

Abstract
TAR DNA-binding protein (TDP-43) is a highly conserved and essential DNA- and RNA-binding protein that controls gene expression through RNA processing, in particular, regulation of splicing. Intracellular aggregation of TDP-43 is a hallmark of amyotrophic lateral sclerosis and ubiquitin-positive frontotemporal lobar degeneration. This TDP-43 pathology is also present in other types of neurodegeneration including Alzheimer's disease. We report here that TDP-43 is a substrate of MEK, a central kinase in the MAPK/ERK signaling pathway. TDP-43 dual phosphorylation by MEK, at threonine 153 and tyrosine 155 (p-T153/Y155), was dramatically increased by the heat shock response (HSR) in human cells. HSR promotes cell survival under proteotoxic conditions by maintaining protein homeostasis and preventing protein misfolding. MEK is activated by HSR and contributes to the regulation of proteome stability. Phosphorylated TDP-43 was not associated with TDP-43 aggregation, and p-T153/Y155 remained soluble under conditions that promote protein misfolding. We found that active MEK significantly alters TDP-43-regulated splicing and that phosphomimetic substitutions at these two residues reduce binding to GU-rich RNA. Cellular imaging using a phospho-specific p-T153/Y155 antibody showed that phosphorylated TDP-43 was specifically recruited to the nucleoli, suggesting that p-T153/Y155 regulates a previously unappreciated function of TDP-43 in the processing of nucleolar-associated RNA. These findings highlight a new mechanism that regulates TDP-43 function and homeostasis through phosphorylation and, therefore, may contribute to the development of strategies to prevent TDP-43 aggregation and to uncover previously unexplored roles of TDP-43 in cell metabolism.
Keywords: amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), neurodegeneration, protein aggregation, RNA binding protein, RNA-protein interaction, TAR DNA-binding protein 43 (TDP-43) (TARDBP), MEK kinase, heat shock response
Introduction
In recent years the role of RNA-binding proteins in neurodegenerative disorders, particularly amytrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD)5 has become the focus of intense research. The TAR DNA-binding protein (TDP-43) forms cytoplasmic aggregates in almost all ALS and ∼50% of FTLD cases (1, 2). TDP-43 pathology is observed in other neurological disorders including Alzheimer's disease (3, 4). The direct role TDP-43 plays in disease is underscored by >40 ALS/FTLD patient-derived autosomal dominant missense mutations in the TDP-43 gene (TARDBP) (5). TDP-43 inclusions coincide with a dramatic reduction of the normal nuclear TDP-43 detection (2), implying a loss of function upon aggregate accumulation. The link between neurodegeneration and TDP-43 function has not been clearly established and the contribution of TDP-43 aggregation to pathogenesis is poorly understood.
TDP-43 is a highly conserved heterogeneous nuclear ribonucleoprotein (hnRNP) with two canonical RNA recognition motifs (RRMs) of which RRM1 is necessary and sufficient to bind RNA. A low complexity sequence, or prion-like domain at the C terminus mediates protein interactions required for RNA processing (6, 7) and is the main driver of protein aggregation (8). Although TDP-43 shuttles between nucleus and cytoplasm, it is predominantly nuclear (9). In the nucleus, TDP-43 shows a diffuse pattern and recruitment to Cajal bodies and gems (10). Under specific cellular stress conditions, such as oxidative stress, TDP-43 localizes to cytoplasmic stress granules (11), which may contribute to pathological aggregate formation (12).
TDP-43 is essential for development and survival in animal models and cultured cells (13–17). In human cells and mouse brain, TDP-43 binds thousands of transcripts with a strong preference for GU-rich sequences and regulates >600 protein-coding genes. TDP-43 controls these genes predominantly through splicing regulation (18–20). Although TDP-43 participates in different mechanisms of RNA processing and RNA transport, the regulation of splicing is its best-characterized function (20, 21). In addition, recent findings show that TDP-43 plays a more global role in gene regulation as an inhibitor of cryptic exon inclusion (22).
Despite evidence that TDP-43 controls a large number of genes and is dynamically distributed in cells, the cellular stimuli and factors that regulate TDP-43 function through posttranslational modifications are still mostly unknown. We characterized new TDP-43 phosphorylation sites found in non-pathological settings and sought to determine the mechanisms that control them. Our findings show that activation of the cellular heat shock response (HSR) dramatically increases TDP-43 phosphorylation at Thr-153 and Tyr-155 through the MAPK/ERK kinase MEK, in the absence of increased TDP-43 aggregation or protein clearance. Increased Thr-153/Tyr-155 phosphorylation in the presence of active MEK and phosphomimetic substitutions at Thr-153/Tyr-155 show decreased splicing regulatory function. Moreover, phosphomimetic substitutions at Thr-153/Tyr-155 decrease RNA binding affinity, suggesting that phosphorylation at these sites modulates TDP-43 activity.
Results
The Heat Shock Response Controls TDP-43 Phosphorylation in RRM1
TDP-43 phosphorylation at threonine 153 and tyrosine 155, located in RRM1 (Fig. 1, A and B), was identified by large scale phosphoproteome mass spectrometry analysis of human HeLa cells (23). These modifications were determined under non-pathological conditions. To characterize these previously unexplored phosphorylation sites, we developed a novel phospho-specific polyclonal antibody, p-T153/Y155-TDP-43, to recognize dual TDP-43 phosphorylation at Thr-153/Tyr-155. The basal levels of the protein recognized by p-T153/Y155-TDP-43 were almost undetectable by immunoblotting in different human cell lines including SH-SY5Y human neuroblastoma cells (Fig. 1, C and D, supplemental Fig. S1). However, we found that heat shock dramatically up-regulated the p-T153/Y155-TDP-43-associated signal. No significant increase was observed under conditions of oxidative stress, DNA damage stress, or transcription inhibition (Fig. 1C). Similar results were obtained with heat shock stressed human HEK-293 (supplemental Fig. S1) and HeLa cells (Fig. 2A). The levels of p-T153/Y155-TDP-43-associated signal were undetectable in control cell lysates with >150 μg of total protein (Fig. 1D, supplemental Fig. S1). In contrast, this signal was observed in heat shock-treated cell lysate starting from ∼20 μg of total protein (Fig. 1D, supplemental Fig. S1). We confirmed antibody specificity for TDP-43 phosphorylation at Thr-153/Tyr-155 through different analyses. The signal associated with p-T153/Y155-TDP-43 following heat shock was significantly reduced upon TDP-43 knockdown (Fig. 2A). Incubation of the antibody with a TDP-43 peptide (amino acids 148–161) phosphorylated at Thr-153 and Tyr-155 resulted in a loss of the p-T153/Y155-TDP-43-associated signal, whereas the non-phosphorylated control peptide had no effect (Fig. 2B). Furthermore, phosphatase treatment of lysate from heat-shocked cells decreased the levels of the p-T153/Y155-TDP-43-associated signal compared with control (Fig. 2C). To further characterize the specificity of our new antibody for dual phosphorylation at Thr-153 and Tyr-155, we determined p-T153/Y155-TDP-43 activity toward single phosphorylation at Thr-153 or Tyr-155. Immunoblotting competition assays were carried out using peptides phosphorylated at single amino acid residues Thr-153 or Tyr-155 (supplemental Fig. S2, A and B). The peptide phosphorylated at Thr-153 (Thr(P)-153) was unable to block recognition of the p-T153/Y155-TDP-43-associated band. Similar high concentrations of the Tyr-155 phosphorylated peptide (Tyr(P)-155, 0.1 μg/ml) showed partial inhibition of p-T153/Y155-TDP-43 activity. However, blocking the antibody with decreasing concentrations of the Tyr(P)-155 peptide showed no significant competing activity as compared with equal concentrations of the p-T153/Y155 dual phosphorylated peptide (supplemental Fig. S2B). Furthermore, we analyzed whether the heat shock-associated signal could be detected by an antibody generated to recognize single phosphorylation at Tyr-155 (p-Y155-TDP-43). This antibody showed no detection of the p-T153/Y155-TDP-43-associated signal under control or heat shock conditions (supplemental Fig. S2C). Collectively, these results strongly support the specificity of the newly developed p-T153/Y155-TDP-43 antibody for dual TDP-43 phosphorylation at Thr-153 and Tyr-155, and show the regulation of TDP-43 posttranslational modification by the HSR.
FIGURE 1.

Heat shock induces TDP-43 phosphorylation at Thr-153 and Tyr-155. A, schematic representation highlighting TDP-43 domains linked to protein activity and structure: nuclear localization sequence (NLS) at the N terminus, RRM 1 and 2, and the low complexity sequence at the C terminus. Threonine 153 and tyrosine 155 (red) are highlighted in the RRM1 insert. B, ribbon and surface representation of TDP-43 RRM1–2 fragment (amino acids 102–269) bound to a UG-rich RNA molecule (gray) based on an NMR structure of the complex (36). Thr-153 and Tyr-155 are highlighted in red. C, immunoblots of SH-SY5Y cells exposed to heat shock, 43 °C for 30 min; treated with sodium arsenite, 0.5 mm for 1 h (NaAsn); H2O2, 100 μm for 5 h; hydroxyurea, 4 mm for 4 h (HU); and actinomycin D, 5 μg/ml for 3 h (Act D). D, immunoblots of increasing cell lysate derived from control and heat shock-treated SH-SY5Y cells. An antibody recognizing total, phospho-independent TDP-43 was used as control. Tubulin and GAPDH were used as loading control.
FIGURE 2.
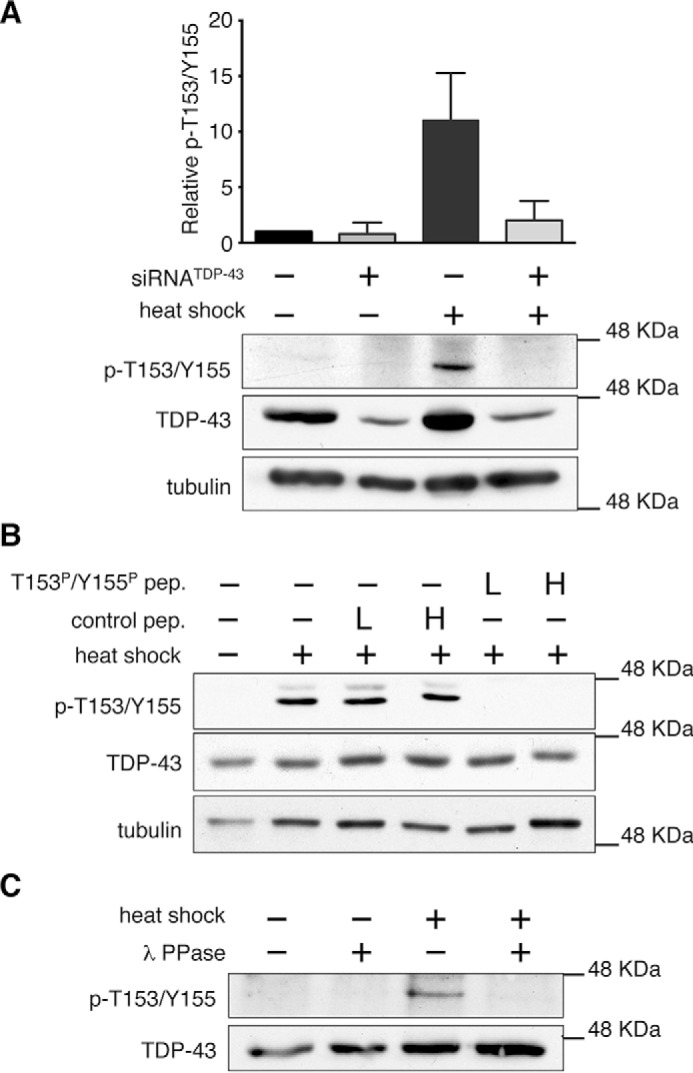
Antibody p-T153/Y155-TDP-43 specifically detects heat shock-mediated TDP-43 phosphorylation. A, immunoblots of HeLa cells treated with TDP-43-specific and control siRNA to compare levels of p-T153/Y155-TDP-43 following heat shock stress. Error bars, S.D. n = 4. B, detection of the heat shock-associated signal in SH-SY5Y cell lysate with p-T153/Y155-TDP-43 antibody blocked with a TDP-43 peptide corresponding to the Thr-153/Tyr-155 region (amino acids 148–161) phosphorylated at Thr-153 and Tyr-155 (T153P/Y155P), or with the corresponding non-phosphorylated peptide, as control. Two concentrations of peptides were used low (L) and high (H), described under “Experimental Procedures.” C, p-T153/Y155-TDP-43 detection of control and λ-phosphatase-treated lysates from control and heat shock-treated SH-SY5Y cells.
To confirm our results and to determine whether phosphorylation at Thr-153/Tyr-155 modifies cellular localization of TDP-43, we characterized the p-T153/Y155-TDP-43 antibody by indirect immunofluorescence analysis. In non-treated cells, the p-T153/Y155-TDP-43-associated signal localized in the nucleolar compartment as dense coil-like structures (Fig. 3). This was observed in different human cell lines, including HeLa, SH-SY5Y (Fig. 3, A and B), U2-OS, and HEK-293 cells (not shown). We confirmed antibody specificity for TDP-43 phosphorylation at Thr-153/Tyr-155 in our immunofluorescence assays by RNAi-mediated knockdown of TDP-43 and competition assays with the Thr-153 and Tyr-155 phosphorylated peptide (amino acids 148–161) as above. Detection of the nucleolar signal was blocked in the presence of the phosphorylated peptide, whereas incubation of the antibody with the non-phosphorylated form of the peptide had no effect (supplemental Fig. S3A). Furthermore, knockdown of TDP-43 via shRNA reduced levels of the nucleolar signal compared with control treated cells (supplemental Fig. S3B). These results suggest that p-T153/Y155 is associated with nucleolar localization of TDP-43 under normal conditions. This is in agreement with previous mass spectrometry detection of TDP-43 in the nucleolar domain of HeLa cells (24). The levels of total TDP-43 recruited to nucleoli appear to be considerably lower compared with the rest of the nucleus, based on the detection of phospho-independent TDP-43 (Fig. 3A). Confocal microscopy analyses of HeLa and SH-SY5Y cells suggest that phosphorylated TDP-43 is part of the dense fibrillar component (DFC) of nucleoli as we observed significant colocalization with fibrillarin, a well established DFC marker (Fig. 3B). Localization of p-T153/Y155 in the nucleolus was maintained following heat shock. Quantification of p-T153/Y155 levels throughout the cell showed an increase in relative fluorescence after heat shock of 50 and 40% in HeLa and HEK-293 cells, respectively (Fig. 3C, supplemental Fig. S4). Further experiments are required to determine whether the increase in phosphorylation during HSR primarily derives from changes in the nucleolar compartment, relative to the rest of the cell.
FIGURE 3.
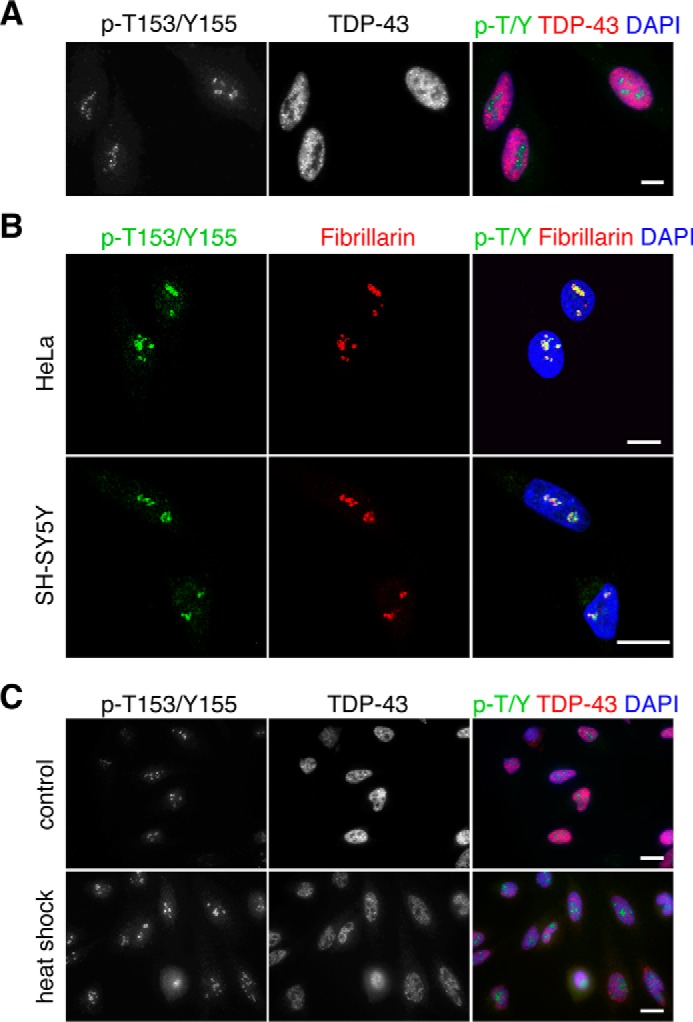
p-T153/Y155-TDP-43 detects nucleolar localization of TDP-43 in the dense coil-like structures. A, fluorescence imaging of non-treated HeLa cells showing total TDP-43 and p-T153/Y155-TDP-43 localization. B, p-T153/Y155 colocalization with the nucleolar marker fibrillarin in HeLa and SH-SY5Y cells as seen by confocal microscopy. C, detection of p-T153/Y155 in HeLa cells upon heat shock compared with control-treated cells. Bars, 10 μm.
TDP-43 Phosphorylation Is Not Associated with Aggregation, Stress Granules, or Protein Clearance
We analyzed TDP-43 aggregation following heat shock to determine whether p-T153/Y155 was associated with changes in protein solubility, and to compare p-T153/Y155 with the two previously determined TDP-43 phosphorylation sites Ser-403/404, Ser-409/410, which are tightly linked to aggregation and pathology (25, 26). Heat shock significantly induced misfolding and accumulation of total TDP-43 in the insoluble fraction following protein extraction from cellular lysate (CL) into RIPA (R) and urea (U) soluble fractions. Immunoblot analysis showed a great reduction of total TDP-43 in the RIPA soluble fraction and a corresponding increase in the urea soluble sample after heat shock (Fig. 4A). In contrast, p-T153/Y155 was present exclusively in the RIPA soluble fraction. Recovery from heat shock by incubation at 37 °C reduced the levels of total TDP-43 in the urea fraction, indicating clearance of misfolded TDP-43 and/or chaperone-assisted refolding of the protein. At the same time, the levels of p-T153/Y155 greatly decreased during recovery. These results are consistent with imaging analysis, which showed no accumulation of p-T153/Y155 into visible aggregates following heat shock (Fig. 3C). Even though total TDP-43 accumulated in the urea fraction following heat shock, we did not observe accumulation of total TDP-43 into cytoplasmic aggregates (Fig. 3C), suggesting that TDP-43 present in the urea fraction after heat shock represents misfolded species that may precede formation of larger, visible aggregates.
FIGURE 4.

p-T153/Y155 is not associated with aggregation, stress granule recruitment, or clearance of TDP-43. A, fractionation of SH-SY5Y cells performed with control, heat shock-treated cells (43 °C for 30 min), and cells allowed to recover at 37 °C for 1 or 2 h (1 hr R, 2 hr R) following heat shock. Immunoblots of the total lysate (CL), RIPA (R), and urea (U) soluble fractions detecting p-T153/Y155 and total TDP-43. Equal volumes of total lysate and RIPA soluble fraction were loaded, whereas the urea fraction is 5-fold more concentrated. B, immunoblots of SH-SY5Y cells treated with the UPS inhibitor MG132 (20 μm for 5 h), heat shock (HS: 43 °C for 30 min), trehalose (100 mm for 24 h), thapsigargin (THP: 1 μm, 2 h), and serum starvation (24 h). C, fluorescence microscopy of control and heat shock-treated HeLa cells detecting p-T153/Y155 and a stress granule marker, TIAR. Arrows indicate TIAR-positive stress granules. D, control and MG132-treated HeLa cells (20 μm,5 h). Arrows indicate TDP-43 cytoplasmic aggregates detected with a phospho-independent antibody. Bars, 10 μm. E, control and trehalose (100 mm, 24 h) treated HeLa cells. Formation of autophagy vesicles (arrow, lower panels) visualized upon transfection with GFP-fused microtubule-associated protein 1A/1B-light chain 3 (LC3). Bars, 25 μm.
To determine whether phosphorylation of Thr-153 and Tyr-155 is associated with pathological inclusions in disease conditions, we analyzed p-T153/Y155 localization in FTLD cases. As positive control for the presence of TDP-43 pathology, we used an antibody recognizing phosphorylated Ser-409/410, which is a well established marker of TDP-43 disease-associated inclusions (25). Immunohistochemical analysis of frontotemporal dementia brain sections showed no significant accumulation of p-T153/Y155 in aggregates (supplemental Fig. S5). These initial observations suggest that, unlike previously identified phosphorylation events (i.e. Ser(P)-403/404 and Ser(P)-409/410), p-T153/Y155 is not linked to the formation of aggregates in FTLD. This is in agreement with our cell-based data suggesting that p-T153/Y155 regulates TDP-43 function and localization and that it is linked to soluble protein under conditions that promote TDP-43 misfolding (Fig. 4A). However, at this time, we cannot exclude the possibility that increased p-T153/Y155 levels, or specific patterns of detection may be observed in different types of TDP-43 proteinopathies.
TDP-43 recruitment to stress granules (SGs) has been observed upon exposure to oxidative and proteotoxic stress (11). To investigate whether phosphorylation mediates recruitment of TDP-43 to SGs upon heat shock, we analyzed the colocalization of p-T153/Y155 with the SG marker T-cell-restricted intracellular antigen-related protein (TIAR). TIAR-positive SGs formed in the cytoplasm following heat shock (Fig. 4C). However, we did not observe recruitment of p-T153/Y155 or phospho-independent TDP-43 to SGs, in agreement with previous reports (27). In summary, our findings indicate that phosphorylation at Thr-153/Tyr-155 is not linked to aggregation or stress granule recruitment of TDP-43. Instead, we find that in conditions that trigger TDP-43 misfolding, such as heat shock, p-T153/Y155 is associated with a soluble conformation of the protein. However, at this time, we cannot exclude the possibility that increased TDP-43 aggregation leads to structures that make the Thr-153/Tyr-155 inaccessible to phosphorylation.
Next, we asked whether increased TDP-43 phosphorylation during HSR occurs upon activation of two pathways that mediate TDP-43 clearance: the ubiquitin proteasome system (UPS) and macroautophagy (autophagy) (28–30). Cells treated with the UPS inhibitor MG132, which induces the accumulation of TDP-43 destined for UPS degradation (28), did not show changes p-T153/Y155 levels (Fig. 4B). Immunofluorescence analysis showed the accumulation of cytoplasmic aggregates of total TDP-43 following MG132 treatment (Fig. 4D), as previously shown (29). However, we observed no colocalization of p-T153/Y155 with these aggregates. We then asked whether p-T153/Y155 mediates autophagy-associated degradation of TDP-43. Cells were exposed to conditions that either increase autophagy (trehalose, serum starvation), or prevent autophagosome-lysosome fusion (thapsigargin). The levels of p-T153/Y155 were unaffected under these conditions (Fig. 4B). Moreover, we observed no colocalization of p-T153/Y155 with microtubule-associated protein light chain 3 (LC-3), a marker of autophagy vesicles (Fig. 4E). These results indicate that phosphorylation at Thr-153/Tyr-155 is not linked to known pathways of TDP-43 clearance.
MEK Regulates TDP-43 Phosphorylation at Thr-153/Tyr-155
The dual specificity mitogen-activated protein/ERK kinase (MEK) (isoforms 1/2, MAP2K1/2) is a prime candidate to phosphorylate at Thr-153/Tyr-155 based on prediction analysis of consensus sequence. Amino acid sequence alignment of the Thr-153/Tyr-155 region with the dual phosphorylation site of the well established MEK substrates ERK1/2 (extracellular signal-regulated kinase) is shown in Fig. 5A. The phosphorylation sites show conservation of the TEY motif, which is unique for MEK substrates compared with dual phosphorylation consensus sequences of other kinases in the MAPK family. The glutamic acid residue in the motif is critical for efficient ERK1/2 phosphorylation by MEK (31). Heat shock treatment dramatically increased activation and phosphorylation of MEK (Fig. 5B), in agreement with previous results (32). This triggered phosphorylation and activation of ERK as well as phosphorylation of a downstream substrate MSK1 (Fig. 5B). We investigated MEK phosphorylation of TDP-43 by use of specific kinase inhibitors, PD184352 and PD98059. Inhibitor treatment prior to heat shock blocked Thr-153/Tyr-155 phosphorylation (Fig. 5C). However, inhibition of the downstream kinase ERK with FR180204 did not decrease Thr-153/Tyr-155 phosphorylation (Fig. 5C). Addition of okadaic acid, which blocks MEK inactivation by inhibiting protein phosphatase PP1/2A, increased p-T153/Y155 levels under control and heat shock conditions (Fig. 5D). This effect was blocked by addition of the MEK inhibitor PD184352. Furthermore, overexpression of a constitutively active mutant of MEK1, GFP-MEK1_DD (S218D/S222D), robustly increased TDP-43 phosphorylation at Thr-153/Tyr-155 in non-treated cells (Fig. 5E). We also observed increased phosphorylation of a hemagglutinin (HA)-tagged TDP-43 expressed in the presence of the constitutively active MEK compared with control (supplemental Fig. S6). Collectively, these findings strongly suggest that TDP-43 is a novel substrate of MEK and that heat shock is not a prerequisite for Thr-153/Tyr-155 phosphorylation by constitutively active MEK.
FIGURE 5.

TDP-43 is specifically phosphorylated by MEK at Thr-153/Tyr-155. A, human TDP-43 amino acid sequence surrounding Thr-153 and Tyr-155 (underlined) aligned with the human ERK1 activation loop sequence where MEK phosphorylates at Thr-202 and Tyr-204 (underlined). B, immunoblots of heat shock and control treated HEK-293 and SH-SY5Y cells. The levels of phospho-independent proteins are shown and tubulin was used as loading control. C, SH-SY5Y cells treated with specific MEK inhibitors PD184352 (10 μm) and PD98059 (50 μm), and with a specific inhibitor of the downstream kinase ERK FR180204 (30 μm). Cells were exposed to heat shock following 1 h of inhibitor treatment. D, SH-SY5Y cells treated with the protein phosphatase PP1/2A inhibitor, okadaic acid (0.5 μm), combined with the MEK inhibitor PD184352 for 1 h before heat shock. E, p-T153/Y155 levels analyzed upon expression of the constitutively active GFP-MEK_DD mutant and a GFP control construct in SH-SY5Y cells in the absence of heat shock.
MEK Phosphorylation at Thr-153 and Tyr-155 Reduces TDP-43 regulation of Splicing
To explore the effect of Thr-153/Tyr-155 phosphorylation on TDP-43 function, we used a reporter of splicing in cells. The well established cystic fibrosis transmembrane conductance regulator (CFTR) exon 9 mini-gene reporter (6, 33) was used in combination with a Thr-153/Tyr-155 phosphomimetic TDP-43 variant (T153E/Y155E). TDP-43 prevents exon 9 splicing through binding to a GU-rich sequence upstream of the 3′ splice site (Fig. 6A) (34). Direct binding of TDP-43 to this sequence recruits additional hnRNPs, in particular hnRNP A2, preventing recognition of the 3′ splice site by steric hindrance (6). Under control conditions, exon 9 showed 26% inclusion and, as expected, siRNA-mediated knockdown of TDP-43 greatly increased exon inclusion (supplemental Fig. S7A) (30). Splicing inhibition was re-established upon expression of siRNA-resistant wild-type TDP-43 (WTsiRes) (6, 33). The double mutant Phe-147 and Phe-149 to Leu (F147L/P149L) was used as control in our assays because of its inability to regulate splicing due to greatly reduced RNA binding (35, 36). As expected, F147L/F149L showed ∼50% loss in activity compared with wild-type (Fig. 6B). We found that the phosphomimetic T153E/Y155E substitutions caused a 30% loss in splicing inhibition compared with wild-type and T153E/Y155A, where the phosphosites were mutated to alanine to prevent phosphorylation (Fig. 6B). Similar assays were carried out to further analyze the effect of Thr-153/Tyr-155 and MEK regulation on TDP-43 activity. Fig. 6C shows CFTR exon 9 splicing activity of control and siRNA-treated cells as a function of MEK_DD expression. The relative TDP-43 activity shown in Fig. 6C is a measure of CFTR exon 9 inclusion levels in each condition compared with WTsiRes cells. In control cells, MEK_DD reduced inhibition of CFTR exon 9 splicing by ∼30% and this effect was lost upon TDP-43 knockdown. These results suggest that MEK_DD phosphorylation of endogenous TDP-43 decreases splicing regulatory activity, as seen with the phosphomimetic mutation (Fig. 6B). Accordingly, the effect of MEK_DD expression on reporter exon splicing was re-established in the presence of WTsiRes TDP-43, where MEK_DD reduced splicing regulation by ∼50%. In contrast, this effect of MEK_DD was significantly diminished upon expression of the siRes, non-phosphorylatable, T153A/Y155A TDP-43 mutant. Collectively, our observations suggest that MEK phosphorylation at Thr-153/Tyr-155 inhibits the TDP-43-mediated regulation of splicing.
FIGURE 6.
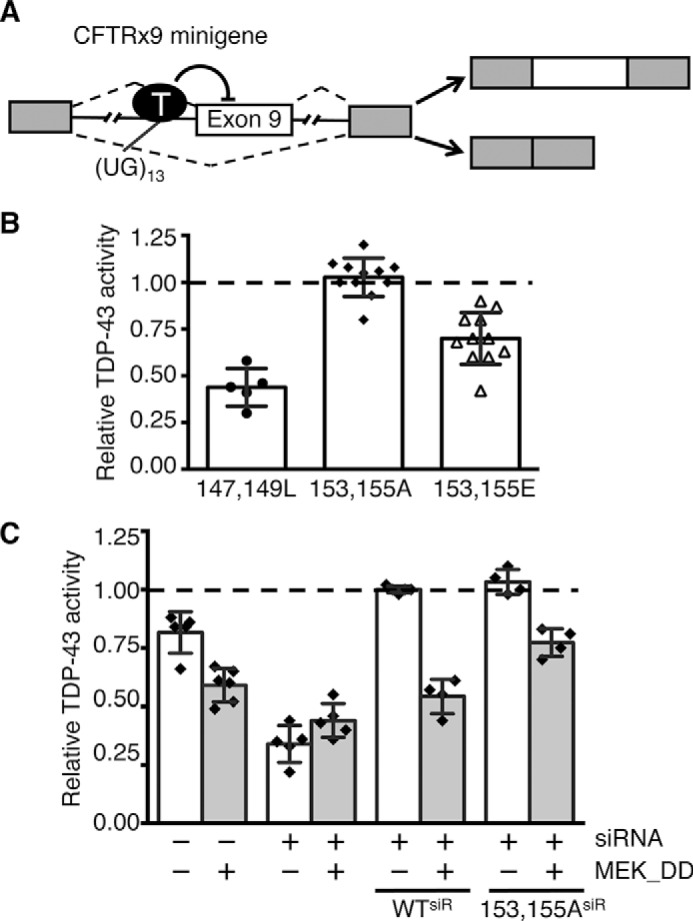
Phosphomimetic substitutions at Thr-153/Tyr-155 modulate TDP-43 activity. A, schematic representation of the CFTR exon 9 mini-gene cellular reporter of TDP-43 activity. Exons and intron regions are represented as boxes and lines, respectively. TDP-43 binding to UG repeats (UG13) near the 3′ splice site inhibits exon 9 inclusion. The two major products of splicing are shown to the right. B, RNAi-resistant (siRes) TDP-43 mutants were expressed in siRNA-treated HeLa cells. Relative TDP-43 activity was calculated comparing wild-type siRes. The effect of the RNA binding-deficient mutant, F147L/F149L, was used as control. Error bars, S.D., n > 5. C, constitutively active MEK_DD, or control GFP vector, expressed in siRNA or control treated cells. Reporter activity was analyzed relative to siRes wild-type (WTsiR). Values represent the averages of independent transfection experiments. Error bars, S.D., n ≥ 4.
To elucidate whether the reduced splicing regulatory activity seen with T153E/Y155E may be caused by changes in RNA binding affinity, we generated recombinant TDP-43 using a bacterial expression system. We expressed and purified TDP-43 fused to a His-SUMO tag at the N terminus to improve yield and protein solubility. Next, we developed a fluorescence-based RNA binding assay to measure the apparent dissociation constant (Kd(app)) of TDP-43-RNA interactions as an alternative to the electromobility shift assay (EMSA). This new method was based on our observations that the intrinsic fluorescence of TDP-43 dramatically decreases upon RNA binding (approximately 50%). Using this assay we obtained Kd(app) = 2.3 ± 0.7 nm for wild-type TDP-43 binding to A(GU)6 with a 1:1 stoichiometry (Fig. 7A, Table 1), in close agreement with values obtained by EMSA (37). As expected, mutations of Phe-147 and Phe-149 (F147L/P149L), previously shown to make direct contacts with GU repeats (35, 36), showed a great reduction in binding affinity (Table 1). We investigated the contribution of tryptophan residues to the changes in TDP-43 intrinsic fluorescence associated with RNA binding to further validate our assays. Removal of the C-terminal (ΔC) or both N- and C-terminal domains (fragment 102–269) did not significantly alter affinity or decrease the change in fluorescence compared with full-length TDP-43 (Table 1). These results indicate that Trp residues in the N and C terminus did not contribute to the fluorescence change linked to RNA binding. Moreover, they are in agreement with previous work showing that the carboxyl- and amino-terminal domains of TDP-43 do not contribute to RNA binding (35). We then substituted the remaining tryptophans at positions 113 and 172, found in RRM1. Substitution of Trp-113 to Ala (W113A) results in the loss of the fluorescence signal change as a function of RNA, whereas W113F reduced the fluorescence change by 50%, but had a modest effect on the Kd(app) (Table 1). Mutations of Trp-172 to either phenylalanine or alanine did not affect binding affinity or fluorescence change in our assays (Table 1). Our findings strongly suggest that changes surrounding the Trp-113 region are principally responsible for the observed decrease in intrinsic fluorescence upon RNA binding. Trp-113 makes sequence-specific contacts with GU-rich RNA and substitutions of this residue reduce binding affinity (36, 37). These interactions, according to our data, result in dramatic changes in intrinsic fluorescence that may be exploited to accurately measure RNA binding affinity.
FIGURE 7.
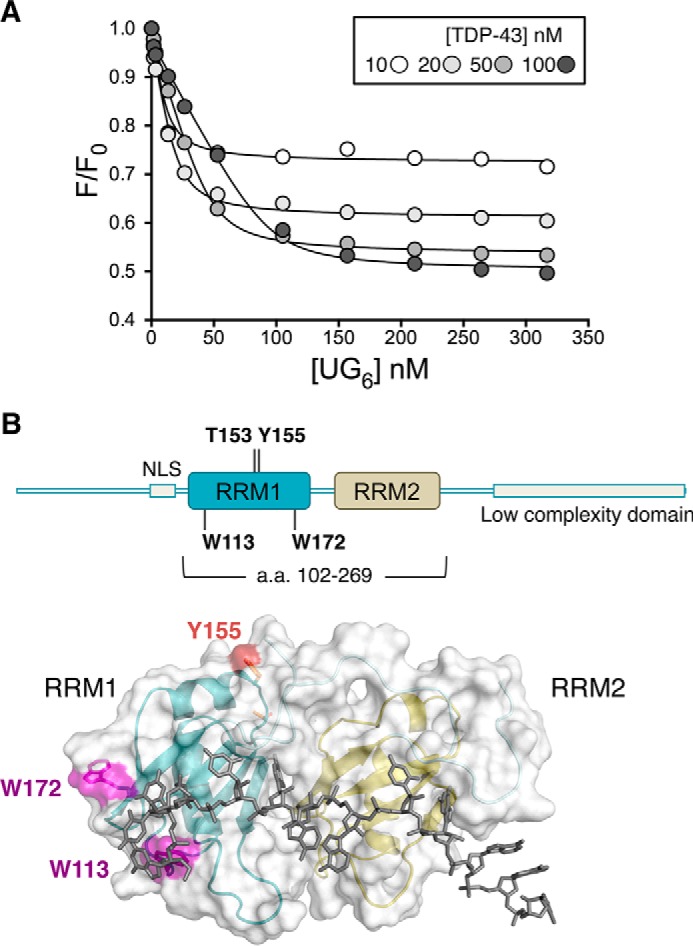
Fluorescence-based assay to measure TDP-43 RNA binding affinity for GU-repeats. A, intrinsic fluorescence (F) of recombinant TDP-43 upon titration of A(UG)6 RNA normalized by the fluorescence in the absence of RNA (F0). Shown are F/F0 values at four different TDP-43 concentrations and data were analyzed according to Equations 1 and 2 under “Experimental Procedures.” The best fit values for the apparent equilibrium dissociation constant, Kd(app), are shown in Table 1. B, schematic representation of full-length TDP-43 highlighting Trp-113 and Trp-172 in RRM1. Lower panel shows the surface and ribbon representation of the 102–269 fragment bound to GU-rich RNA (36). Tryptophan residues in RRM1 are highlighted in magenta.
TABLE 1.
Binding affinities of wild-type and mutant TDP-43
The apparent equilibrium dissociation constant Kd(app) measured for recombinant TDP-43 binding to A(GU)6 RNA.
| Kd(app) | |
|---|---|
| nm | |
| Wild-type | 2.3 ± 0.5 |
| ΔC (aa 1–261) | 2.8 ± 0.6 |
| RRM1–2 (aa 102–269) | 2.2 ± 0.5 |
| RRM1–2 W113A | NDa |
| RRM1–2 W113F | 6.0 ± 0.6 |
| RRM1–2 W172F | 2.0 ± 0.4 |
| RRM1–2 W172A | 5.0 ± 0.6 |
| F147/149L | NDa |
| T153A/Y155A | 3.1 ± 0.6 |
| T153E/Y155E | 10 ± 3 |
a ND, not determined.
Analysis of T153E/Y155E using the fluorescence-based assay showed a 5-fold decrease in A(GU)6 binding affinity in the presence of the phosphomimetic substitutions compared with wild-type, whereas the corresponding alanine mutations did not affect binding (Table 1). These results are consistent with the reduced splicing regulatory function seen in the presence of the phosphomimetic substitutions and strongly suggest that phosphorylation modulates TDP-43 activity.
Discussion
TDP-43 regulates hundreds of genes through different RNA processing mechanisms, in particular through the regulation of splicing (18, 19, 22). TDP-43 cellular localization is highly dynamic and defects in nuclear import are linked to aggregation and neurotoxicity (9, 38, 39). How these various TDP-43 functions are controlled in cells is largely unknown. Previously determined TDP-43 phosphorylation sites at Ser-403/404 and Ser-409/410 are poorly associated with the control of TDP-43 function and are mostly markers of pathological aggregates (25, 26). Here we report dual phosphorylation of TDP-43 at Thr-153/Tyr-155 and provide strong evidence that it is specifically controlled by the MAPK/ERK kinase MEK. This phosphorylation is dramatically up-regulated by the HSR in human cells. The increase in p-T153/Y155 is not associated with greater aggregation or protein clearance under the conditions tested. We find, instead, that p-T153/Y155 remains soluble under conditions that promote TDP-43 misfolding. In support of a functional role of p-T153/Y155, our results show that increased MEK activity significantly alters TDP-43-mediated control of exon splicing in cells. Our observations suggest that the effect of MEK is mediated by phosphorylation at Thr-153/Tyr-155. The decrease in TDP-43 activity may be at least partly explained by a reduction in RNA binding as we find that T153E/Y155E binds GU-rich RNA with 5-fold lower affinity. Future studies should determine whether phosphorylation changes RNA sequence specificity, or whether it affects protein interactions required for TDP-43 regulation of splicing, such as binding to hnRNP A2 (6).
The dramatic up-regulation of TDP-43 phosphorylation by heat shock, along with recent findings on the reduction of TDP-43 aggregation upon heat shock factor 1 (HSF1) activation (40, 41), point to a previously unappreciated role of HSR in TDP-43 homeostasis and posttranslational regulation. In addition to hyperthermic stress, HSR is activated by other types of insults that compromise protein homeostasis (42, 43), and defects in this response are commonly found in human disease including neurodegeneration. Therapies targeting HSR pathways, such as those showing promising effects in ALS (44), may be designed to specifically target TDP-43. We posit that p-T153/Y155 might mediate interactions with chaperones/heat shock proteins that prevent TDP-43 misfolding and aggregation, such as DNAJB2a and Hsp70 (40). Alternatively, it may be associated with an HSR-mediated clearance pathway, different from UPS and autophagy, which prevents aggregate accumulation. We also propose the alternative, tantalizing scenario that TDP-43 might regulate the expression of the HSR target genes whose levels are controlled by alternative splicing during the proteotoxic stress response (45). Based on our findings, this HSR-specific function of TDP-43 may be controlled by phosphorylation at Thr-153/Tyr-155.
MEK is activated upon heat shock and was recently shown to maintain protein homeostasis, at least in part, by phosphorylating and activating HSF1 (32). Previously, the only other recognized MEK substrate was ERK. Now, we provide evidence that TDP-43 is a novel MEK substrate in the presence and absence of heat shock. MEK may not be the only kinase to phosphorylate TDP-43 at Thr-153/Tyr-155, however, we observed no significant reduction of HSR-induced p-T153/Y155 levels upon specific inhibition of MAPK-related dual specificity kinases, namely JNK and p38 kinase (supplemental Fig. S8). Our evidence suggests that cellular processes associated with active MEK, such as proliferation and differentiation, may also induce p-T153/Y155 and regulate TDP-43 function. Based on our findings that p-T153/Y155 is associated with TDP-43 solubility in conditions that trigger protein misfolding, future work should determine whether MEK activation prevents TDP-43 aggregation and blocks TDP-43-associated neurotoxicity.
Our findings showing the specific localization of phosphorylated TDP-43 suggest that nucleolar TDP-43 recruitment is regulated by phosphorylation and may provide new insight into a previously unrecognized role of TDP-43 in cellular function linked to nucleolar RNA processing. In the C9ORF72 hexanucleotide repeat expansion, which is the most common cause of familial ALS and FTD (46, 47), the nucleolar compartment is disrupted by the generation of arginine-rich (GR and PR) dipeptide repeats (48–50). GR and PR are toxic, accumulate in nucleoli, and bind to proteins containing low complexity sequence domains including TDP-43 (51, 52). These findings strongly suggest that one of the principal pathogenic mechanisms in ALS/FTD is abnormal nucleolar function. Future experiments will determine TDP-43 function, particularly the role of p-T513/Y155 in nucleolar-specific RNA processing, and the effect of Arg-rich dipeptide repeats on this function. This may elucidate a convergent process resulting from TDP-43 and C9ORF72-associated dysfunction in ALS/FTD pathogenesis.
Experimental Procedures
Materials
All reagents are from Sigma unless otherwise specified. The human cell lines: neuroblastoma SH-SY5Y, epithelial HeLa, and embryonic kidney HEK-293 cells were purchased from ATCC.
Plasmid Construction
FLAG-tagged wild-type TDP-43, siRNA-resistant construct for mammalian expression was previously described (7). This served as template for site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Agilent Technologies) to generate TDP-43 mutants for mammalian expression. MEK1_DD-GFP (S218D/S222D) was generated by site-directed mutagenesis using the wild-type MEK1-GFP construct (Addgene, 14746) as template. pEGFP-LC3 vector was purchased from Addgene (24920). Constructs for bacterial TDP-43 expression (SUMO-TDP43) were made by cloning TDP-43 in pET-28b/His-SUMO (53) using pQTDPBam_FW and SacTDPend_RV. This construct was the template to generate TDP-43 mutants for bacterial expression through site-directed mutagenesis as above. pQTDPBam_FW and Sac_269_RV; Bam_102_FW and Sac_269_RV were used to clone ΔC-TDP-43 and fragment 102–269, respectively. Oligonucleotides used for mutagenesis and cloning are described in supplemental Table S1. The pLKO vector for TDP-43 down-regulation was generated by ligation of shT2_FW and shT2_RV annealing product into pLKO.1-puro vector digested with AgeI and EcoRI.
Cell Culture, Fractionation, and Splicing Assays
Human cell lines were grown in growth media, Dulbecco's modified Eagle's medium, 4,500 mg/liter of glucose, l-glutamine, and sodium bicarbonate, and supplemented with filtered fetal bovine serum at 10%. For the soluble and insoluble fractions cell pellets were resuspended in RIPA buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 1% Nonidet P-40, 5 mm EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1× protease inhibitor mixture, 1× Phostop Phosphatase inhibitor mixture (Roche Applied Science)). Lysates were sonicated using a Bioruptor Pico (Diagenode) using 10, 30-s on/30-s off, cycles at 4 °C and centrifuged at 40,000 × g for 30 min at 4 °C. The pellet was rinsed with RIPA buffer and the final pellet was resuspended in urea buffer (7 m urea, 2 m thiourea, 4% CHAPS, and 30 mm Tris, pH 8.5). Stable HEK-293 cells expressing HA-tagged TDP-43 under tetracycline induction (7) were used for immunoprecipitation experiments as previously described (54). RNAi-mediated down-regulation of TDP-43 was carried out as previously described (33) and siCONTROL Nontargetting siRNA#1 (Dharmacon) was used as control. shRNA-mediated down-regulation of TDP-43 was carried out by generating lentiviral particles in HEK-293T cells using a pLKO vector system (pCMV8.2ΔR, pCMV-VSV-G, generous gifts of Susana Gonzalo's lab, St. Louis University). shRNA luciferase lentiviral particles were generated for control transduction. HEK-293T cells were transfected with Lipofectamine 2000 (Life Technologies) in Opti-MEM Reduced Serum Media (Life Technologies) according to manufacturer's protocols. After 6 h, media was replaced with growth media and cells were incubated at 37 °C, 5% CO2 for 48 h. After 48 h, the lentivirus-containing media was collected and filter sterilized with a 0.45-μm filter. HeLa cells were transduced with control and TDP-43-shRNA lentiviral particles with Polybrene transfection reagent. TDP-43 knockdown clones were generated by puromycin selection (1 μg/ml) 48 h after transduction. TDP-43 down-regulation was confirmed by immunoblotting.
Splicing assays were carried out in HeLa cells transiently transfected with Lipofectamine 2000 and Oligofectamine in the case of siRNA (Life Technologies) according to the manufacturer's protocols. Assays were performed using the CFTR reporter mini-gene as previously described (6, 33). Briefly, following two transfections with control or siRNA TDP-43 (0.1 nmol) in a 6-well format, cells were co-transfected with FLAG-TDP43 siRNA-resistant constructs of wild-type or mutant TDP-43 and the CFTR reporter (0.3 μg each) (17). RNA was extracted after 24 h and RT-PCR analysis was performed to compare the levels of exon inclusion/exclusion. PCR products were quantified with ImageJ.
Phosphatase Treatment
Cell lysates containing ∼30 μg of total protein were incubated with 400 units of λ-protein phosphatase (New England Biolabs) for 1 h at 37 °C according to the manufacturer's instructions.
Peptide Competition
p-T153/Y155 antibody was incubated with a peptide corresponding to amino acids 148–161 of TDP-43 phosphorylated at positions Thr-153 and/or Tyr-155. Non-phosphorylated peptide was used as a control. Prior to immunoblotting or immunofluorescence 1 mg/ml of p-T153/Y155-TDP-43 antibody was incubated with 0.02 μg/ml (low) and 1 μg/ml (high) peptide for 1 h at 25 °C.
Immunoblotting and Immunofluorescence Microscopy
Cell pellets were resuspended in lysis buffer (0.25 m NaCl, 15 mm Hepes, pH 7.5, 0.5% Nonidet P-40, 10% glycerol, 1× protease inhibitor mixture, 1× Phostop Phosphatase inhibitor mixture (Roche Applied Science)). Approximately 30 μg of total protein were loaded for immunoblot analyses, unless otherwise specified. Samples were measured by Bradford assays. Proteins were separated by SDS-PAGE and transferred to nitrocellulose (0.22 μm, GE Healthcare). Membranes were blocked with 5% BSA/TBS-Tween and immunodetection was carried out by enhanced chemiluminescence (ECL, Pierce, ThermoFisher Scientific). Antibodies against phospho-independent TDP-43 were used to compare p-T153/Y155 levels with total TDP-43. Either tubulin or GAPDH was used as the loading control. Indirect immunofluorescence was carried out as previously described (17). Cells were observed on a Leica DM5000B microscope equipped with the DFC350FX digital camera (Leica Microsystems Inc.). A ×63/1.4 oil immersion objective was used for confocal microscopy studies on a TCS SP5 microscope (Leica) using the LAS AF software.
Immunohistochemistry
We studied FTLD cases with TDP-43 pathology and age-matched controls from the Northwestern Neuropathology Core Tissue Bank (NADC AG13854). Frontal cortex and dentate gyrus sections were immunostained according to previously described protocols (55) with p-T153/Y155-TDP-43. pS409/410 (Cosmo Bio USA) antibody was used as control.
Expression and Purification of Recombinant TDP-43
Recombinant TDP-43 was expressed in BL21(DE3) Escherichia coli cells from the SUMO-TDP43 plasmid. Induction was carried out with 0.3 mm isopropyl β-d-thiogalactopyranoside and cells were grown at 25 °C for 4 h. Cell pellets were resuspended in lysis buffer (500 mm NaCl, 50 mm Tris, pH 8.0, 1 mm Tris(2-carboxyethyl)phosphine, 10% sucrose, 10% glycerol, 5 mm CHAPS, 0.1% sodium deoxycholate, EDTA-free protease inhibitor mixture, 2.5 μg/ml of lysozyme, 10 μg/ml of DNase I). Lysates were centrifuged for 20 min at 20,000 × g at 4 °C following sonication on ice. Protein was isolated on nickel-nitrilotriacetic acid resin (McLab) by affinity chromatography (AKTA-Start, GE Healthcare). The column was washed with Buffer A: 500 mm NaCl, 50 mm Tris, pH 8.0, 10% sucrose, 10% glycerol, 50 mm imidazole. Recombinant TDP-43 was eluted with Buffer A and 300 mm imidazole. The SUMO tag was removed with Ulp1 protease at a 1:1,000 (w/w) ratio as described (53). Ulp1 was generously provided by Sergey Korolev's group. All steps were carried out at 4 °C.
Fluorescence Titration RNA Binding Assays
TDP-43 intrinsic fluorescence was measured upon titration with increasing concentrations of A(GU)6 RNA on a Fluoromax-4 spectrofluorometer (Horiba Scientific). Fluorescence readings were performed in a 1-ml cuvette at four different recombinant TDP-43 concentrations in the range of 10–100 nm in the presence of 200 mm NaCl, 20 mm Tris, pH 8.0, 0.1% PEG-8000, 2% glycerol, at 25 °C. Protein was excited at 280 nm (slits 5/10, exposure time of 0.02 s), and emission was recorded at 340 nm. We observed no significant photobleaching under these conditions. Data were analyzed with Origin (version 8.1, OriginLab) according to Equation 1,
| (Eq. 1) |
where F is fluorescence, F0 is F in the absence of RNA, ΔFmax is the total change in F at saturation.
| (Eq. 2) |
Equation 2 describes the fractional saturation in terms of the apparent equilibrium dissociation constant Kd(app), the total concentration of TDP-43 (Tt), total RNA concentration (Rt), and TDP-43:RNA binding stoichiometry, N. Four TDP-43-RNA binding curves were analyzed simultaneously to obtain Kd(app) and N.
Antibodies
Immunoblots and indirect immunofluorescence were performed with: rabbit polyclonal and mouse monoclonal anti-TDP-43 (ProteinTech 10782-2-AP, Abcam, ab109535), anti-TIAR (BD Biosciences, 610352), anti-fibrillarin (Abcam, ab4566), mouse anti-tubulin (Andres Muro, Trieste, Italy), anti-GAPDH (Abcam, ab181602), anti-MEK (Abcam, ab178876), anti-phospho-MEK1/2 (Cell Signaling, number 9154), anti-ERK1/2 (Cell Signaling, number 4695), anti-phospho-ERK1/2 (Cell Signaling, number 4370), anti-MSK1 (Cell Signaling, number 3489), and anti-phospho-MSK1 (Cell Signaling, number 9595). The rabbit polyclonal antibodies recognizing p-T153/Y155 and p-Y155-TDP-43 were produced by 21st Century Biochemicals, MA. Peptide including TDP-43 amino acids 148–161 phosphorylated at either Thr-153/Tyr-155 or Tyr-155 served as epitope for antibody production. Antibody specificity was enhanced by affinity purification to select for binding to phosphorylated Thr-153/Tyr-155 or Tyr-155, but not the corresponding non-phosphorylated peptides.
Author Contributions
W. L., A. N. R., B. L., P. S., E. E. F., C. R. K., R. L. F., E. H. B., and Y. M. A. designed the research, performed the experiments and analyzed the data. Y. M. A. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Enrico Di Cera and collegues for helpful discussions on the design and analysis of the fluorescent-based RNA binding assays; Olga Koroleva for valuable advice on recombinant protein production; Tom Misteli for comments and suggestions on the manuscript; and Alessandro Vindigni for comments and helpful discussions.
This work was supported in part by the National Institutes of Health, NINDS Grant K01 NS082391 and St. Louis University Presidential Research Fund Grant 230175. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S8 and Table S1.
- FTLD
- frontotemporal lobar degeneration
- RRM
- RNA recognition motif
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- TDP-43
- TAR DNA-binding protein
- HSR
- heat shock response
- DFC
- dense fibrillar component
- SG
- stress granule
- TIAR
- T-cell-restricted intracellular antigen-related protein
- UPS
- ubiquitin proteasome system
- CFTR
- cystic fibrosis transmembrane conductance regulator
- HSF
- heat shock factor.
References
- 1. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., and Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 2. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., and Lee V. M. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 3. Uryu K., Nakashima-Yasuda H., Forman M. S., Kwong L. K., Clark C. M., Grossman M., Miller B. L., Kretzschmar H. A., Lee V. M., Trojanowski J. Q., and Neumann M. (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 67, 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Josephs K. A., Murray M. E., Whitwell J. L., Parisi J. E., Petrucelli L., Jack C. R., Petersen R. C., and Dickson D. W. (2014) Staging TDP-43 pathology in Alzheimer's disease. Acta Neuropathol. 127, 441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buratti E. (2015) Functional significance of TDP-43 mutations in disease. Adv. Genet. 91, 1–53 [DOI] [PubMed] [Google Scholar]
- 6. D'Ambrogio A., Buratti E., Stuani C., Guarnaccia C., Romano M., Ayala Y. M., and Baralle F. E. (2009) Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 37, 4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ayala Y. M., De Conti L., Avendaño-Vazquez S. E., Dhir A., Romano M., D'Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E., and Baralle F. E. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fuentealba R. A., Udan M., Bell S., Wegorzewska I., Shao J., Diamond M. I., Weihl C. C., and Baloh R. H. (2010) Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 285, 26304–26314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., and Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 10. Tsuiji H., Iguchi Y., Furuya A., Kataoka A., Hatsuta H., Atsuta N., Tanaka F., Hashizume Y., Akatsu H., Murayama S., Sobue G., and Yamanaka K. (2013) Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 5, 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dewey C. M., Cenik B., Sephton C. F., Johnson B. A., Herz J., and Yu G. (2012) TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res. 1462, 16–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Y. R., King O. D., Shorter J., and Gitler A. D. (2013) Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feiguin F., Godena V. K., Romano G., D'Ambrogio A., Klima R., and Baralle F. E. (2009) Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592 [DOI] [PubMed] [Google Scholar]
- 14. Schmid B., Hruscha A., Hogl S., Banzhaf-Strathmann J., Strecker K., van der Zee J., Teucke M., Eimer S., Hegermann J., Kittelmann M., Kremmer E., Cruts M., Solchenberger B., Hasenkamp L., van Bebber F., et al. (2013) Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. U.S.A. 110, 4986–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sephton C. F., Good S. K., Atkin S., Dewey C. M., Mayer P. 3rd, Herz J., and Yu G. (2010) TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 285, 6826–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiang P. M., Ling J., Jeong Y. H., Price D. L., Aja S. M., and Wong P. C. (2010) Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. U.S.A. 107, 16320–16324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ayala Y. M., Misteli T., and Baralle F. E. (2008) TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. U.S.A. 105, 3785–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Polymenidou M., Lagier-Tourenne C., Hutt K. R., Huelga S. C., Moran J., Liang T. Y., Ling S. C., Sun E., Wancewicz E., Mazur C., Kordasiewicz H., Sedaghat Y., Donohue J. P., Shiue L., Bennett C. F., Yeo G. W., and Cleveland D. W. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tollervey J. R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., König J., Hortobágyi T., Nishimura A. L., Zupunski V., Patani R., Chandran S., Rot G., Zupan B., Shaw C. E., and Ule J. (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buratti E., and Baralle F. E. (2010) The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 7, 420–429 [DOI] [PubMed] [Google Scholar]
- 21. Alami N. H., Smith R. B., Carrasco M. A., Williams L. A., Winborn C. S., Han S. S., Kiskinis E., Winborn B., Freibaum B. D., Kanagaraj A., Clare A. J., Badders N. M., Bilican B., Chaum E., Chandran S., et al. (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ling J. P., Pletnikova O., Troncoso J. C., and Wong P. C. (2015) TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 24. Boisvert F. M., Ahmad Y., Gierlinski M., Charriere F., Lamont D., Scott M., Barton G., and Lamond A. I. (2012) A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell Proteomics 11, M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neumann M., Kwong L. K., Lee E. B., Kremmer E., Flatley A., Xu Y., Forman M. S., Troost D., Kretzschmar H. A., Trojanowski J. Q., and Lee V. M. (2009) Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F., Morita M., Nakano I., Oda T., Tsuchiya K., and Akiyama H. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Udan-Johns M., Bengoechea R., Bell S., Shao J., Diamond M. I., True H. L., Weihl C. C., and Baloh R. H. (2014) Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 23, 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scotter E. L., Vance C., Nishimura A. L., Lee Y. B., Chen H. J., Urwin H., Sardone V., Mitchell J. C., Rogelj B., Rubinsztein D. C., and Shaw C. E. (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 127, 1263–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang C. C., Bose J. K., Majumder P., Lee K. H., Huang J. T., Huang J. K., and Shen C. K. (2014) Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 127, 3024–3038 [DOI] [PubMed] [Google Scholar]
- 30. Wang I. F., Guo B. S., Liu Y. C., Wu C. C., Yang C. H., Tsai K. J., and Shen C. K. (2012) Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U.S.A. 109, 15024–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Butch E. R., and Guan K. L. (1996) Characterization of ERK1 activation site mutants and the effect on recognition by MEK1 and MEK2. J. Biol. Chem. 271, 4230–4235 [DOI] [PubMed] [Google Scholar]
- 32. Tang Z., Dai S., He Y., Doty R. A., Shultz L. D., Sampson S. B., and Dai C. (2015) MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160, 729–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ayala Y. M., Pagani F., and Baralle F. E. (2006) TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett. 580, 1339–1344 [DOI] [PubMed] [Google Scholar]
- 34. Buratti E., Dörk T., Zuccato E., Pagani F., Romano M., and Baralle F. E. (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buratti E., and Baralle F. E. (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- 36. Lukavsky P. J., Daujotyte D., Tollervey J. R., Ule J., Stuani C., Buratti E., Baralle F. E., Damberger F. F., and Allain F. H. (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449 [DOI] [PubMed] [Google Scholar]
- 37. Ayala Y. M., Pantano S., D'Ambrogio A., Buratti E., Brindisi A., Marchetti C., Romano M., and Baralle F. E. (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 348, 575–588 [DOI] [PubMed] [Google Scholar]
- 38. Bentmann E., Neumann M., Tahirovic S., Rodde R., Dormann D., and Haass C. (2012) Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., and Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen H. J., Mitchell J. C., Novoselov S., Miller J., Nishimura A. L., Scotter E. L., Vance C. A., Cheetham M. E., and Shaw C. E. (2016) The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain 149, 1417–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin P. Y., Folorunso O., Taglialatela G., and Pierce A. (2016) Overexpression of heat shock factor 1 maintains TAR DNA binding protein 43 solubility via induction of inducible heat shock protein 70 in cultured cells. J. Neurosci. Res. 94, 671–682 [DOI] [PubMed] [Google Scholar]
- 42. Lindquist S. (1986) The heat-shock response. Annu. Rev. Biochem. 55, 1151–1191 [DOI] [PubMed] [Google Scholar]
- 43. Morimoto R. I. (2011) The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harbor Symp. Quant. Biol. 76, 91–99 [DOI] [PubMed] [Google Scholar]
- 44. Kalmar B., Lu C. H., and Greensmith L. (2014) The role of heat shock proteins in amyotrophic lateral sclerosis: the therapeutic potential of Arimoclomol. Pharmacol. Ther. 141, 40–54 [DOI] [PubMed] [Google Scholar]
- 45. Shalgi R., Hurt J. A., Lindquist S., and Burge C. B. (2014) Widespread inhibition of posttranscriptional splicing shapes the cellular transcriptome following heat shock. Cell Rep. 7, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 46. DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G. Y., Karydas A., et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Renton A. E., Majounie E., Waite A., Simón-Sanchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gendron T. F., Bieniek K. F., Zhang Y. J., Jansen-West K., Ash P. E., Caulfield T., Daughrity L., Dunmore J. H., Castanedes-Casey M., Chew J., Cosio D. M., van Blitterswijk M., Lee W. C., Rademakers R., Boylan K. B., Dickson D. W., and Petrucelli L. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zu T., Liu Y., Bañez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T. M., Harms M. B., Falchook A. E., Subramony S. H., Ostrow L. W., Rothstein J. D., Troncoso J. C., and Ranum L. P. (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U.S.A. 110, E4968–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mori K., Weng S. M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H. A., Cruts M., Van Broeckhoven C., Haass C., and Edbauer D. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 [DOI] [PubMed] [Google Scholar]
- 51. Lee K. H., Zhang P., Kim H. J., Mitrea D. M., Sarkar M., Freibaum B. D., Cika J., Coughlin M., Messing J., Molliex A., Maxwell B. A., Kim N. C., Temirov J., Moore J., Kolaitis R. M., et al. (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788.e717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin Y., Mori E., Kato M., Xiang S., Wu L., Kwon I., and McKnight S. L. (2016) Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167, 789–802.e712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yunus A. A., and Lima C. D. (2009) Purification of SUMO conjugating enzymes and kinetic analysis of substrate conjugation. Methods Mol. Biol. 497, 167–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Glatter T., Wepf A., Aebersold R., and Gstaiger M. (2009) An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol. 5, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bigio E. H., Mishra M., Hatanpaa K. J., White C. L. 3rd, Johnson N., Rademaker A., Weitner B. B., Deng H. X., Dubner S. D., Weintraub S., and Mesulam M. (2010) TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol. 120, 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.