκB (sc-372), p50 NFκB (sc-7178), IκBα (sc-371), histone H3 (sc-8654), Lys-9/14-acetylated histone H3 (sc-8655), IKKα (sc-7218), IKKβ (sc-8014), IKKϵ (sc-376114), IL-8/CXCL8 (sc-7922), CXCR1 (sc-7303), and CXCR2 (sc-7304) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody against TGFβ1 (MAB1835) was from R&D Systems (Minneapolis, MN). The antibody against Lys-314/315-acetylated p65 (HW136) was purchased from Signalway Antibody (College Park, MD). The anti-murine neutrophil [7/4] antibody (ab53457) was from Abcam (Cambridge, MA). The antibody against actin was purchased from Sigma. The HRP-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Vorinostat was obtained from Selleck Chemicals (Houston, TX). Romidepsin was from ChemieTek (Indianapolis, IN). The IKK inhibitor Bay 117085 was purchased from Cayman Chemicals (Ann Arbor, MI). All other reagents were of molecular biology grade and were obtained from Sigma.
Cell Culture
Human ovarian cancer SKOV3, CAOV3, and OVCAR3 cells were obtained from the ATCC (Manassas, VA). Cells were cultured (5 × 105 cells/ml) in 6-well plates in RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen) and antibiotics at 37 °C with 5% CO2 as described previously (12, 13). For in vitro experiments, vorinostat, romidepsin, apicidin, Bay 117085, and SB 203580 were dissolved in DMSO, and an equivalent DMSO volume was used as a solvent control. Cell viability was measured by using trypan blue exclusion.
Transfection with siRNA
Human IL-8/CXCL8 (sc-39631 and sc-156051) and non-silencing (sc-37007) siRNAs were obtained from Santa Cruz Biotechnology. The sc-39631 IL-8 siRNA oligonucleotides were 5′-GGGUGCAGAGGGUUGUGGAGAtt-3′ (sense) and 5′-UCUCCACAACCCUCUGCACCCtt-3′ (antisense). The sc-156051 IL-8 siRNA was a pool of three different siRNA duplexes: sc-156051A, 5′-GUCAGUGCAUAAAGACAUAtt-3′ (sense) and 5′-UAUGUCUUUAUGCACUGACtt-3′ (antisense); sc-156051B, 5′-GAAGAGGGCUGAGAAUUC-Att-3′ (sense) and 5′-UGAAUUCUCAGCCCUCUU-Ctt-3′ (antisense); and sc-156051C, 5′-CAAG-AGAAUAUCCGAACUUtt-3′ (sense) and 5′-AAG-UUCGGAUAUUCUCUUGtt-3′ (antisense). Prior to transfection, cells were seeded into a 6-well plate and incubated in a humidified 5% CO2 atmosphere at 37 °C in antibiotic-free RPMI medium supplemented with 10% FBS for 24 h to about 80% confluence. For each transfection, 80 pmol of either non-silencing siRNA control or specific siRNA was used. Cells were transfected for 7 h in siRNA transfection medium with siRNA transfection reagent according to the instructions of the manufacturer (Santa Cruz Biotechnology). After transfection, fresh medium with antibiotics was added, and the cells were grown for 24 h before treatment.
Apoptosis, Colony Formation, Cell Proliferation, and IL-8/CXCL8 Neutralizing Assays
Apoptosis was evaluated using a cell death detection ELISA kit that quantifies the release of nucleosomes into the cytoplasm (Cell Death Detection ELISAPLUS, Roche) as described previously (13). For the colony formation assay, cells were seeded into 6-well plates at a density of 2000 cells/well 24 h after transfection and treated with vorinostat for 48 h. The medium was changed every 3 days. After incubation at 37 °C for 14 days, the cells were washed twice with PBS, fixed with methanol, stained with crystal violet, and counted under a microscope. The colony formation rate was calculated as the percentage of plated cells that formed colonies.
Cell proliferation was measured by CellTiter 96 One Solution cell proliferation assay (Promega, Madison, WI). Transfected cells were seeded into 96-well plates at a density of 5000 cells/100 μl of medium and incubated with 1.5 μm vorinostat or control DMSO at 37 °C. At the indicated time points, 20 μl of CellTiter 96 One Solution reagent was added to each well and incubated for 4 h at 37 °C, and absorbance at 490 nm was measured.
For the CXCL8 neutralization experiments, SKOV3 cells were incubated for 48 h with 0, 2, and 5 μm vorinostat in the presence of 2 μg/ml anti-human IL-8/CXCL8 monoclonal IgG1 antibody (MAB208, Novus, Littleton, CO) or control mouse IgG1 (MAB002, R&D Systems), and cell viability and proliferation were measured as described above. For the reconstitution experiments, cells were incubated for 48 h with 0 and 5 μm vorinostat in the presence of control mouse IgG1 or IL-8/CXCL8 mouse IgG1 as described above, with and without 0.5 μg/ml exogenously added recombinant human IL-8/CXCL8 (208-IL-010, Novus) or CXCL5 (NBP2-34970, Novus) proteins, and cell proliferation was measured as described above.
Ovarian Cancer Xenografts
All animal procedures were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of St. John's University. Six-week-old female athymic nude mice (Taconic Farms) were used for tumor xenograft experiments. Mice were maintained on an alternating 12-h light/dark cycle with ad libitum water and rodent chow as described previously (13). Mice were injected subcutaneously in the right flank with 5 × 106 SKOV3 cells in 200 μl of serum-free RPMI medium. When the average tumor size reached about 70 mm3, the mice were randomly divided into four groups. The control group received the control vehicle (PBS), injected i.p. every other day for 4 consecutive weeks. The IKK inhibitor group received Bay 117085 (5 mg/kg) prepared in PBS, injected i.p. every other day. The vorinostat group received vorinostat (50 mg/kg), injected i.p. every other day. The combination group received Bay 117085 and vorinostat, injected i.p. every other day for 4 consecutive weeks. Mice were weighed, and tumor volume was monitored by caliper measurement every other day. Tumor volumes were calculated using the following formula: volume = length × width2 × 0.5.
At the end point, mice were anesthetized using isoflurane inhalant gas, and ∼1 ml of blood was collected from each mouse by cardiac puncture. The blood samples were centrifuged at room temperature in heparinized tubes to prepare plasma samples, which were stored at −80 °C for future chemokine analysis. Immediately after blood collection, mice were euthanized using carbon dioxide, and tumors were excised, measured, and weighed. Tumors were then cut into two parts: one part was stored in RNAlater solution for RNA extraction and gene expression analysis, and the other part was processed for preparation of whole cell extracts (WCEs) for Western blotting analysis as described previously (13).
ELISA
Human IL-8/CXCL8, TNFα, IL-6, and TGFβ1 release was measured by commercially available ELISA kits (R&D Systems) as described previously (12, 13).
Real-time RT-PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA). The iScript one-step RT-PCR kit with SYBR Green (Bio-Rad) was used as a supermix, and 20 ng/μl RNA was used as template on a Bio-Rad MyIQ single color real-time PCR detection system. The primers used for quantification of human IL-8/CXCL8, TNFα, IL-6, CXCL5, TGFβ1, cIAP-1, Bcl-2, p65, p50, IκBα, CXCR-1, CXCR2, IKKα, IKKβ, IKKϵ, and actin mRNA were purchased from SA Biosciences (Frederick, MD). The mRNA values are expressed as a percentage of untreated (UT) samples, which were arbitrarily set as 100% (12).
Western Blotting Analysis
WCEs, nuclear extracts (NE), and cytoplasmic extracts (CEs) were prepared as described previously (12, 40). Denatured proteins were separated on 10–14% SDS gels and analyzed by immunoblotting as described previously (12, 40).
ChIP
ChIP analysis was performed as described previously (12, 13). Briefly, proteins and DNA were cross-linked by formaldehyde, and cells were washed and sonicated. The lysates were centrifuged (15,000 × g, 10 min, 4 °C), and the supernatant extracts were diluted with ChIP dilution buffer and precleared with protein A/G-agarose (Santa Cruz Biotechnology) for 2 h at 4 °C. Immunoprecipitations were performed overnight at 4 °C. Following immunoprecipitation, the samples were incubated with protein A/G-agarose (1 h, 4 °C), and the immune complexes were collected by centrifugation (150 × g, 5 min, 4 °C), washed, and extracted with 1% SDS-0.1 M NaHCO3. After reversing the cross-linking, proteins were digested with proteinase K, and the samples were extracted with phenol/chloroform followed by precipitation with ethanol. The pellets were resuspended in nuclease-free water and subjected to real-time PCR. Immunoprecipitated DNA was analyzed by real-time PCR (25-μl reaction mixture) using iQ SYBR Green Supermix and the Bio-Rad MyIQ single color real-time PCR detection system. Each immunoprecipitation was performed at least three times using different chromatin samples, and the occupancy was calculated using human IGX1A negative control primers (SA Biosciences), which detect a specific genomic ORF-free DNA sequence that does not contain a binding site for any known transcription factors. The results were calculated as -fold difference in occupancy of the particular protein at the particular locus in comparison with the IGX1A locus. The primers used for real-time PCR were as follows: IL-8/CXCL8, 5′-GGGCCATCAGTTGCA-AATC-3′ (forward) and 5′-GCTTGTGTGCTCT-GCTGTCTC-3′ (reverse); TNFα, 5′-CGCTTCCT-CCAGATGAGCTT-3′ (forward) and 5′-TGCTG-TCCTTGCTGAGGGA-3′ (reverse); and IL-6, 5′-CCT-CAGACATCTCCAGTCCT-3′ (forward) and 5′-AATGACGACCTAAGCTGCAC-3′ (reverse).
Statistical Analysis
The results represent at least three independent experiments. Numerical results are presented as means ± S.E. Data were analyzed by using an InStat software package (GraphPad, San Diego, CA). Statistical significance was evaluated using Mann-Whitney U test, and p < 0.05 was considered significant. Levels of significance are indicated as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author Contributions
H. R. G. conceived the project, conducted the experiments, and analyzed the results. Y. Z., M. M. U., B. S., P. B., and A. V. developed the methods and analyzed the data. I. V. conceived the project, analyzed the data, wrote the paper, and coordinated the study.
This work was supported by St. John's University and by National Institutes of Health Grants CA173452 and CA202775 (to I. V.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EOC
- epithelial ovarian cancer
- OC
- ovarian cancer
- IKK
- IκB kinase
- HDAC
- histone deacetylase
- HDI
- histone deacetylase inhibitor
- WCE
- whole cell extract
- UT
- untreated
- NE
- nuclear extract
- CE
- cytoplasmic extract.
References
- 1. Xu L., and Fidler I. J. (2000) Interleukin 8: an autocrine growth factor for human ovarian cancer. Oncol. Res. 12, 97–106 [DOI] [PubMed] [Google Scholar]
- 2. Merritt W. M., Lin Y. G., Spannuth W. A., Fletcher M. S., Kamat A. A., Han L. Y., Landen C. N., Jennings N., De Geest K., Langley R. R., Villares G., Sanguino A., Lutgendorf S. K., Lopez-Berestein G., Bar-Eli M. M., and Sood A. K. (2008) Effect of interleukin-8 gene silencing with liposome-encapsulated small interfering RNA on ovarian cancer cell growth. J. Natl. Cancer Inst. 100, 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang S., Robinson J. B., Deguzman A., Bucana C. D., and Fidler I. J. (2000) Blockade of nuclear factor-κB signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Res. 60, 5334–5339 [PubMed] [Google Scholar]
- 4. Mabuchi S., Ohmichi M., Nishio Y., Hayasaka T., Kimura A., Ohta T., Saito M., Kawagoe J., Takahashi K., Yada-Hashimoto N., Sakata M., Motoyama T., Kurachi H., Tasaka K., and Murata Y. (2004) Inhibition of NFκB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J. Biol. Chem. 279, 23477–23485 [DOI] [PubMed] [Google Scholar]
- 5. Annunziata C. M., Stavnes H. T., Kleinberg L., Berner A., Hernandez L. F., Birrer M. J., Steinberg S. M., Davidson B., and Kohn E. C. (2010) Nuclear factor κB transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer 116, 3276–3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hinz M., and Scheidereit C. (2014) The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 15, 46–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu F., Xia Y., Parker A. S., and Verma I. M. (2012) IKK biology. Immunol. Rev. 246, 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Natoli G. (2012) NF-κB and chromatin: ten years on the path from basic mechanisms to candidate drugs. Immunol. Rev. 246, 183–192 [DOI] [PubMed] [Google Scholar]
- 9. Huang B., Yang X. D., Lamb A., and Chen L. F. (2010) Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell Signal. 22, 1282–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oeckinghaus A., and Ghosh S. (2009) The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1, a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Viatour P., Merville M. P., Bours V., and Chariot A. (2005) Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 30, 43–52 [DOI] [PubMed] [Google Scholar]
- 12. Singha B., Gatla H. R., Manna S., Chang T. P., Sanacora S., Poltoratsky V., Vancura A., and Vancurova I. (2014) Proteasome inhibition increases recruitment of IκB kinase β (IKKβ), S536P-p65, and transcription factor EGR1 to interleukin-8 (IL-8) promoter, resulting in increased IL-8 production in ovarian cancer cells. J. Biol. Chem. 289, 2687–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singha B., Gatla H. R., Phyo S., Patel A., Chen Z. S., and Vancurova I. (2015) IKK inhibition increases bortezomib effectiveness in ovarian cancer. Oncotarget 6, 26347–26358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Singha B., Gatla H. R., and Vancurova I. (2015) Transcriptional regulation of chemokine expression in ovarian cancer. Biomolecules 5, 223–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marks P., Rifkind R. A., Richon V. M., Breslow R., Miller T., and Kelly W. K. (2001) Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 1, 194–202 [DOI] [PubMed] [Google Scholar]
- 16. Khabele D., Son D. S., Parl A. K., Goldberg G. L., Augenlicht L. H., Mariadason J. M., and Rice V. M. (2007) Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol. Ther. 6, 795–801 [DOI] [PubMed] [Google Scholar]
- 17. Takai N., and Narahara H. (2009) Histone deacetylase inhibitor therapy in epithelial ovarian cancer. J. Oncol. 10.1155/2010/458431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khabele D. (2014) The therapeutic potential of class I selective histone deacetylase inhibitors in ovarian cancer. Front. Oncol. 4, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marks P. A., Richon V. M., and Rifkind R. A. (2000) Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 92, 1210–1216 [DOI] [PubMed] [Google Scholar]
- 20. Johnstone R. W. (2002) Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–299 [DOI] [PubMed] [Google Scholar]
- 21. Marks P. A., and Breslow R. (2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 [DOI] [PubMed] [Google Scholar]
- 22. Grant S., Easley C., and Kirkpatrick P. (2007) Vorinostat. Nat. Rev. Drug Discov. 6, 21–22 [DOI] [PubMed] [Google Scholar]
- 23. Fantin V. R., and Richon V. M. (2007) Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin. Cancer Res. 13, 7237–7242 [DOI] [PubMed] [Google Scholar]
- 24. Zhang C., Richon V., Ni X., Talpur R., and Duvic M. (2005) Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J. Invest. Dermatol. 125, 1045–1052 [DOI] [PubMed] [Google Scholar]
- 25. Chang T. P., Poltoratsky V., and Vancurova I. (2015) Bortezomib inhibits expression of TGF-β1, IL-10, and CXCR4, resulting in decreased survival and migration of cutaneous T Cell lymphoma cells. J. Immunol. 194, 2942–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Venkatakrishnan G., Salgia R., and Groopman J. E. (2000) Chemokine receptors CXCR-1/2 activate mitogen-activated protein kinase via the epidermal growth factor receptor in ovarian cancer cells. J. Biol. Chem. 275, 6868–6875 [DOI] [PubMed] [Google Scholar]
- 27. Ivarsson K., Ekerydh A., Fyhr I. M., Janson P. O., and Brännström M. (2000) Upregulation of interleukin-8 and polarized epithelial expression of interleukin-8 receptor A in ovarian carcinomas. Acta Obstet. Gynecol. Scand. 79, 777–784 [PubMed] [Google Scholar]
- 28. Maxwell P. J., Gallagher R., Seaton A., Wilson C., Scullin P., Pettigrew J., Stratford I. J., Williams K. J., Johnston P. G., and Waugh D. J. (2007) HIF-1 and NF-κB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 26, 7333–7345 [DOI] [PubMed] [Google Scholar]
- 29. Richmond A. (2002) NF-κB, chemokine gene transcription and tumour growth. Nat. Rev. Immunol. 2, 664–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Balkwill F. (2004) Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550 [DOI] [PubMed] [Google Scholar]
- 31. Waugh D. J., and Wilson C. (2008) The interleukin-8 pathway in cancer. Clin. Cancer Res. 14, 6735–6741 [DOI] [PubMed] [Google Scholar]
- 32. Lazennec G., and Richmond A. (2010) Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol. Med. 16, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campbell L. M., Maxwell P. J., and Waugh D. J. (2013) Rationale and means to target pro-inflammatory interleukin-8 (CXCL8) signaling in cancer. Pharmaceuticals 6, 929–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strait K. A., Warnick C. T., Ford C. D., Dabbas B., Hammond E. H., and Ilstrup S. J. (2005) Histone deacetylase inhibitors induce G2-checkpoint arrest and apoptosis in cisplatinum-resistant ovarian cancer cells associated with overexpression of the Bcl-2-related protein Bad. Mol. Cancer Ther. 4, 603–611 [DOI] [PubMed] [Google Scholar]
- 35. Chen M. Y., Liao W. S., Lu Z., Bornmann W. G., Hennessey V., Washington M. N., Rosner G. L., Yu Y., Ahmed A. A., and Bast R. C. Jr. (2011) Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 117, 4424–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson A. J., Holson E., Wagner F., Zhang Y. L., Fass D. M., Haggarty S. J., Bhaskara S., Hiebert S. W., Schreiber S. L., and Khabele D. (2011) The DNA damage mark pH2AX differentiates the cytotoxic effects of small molecule HDAC inhibitors in ovarian cancer cells. Cancer Biol. Ther. 12, 484–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y., Yang J., Gao Y., Du Y., Bao L., Niu W., and Yao Z. (2005) Regulatory effect of e2, IL-6 and IL-8 on the growth of epithelial ovarian cancer cells. Cell Mol. Immunol. 2, 365–372 [PubMed] [Google Scholar]
- 38. Yang G., Rosen D. G., Liu G., Yang F., Guo X., Xiao X., Xue F., Mercado-Uribe I., Huang J., Lin S. H., Mills G. B., and Liu J. (2010) CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin. Cancer Res. 16, 3875–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stronach E. A., Cunnea P., Turner C., Guney T., Aiyappa R., Jeyapalan S., de Sousa C. H., Browne A., Magdy N., Studd J. B., Sriraksa R., Gabra H., and El-Bahrawy M. (2015) The role of interleukin-8 (IL-8) and IL-8 receptors in platinum response in high grade serous ovarian carcinoma. Oncotarget 6, 31593–31603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galdieri L., Gatla H., Vancurova I., and Vancura A. (2016) Activation of AMP-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J. Biol. Chem. 291, 25154–25166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yin M. J., Yamamoto Y., and Gaynor R. B. (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature 396, 77–80 [DOI] [PubMed] [Google Scholar]
- 42. Sanacora S., Urdinez J., Chang T. P., and Vancurova I. (2015) Anticancer drug bortezomib increases IL-8 expression in human monocytes. Biochem. Biophys. Res. Commun. 460, 375–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen L. F., Mu Y., and Greene W. C. (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 21, 6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buerki C., Rothgiesser K. M., Valovka T., Owen H. R., Rehrauer H., Fey M., Lane W. S., and Hottiger M. O. (2008) Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 36, 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rothgiesser K. M., Fey M., and Hottiger M. O. (2010) Acetylation of p65 at lysine 314 is important for late NF-κB-dependent gene expression. BMC Genomics 10.1186/1471-2164-11-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nomiyama H., Osada N., and Yoshie O. (2010) The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 21, 253–262 [DOI] [PubMed] [Google Scholar]
- 47. Cerretti D. P., Nelson N., Kozlosky C. J., Morrissey P. J., Copeland N. G., Gilbert D. J., Jenkins N. A., Dosik J. K., and Mock B. A. (1993) The murine homologue of the human interleukin-8 receptor type B maps near the Ity-Lsh-Bcg disease resistance locus. Genomics 18, 410–413 [DOI] [PubMed] [Google Scholar]
- 48. Bozic C. R., Gerard N. P., von Uexkull-Guldenband C., Kolakowski L. F. Jr., Conklyn M. J., Breslow R., Showell H. J., and Gerard C. (1994) The murine interleukin 8 type B receptor homologue and its ligands: expression and biological characterization. J. Biol. Chem. 269, 29355–29358 [PubMed] [Google Scholar]
- 49. Fan X., Patera A. C., Pong-Kennedy A., Deno G., Gonsiorek W., Manfra D. J., Vassileva G., Zeng M., Jackson C., Sullivan L., Sharif-Rodriguez W., Opdenakker G., Van Damme J., Hedrick J. A., Lundell D., et al. (2007) Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J. Biol. Chem. 282, 11658–11666 [DOI] [PubMed] [Google Scholar]
- 50. Modesitt S. C., Sill M., Hoffman J. S., Bender D. P., and Gynecologic Oncology Group (2008) A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol. Oncol. 109, 182–186 [DOI] [PubMed] [Google Scholar]
- 51. Grassadonia A., Cioffi P., Simiele F., Iezzi L., Zilli M., and Natoli C. (2013) Role of hydroxamate-based histone deacetylase inhibitors (Hb-HDACIs) in the treatment of solid malignancies. Cancers 5, 919–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matulonis U., Berlin S., Lee H., Whalen C., Obermayer E., Penson R., Liu J., Campos S., Krasner C., and Horowitz N. (2015) Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother. Pharmacol. 76, 417–423 [DOI] [PubMed] [Google Scholar]
- 53. Mayo M. W., Denlinger C. E., Broad R. M., Yeung F., Reilly E. T., Shi Y., and Jones D. R. (2003) Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-κB through the Akt pathway. J. Biol. Chem. 278, 18980–18989 [DOI] [PubMed] [Google Scholar]
- 54. Rundall B. K., Denlinger C. E., and Jones D. R. (2004) Combined histone deacetylase and NF-κB inhibition sensitizes non-small cell lung cancer to cell death. Surgery 136, 416–425 [DOI] [PubMed] [Google Scholar]
- 55. Dai Y., Rahmani M., Dent P., and Grant S. (2005) Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-κB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell Biol. 25, 5429–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duan J., Friedman J., Nottingham L., Chen Z., Ara G., and Van Waes C. (2007) Nuclear factor-κB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 6, 37–50 [DOI] [PubMed] [Google Scholar]
- 57. Ashburner B. P., Westerheide S. D., and Baldwin A. S. (2001) The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell Biol. 21, 7065–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chavey C., Mühlbauer M., Bossard C., Freund A., Durand S., Jorgensen C., Jobin C., and Lazennec G. (2008) Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-κB pathway in breast cancer. Mol. Pharmacol. 74, 1359–1366 [DOI] [PubMed] [Google Scholar]
- 59. Dai Y., Chen S., Wang L., Pei X. Y., Funk V. L., Kramer L. B., Dent P., and Grant S. (2011) Disruption of IκB kinase (IKK)-mediated RelA serine 536 phosphorylation sensitizes human multiple myeloma cells to histone deacetylase (HDAC) inhibitors. J. Biol. Chem. 286, 34036–34050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sonnemann J., Hüls I., Sigler M., Palani C. D., Hong le T. T., Völker U., Kroemer H. K., and Beck J. F. (2008) Histone deacetylase inhibitors and aspirin interact synergistically to induce cell death in ovarian cancer cells. Oncol. Rep. 20, 219–224 [PubMed] [Google Scholar]
- 61. Takada Y., Bhardwaj A., Potdar P., and Aggarwal B. B. (2004) Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-κB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 23, 9247–9258 [DOI] [PubMed] [Google Scholar]