

Abstract
The prototypical PrP prion diseases are invariably fatal, and the search for agents to treat them spans more than 30 years, with limited success. However, in the last few years, the application of high-throughput screening, medicinal chemistry, and pharmacokinetic optimization has led to important advances. The PrP prion inoculation paradigm provides a robust assay for testing therapeutic efficacy, and a dozen compounds have been reported that lead to meaningful extension in survival of prion-infected mice. Here, we review the history and recent progress in the field, focusing on studies validated in animal models. Based on screens in cells infected with mouse-passaged PrP prions, orally available compounds were generated that double or even triple the survival of mice infected with the same prion strain. Unfortunately, no compounds have yet shown efficacy against human prions. Nevertheless, the speed of the recent advances brings hope that an effective therapeutic can be developed. A successful treatment for any neurodegenerative disease would be a major achievement, and the growing understanding that the more common neurodegenerative diseases, including Alzheimer's and Parkinson's, progress by an analogous prion mechanism serves to highlight the importance of antiprion therapeutics.
The use of high-throughput screening, medicinal chemistry, and pharmacokinetic optimization has led to therapeutics for prion-infected mice. These advances bring hope that effective therapeutics can be developed for humans as well.
BACKGROUND
Human PrP Prion Diseases
The prototypical PrP prion diseases are rapidly progressive and fatal neurodegenerative disorders. They are caused by the conformational change of the cellular prion protein (PrPC) into an alternative form, termed PrPSc, which then templates further misfolding of PrPC. This induced conformational self-propagation is now understood to play a central role in many neurodegenerative diseases including Alzheimer's and Parkinson's (Prusiner 2012; Walker and Jucker 2015). Unique among neurodegenerative disorders, PrP prion diseases are also found in commercially important animal species, including scrapie in sheep, chronic wasting disease (CWD) in deer, and bovine spongiform encephalopathy (BSE) in cattle.
Most human PrP prion diseases have no clear origin and are considered sporadic. They occur from an initial stochastic conversion of PrPC to PrPSc, which typically leads to sporadic Creutzfeldt–Jakob disease (sCJD). A polymorphism at residue 129 of human PRNP results in methionine (M) or valine (V), with an allele frequency of 0.6/0.4. Heterozygosity is protective; MM and VV homozygous individuals account for almost 90% of sCJD cases (Parchi et al. 1999). More than 60 mutations have been identified in the open reading frame of the PRNP gene that encodes PrP (Minikel et al. 2016), which increase the likelihood of stochastic PrPC to PrPSc conversion and account for the ∼15% of CJD cases with a genetic etiology. Different mutations produce distinct phenotypes, such as with Gerstmann–Sträussler–Scheinker syndrome, fatal familial insomnia, or familial CJD. Intriguingly, a relatively small number of PrP prion disease cases have been identified with an infectious etiology, in which the refolding cascade is initiated by exogenous PrPSc. Infection may occur from consumption of prion-infected tissue, as with endocannibalism, which led to the spread of kuru among the Fore people of Papua New Guinea (Collinge et al. 2006), and BSE-contaminated cattle, which led to the variant CJD epidemic in Europe (Diack et al. 2014). Although variant CJD only manifests clinically in MM homozygous individuals, estimates of nonsymptomatic carriers are as high as one in 2000 in the British population (Gill et al. 2013). In addition, a number of well-documented CJD cases have an iatrogenic origin, the vast majority of these stemming from the use of cadaver-derived tissues that, without clinicians’ knowledge, contained CJD prions. Additionally, a handful of cases have also occurred from the reuse of incompletely decontaminated surgical instruments (Haïk and Brandel 2014).
Experimental Models
The observation that the neuropathology of kuru resembled that of CJD (Klatzo et al. 1959) and scrapie (Hadlow 1959) led Hadlow to suggest that kuru may be transmissible to primates, as sheep scrapie had been shown to be transmissible to goats (Cuillé and Chelle 1939). Inoculation experiments with human PrP prions were subsequently performed by Carleton Gajdusek and colleagues, demonstrating that kuru (Gajdusek et al. 1966) and CJD (Gibbs et al. 1968) could be transmitted experimentally.
However, it was the transmission of sheep scrapie to mice (Chandler 1961) by intracerebral inoculation of brain homogenate that generated experimentally tractable models for the study of prions in the laboratory. The Chandler isolate was transferred to the Rocky Mountain Laboratories and was serially passaged in mice and widely distributed (it has subsequently been referred to by the acronym RML). Intracerebral inoculation of prion-infected brain homogenate was shown to be the most rapid model of transmission. However, even then the disease is characterized by an asymptomatic phase that can last many months, followed by a rapidly progressive illness. Despite these long incubation periods, disease onset is highly synchronous within a cohort of mice inoculated at the same time. Mice typically show clinical signs within a few days of one another. Transmission of prions between species is generally inefficient, often accompanied by extended incubation periods and only a portion of animals succumbing to disease. The self-templating mechanism of prion propagation implies that prions have the primary sequence of the species in which they were last passaged. Subsequent serial transmission, in which the inoculum and host PrP sequence are the same, typically progresses faster, with all animals succumbing to disease.
Prion-infected mice are characterized by multiple metrics: incubation period (time to onset of clinical disease); biochemical resistance of brain tissue to proteinase K (PK) digestion and guanidine hydrochloride (GdnHCl) denaturation; and neuropathologically by intensity and distribution of vacuolation and by immunohistochemistry for PrP and the glial fibrillary acidic protein (GFAP). One early finding of mouse-passaged prions was that there appeared to be multiple prion “strains,” each producing a characteristic incubation period and neuropathological and biochemical signatures that remained constant upon serial transmission. For many years, this phenomenon was used to justify a nucleic acid component to the prion, but none was ever found (Safar et al. 2005). Strains are now understood to arise from alternative conformations of PrPSc (Bessen and Marsh 1994; Telling et al. 1996). Although some strain characteristics are retained upon interspecies transmission, others can differ.
The infection of mouse neuroblastoma cells with mouse-passaged sheep scrapie (Race et al. 1987; Butler et al. 1988; Race et al. 1988) provided tools that could be used in assays. Subsequently, other prion-infected mouse cell lines were developed, including GT1 (Schätzl et al. 1997), 3T3 (Vorberg et al. 2004), and LD9 and CAD5 (Mahal et al. 2007); however, in all cases, cell lines are limited in the range of prion strains that they propagate.
Although a handful of mouse-passaged human prion strains were developed in mice and used to infect cells, none were shown to fully maintain the properties of the original human prions (Giles et al. 2017). No cell line has yet been developed for the direct propagation of human prions, but transgenic (Tg) mouse models have been established. Mice expressing transgenes chimeric for human and mouse PrP are susceptible to CJD prions, whereas in those with a human PrP transgene, CJD infection was only successful when the endogenous mouse PrP was ablated (Telling et al. 1994, 1995). These initial chimeric lines retained nine human PrP residues, and systematic reversion of these residues enabled the development of Tg mice with shorter incubation periods for CJD prions (Korth et al. 2003; Giles et al. 2010, 2012).
HISTORY OF PRION THERAPEUTICS
Because the central nervous system (CNS) is the site of cellular dysfunction in prion diseases, potential therapeutic compounds must either cross the blood–brain barrier (BBB) or be delivered by intraventricular or intrathecal means.
Polyanions
Early attempts to treat prion diseases used a variety of large polyanionic compounds, including the polyoxometalate HPA-23 (Kimberlin and Walker 1983) and the sulfated glycans dextran sulfate 500 (DS500) and pentosan polysulfate (PPS) (Ehlers and Diringer 1984). In all cases, survival was only extended in peripherally inoculated (but not intracerebrally inoculated) mice and even then only when the treatment was given prophylactically or shortly after inoculation. Additional prophylactic studies were performed with PPS, showing differences in efficacy with inoculum titer, mouse background, and prion strain (Diringer and Ehlers 1991; Farquhar et al. 1999). However, it was only following a comprehensive set of studies using intraventricular delivery of PPS by the Katsumi Doh-ura group (2004) that led to the progression of PPS into the clinic. In a Tg mouse overexpressing hamster PrP, intraventricular PPS administered 10 d after inoculation (∼20% of the untreated incubation period) more than doubled the survival of prion-infected mice. Even an intervention at as late as 70% of the untreated incubation period produced a significant increase in survival (Doh-ura et al. 2004). In the clinical setting, intraventricular PPS was provided on a compassionate basis to a number of CJD patients. Although there was no apparent improvement in the clinical progression of the disease, a number of patients did survive longer than expected (Bone et al. 2008; Tsuboi et al. 2009; Newman et al. 2014). However, extending the late stages of disease when patients were typically in a vegetative state is of questionable benefit.
Small-Molecule Therapeutics
Early therapeutic studies were based on the assumption that the infectious agent in prion diseases was a virus, and the efficacy of various polyanions may have been nonspecific. A more rational route to drug design came with the understanding of the prion concept. Congo red (Fig. 1A) was known to bind to purified prions (Prusiner et al. 1983) and was found to inhibit prion replication in cell culture (Caughey and Race 1992). In prion-inoculated hamsters, Congo red produced a small increase in survival, most notably following peripheral inoculation (Ingrosso et al. 1995). Based on the anionic sulfate esters present on DS500, PPS, and Congo red, phthalocyanine sulfonates were tested and found to be effective in prion-infected cells, as were ferric porphyrin derivatives with two pendant propionate groups (Fig. 1B) (Caughey et al. 1998). These compounds were subsequently shown to extend survival in vivo, but only in peripherally inoculated mice when dosing was started immediately after inoculation (Priola et al. 2000).
Figure 1.

Chemical structures of compounds used in early prion efficacy studies. (A) Congo red, (B) deuteroporphyrin IX 2,4-bis-(ethylene glycol) iron(III), (C) quinacrine, and (D) Compound B.
Because the BBB restricts the access of many molecules to the CNS, we and others screened compounds known to cross the BBB for their efficacy in prion-infected cells, which led to the identification of quinacrine (Fig. 1C) (Doh-ura et al. 2000; Korth et al. 2001). In two mouse neuroblastoma cell lines, the effective concentration for half-maximal inhibition (EC50) of quinacrine was ∼0.3 µM (Doh-ura et al. 2000; Korth et al. 2001). Preliminary studies of quinacrine intervention in prion-infected wild-type (WT) mice showed no extension in survival (Collins et al. 2002; Barret et al. 2003; Doh-ura et al. 2004). Pharmacokinetic analysis subsequently revealed that, depending on dosing paradigm, quinacrine concentrations in the brain only reached ∼1 µM (Yung et al. 2004; Ghaemmaghami et al. 2009; Ahn et al. 2012).
Because quinacrine had been used safely for decades as an antimalarial drug, clinical trials in patients were initiated in parallel with the mouse studies. A trial in France consisted of a series of compassionate use cases (Haïk et al. 2004). A second study in the United Kingdom was an open-label, patient-preference trial (Collinge et al. 2009), and a third in the United States was a placebo-controlled randomized clinical trial (Geschwind et al. 2013). In each case, survival was the end point, but none of the trials yielded a significant difference upon quinacrine treatment.
The restricted penetration of quinacrine through the BBB was determined to be due to active efflux by the P-glycoprotein (P-gp) (Dohgu et al. 2004; Huang et al. 2006). P-gp is also known as multidrug resistance protein 1 (MDR1) in human (of which the mouse has two homologs, Mdr1a and Mdr1b). Short-term dosing of quinacrine in mice in which both Mdr1 proteins had been knocked out, termed Mdr10/0, showed brain levels up to 50-fold higher than WT mice (Huang et al. 2006). Longer-term dosing of quinacrine in Mdr10/0 mice produced brain levels up to 100 µM without any overt signs of toxicity (Ghaemmaghami et al. 2009; Ahn et al. 2012). However, multiple quinacrine treatment paradigms in Mdr10/0 mice failed to extend survival in prion-infected mice (Ghaemmaghami et al. 2009).
More careful analysis suggested a reason for quinacrine's failure. Mdr10/0 mice infected with the RML prion strain were dosed with quinacrine at 40 mg/kg per day starting at 60 days postinoculation (dpi). By 75 dpi, mouse brains had significantly lower levels of PrPSc, as measured by western blot and enzyme-linked immunosorbent assay (ELISA), and greatly reduced GFAP immunoreactivity in brain slices compared with untreated mice. However, the reduction was transient. By 90 dpi, treated and untreated mice were indistinguishable biochemically and neuropathologically, and survival was ultimately not extended (Fig. 2). However, analysis of the brains of quinacrine-treated mice showed subtle differences from controls; there was a change in the susceptibility to denaturation with GdnHCl and in the ability of PrPSc conformers to differentially bind anti-PrP antibodies before and after denaturation as determined by a conformation-dependent immunoassay. Together, these findings suggest that quinacrine treatment may have altered the prion strain (Ghaemmaghami et al. 2009).
Figure 2.

PrPSc levels in the brains of RML-infected Mdr10/0 mice treated with quinacrine at 40 mg/kg per day for 60–90 days postinoculation (n = 3 for each point, shown as mean and standard error). (Figure reprinted from Ghaemmaghami et al. 2009, with permission from PLoS under the Creative Commons Attribution license.)
A series of biaryl hydrazone compounds that showed efficacy inhibiting Alzheimer's disease β-amyloid formation were tested in ScN2a cells, mouse N2a cells infected with the RML prion strain of mouse-passaged sheep scrapie (Butler et al. 1988). One compound, termed Compound B (CmpdB) (Fig. 1D), had cellular EC50 values of 60 pM in ScN2a cells and ∼300 nM in two ScN2a cell lines overexpressing PrP. Long-term dosing with CmpdB doubled survival of RML-infected mice. Lower efficacy was observed against other prion strains in cell culture, and these results were replicated in vivo (Kawasaki et al. 2007).
The studies on CmpdB marked a turning point in prion drug discovery efforts. It represented the first time in which an orally delivered small molecule greatly extended survival of intracerebrally inoculated mice.
HIGH-THROUGHPUT SCREENING
Dividing Cells
In an effort to identify novel inhibitors of prion formation, we designed a phenotypic screen for antiprion compounds because at least three potential mechanisms exist to reduce PrPSc: reduction of PrPC, disaggregation of existing PrPSc, or inhibition of PrPSc formation. We developed an ELISA to measure the amount of PK-resistant PrP in ScN2a cells following incubation with compound at 5 µM and counterscreened for cell toxicity.
Using a 96-well format, we screened a diverse library of approximately 10,000 compounds. Primary hits were defined as compounds that reduced PrPSc by >50% while reducing cell viability by <30%. After retesting compounds, we identified 121 confirmed hits, which fell into four structural classes: 2-aminothiazoles (2-AMT), quinazolines, hydroxyquinolines, and benzoxazoles. Other commercially available 2-AMT analogs were also efficacious in the cell assay (Ghaemmaghami et al. 2010a). We then refined the assay using an N2a cell line overexpressing mouse PrP, termed clone 3 (Ghaemmaghami et al. 2010b), that was infected with the RML prion strain and serially passaged. Prion-infected cells are referred to as ScN2a-cl3. EC50 values determined in ScN2a-cl3 cells were typically ∼10-fold higher than those in ScN2a cells and thus represent a more stringent test of antiprion action (Gallardo-Godoy et al. 2011). Utilizing the ScN2a-cl3 cell assay, we screened an additional approximately 50,000 compounds and identified 10 new chemical scaffolds (Silber et al. 2013a).
Additional High-Throughput Screens
To complement the screen of dividing ScN2a-cl3 cells, we arrested cell division by adding sodium butyrate. From a screen of approximately 50,000 compounds in stationary phase ScN2a-cl3 cells, we identified only 55 confirmed hits, representing three chemical lead series: piperazines, indoles, and ureas (Silber et al. 2013a). Unfortunately, the majority of hits were weak: Out of the six confirmed hits tested for potency, only one produced an EC50 value less than 10 µM, which was weakly potent with an EC50 of 7.5 µM (Silber et al. 2013a). In the stationary phase, cell catabolism is the sole route for prion clearance because PrPSc is not reduced by cell division (if only compounds working by this mechanism were identified, it would explain the lower hit rate in these cells).
Mice heterozygous for the prion protein have dramatically extended incubation periods (Büeler et al. 1994), so even a modest reduction in PrPC levels should have a large impact on survival. We selected human glioblastoma (T98G) and neuroblastoma (IMR32) cell lines, and screened a library of approximately 45,000 compounds for those that led to a reduction of cell surface PrPC by 30% or more. We identified 138 confirmed hits in T98G cells and 114 confirmed hits in IMR32 cells (Silber et al. 2014). To complement confirmed hits from the PrPC reduction screen, we identified structurally related analogs, and more than 300 compounds were tested for full dose–response curves in both T98G and IMR32 cell lines. Only studies in T98G cells identified compounds that reduced PrPC without negatively affecting cell viability. Of 32 hits with EC50 values less than 10 µM, representing six chemical scaffolds, 28 were evaluated in vivo in pharmacokinetic studies. In all cases, brain levels were only detectable following intraperitoneal but not oral dosing (Silber et al. 2014).
STRUCTURE–ACTIVITY RELATIONSHIPS AND PHARMACOKINETICS
2-Aminothiazoles
EC50 values determined using the ScN2a-cl3 dividing cell assay were reproducible, allowing structure–activity relationships (SARs) to be assigned with some confidence (Gallardo-Godoy et al. 2011). Based on guidelines for potential CNS activity of small molecules (Hitchcock and Pennington 2006), we undertook a systematic exploration of 2-AMT SARs. In addition to understanding tolerated and preferred modifications to the A- and C-rings (Fig. 3), selected compounds were tested in vitro to measure microsomal stability and to determine whether they were substrates of P-gp. A lead compound with a cellular EC50 value in the ScN2a-cl3 assay of ∼1 µM was shown to reach a maximal concentration (Cmax) of ∼25 µM in the brains of treated mice (Gallardo-Godoy et al. 2011).
Figure 3.

Summary of preliminary structure–activity relationships (SARs) for 2-AMT analogs. The three rings are arbitrarily denoted A, B, and C for convenience. (Figure reprinted with Gallardo-Godoy et al. 2011, with permission from American Chemical Society © 2011.)
A series of optimized 2-AMTs were subsequently analyzed by a battery of in vitro assays and in vivo pharmacokinetic studies. Solubility, microsomal stability, cell permeability, cytochrome P450 phenotyping, and free fraction determinations were performed. For single-dose pharmacokinetics, brain and plasma compound levels were determined at multiple time points following gavage of compounds dosed at 10 mg/kg or intravenous dosing at 1 mg/kg. In addition to the Cmax value, the area under the concentration–time curve to the last determined point (AUClast) was determined as a measure of exposure. Compounds can have similar Cmax values, but depending on the rate of clearance, they can have different AUClast values. A compound with a slower clearance and thus larger AUClast has a more sustained exposure. Of the 27 2-AMTs tested in a single-dose pharmacokinetic study, 10 with good ratios of AUClast to their respective EC50 values were advanced to longer-term feeding studies. We used a paradigm of mixing compounds with a complete liquid diet because this represents the least invasive way to dose animals for extended periods of many months. Initial feeding studies were performed for 3 d, with samples collected 3 h after the end of the last dark cycle, and thus represent pseudo steady-state rather than peak concentrations. These studies identified two compounds, IND24 and IND81 (Fig. 4), as lead candidates for efficacy studies (Silber et al. 2013b).
Figure 4.

Chemical structures and pharmacokinetic parameters of 2-AMT compounds, including brain Cmax and AUClast following a 10 mg/kg oral gavage.
In parallel with extensive in vivo experiments, we continued to optimize the pharmacotherapeutic properties of 2-AMTs and were able to increase potency in the cell assay while maintaining the good pharmacokinetic profile of earlier leads. In one series, containing a phenoxazole C-ring analogous to CmpdB, we identified two compounds, IND125 and IND126, with submicromolar potency in the ScN2a-cl3 cell assay (Fig. 4) (Giles et al. 2015). Quantum mechanical calculations of IND24 suggested that although the A-B ring system was coplanar the lowest energy conformation of the terminal phenyl attached to the A-ring was out of plane, suggesting it may have less rigid requirements (Li et al. 2013c). To explore this further, we tested diverse heteroaryl and heterocyclic substitutions at this terminal ring, which yielded the pyridine-3-yl analog of IND24, IND114338 (Fig. 4) with an EC50 of 68 nM, and good brain exposure following oral dosing (Li et al. 2013c). In parallel, we explored SARs around the C-ring and found that small aliphatic amides, particularly cyclopropylamide, were effective, ultimately leading to IND126461 (Fig. 4) with 70 nM potency in the cell assay (Li et al. 2013c).
Aryl Piperazines
From the high-throughput screening (HTS) of more than 50,000 compounds, we identified 881 aryl piperazine analogs, 108 of which reduced PrPSc levels in ScN2a-cl3 cells by ≥30%. The most active of these were N-aryl piperazines with a para-methyl ketone group, whereas compounds with other electron-rich or electron-deficient substituents were inactive. In SAR studies, we first sought to replace the ketone with a 1,3-oxazole, which could similarly act as a hydrogen bond acceptor. In parallel, we synthesized the analogous benzoxazoles. All compounds with the oxazole functionality were weakly active, whereas the benzoxazoles had good potency in the ScN2a-cl3 cell assay (Li et al. 2013a). Further optimization led to a range of aryl piperazines with high potency and good brain exposure and metabolic stability (Table 1).
Table 1.
Antiprion potency and pharmacokinetic parameters of aryl piperazines
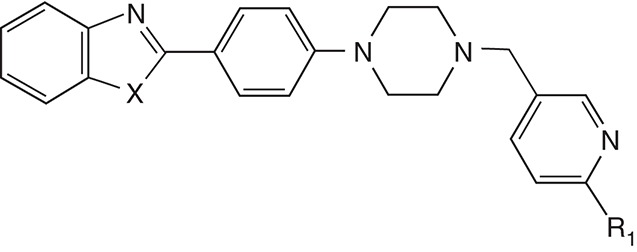
| Compound | X | R1 | EC50 (µM) | Cmax (µM) | AUC (µM·h) | Microsomal stability, t1/2 (min) | |
|---|---|---|---|---|---|---|---|
| Mouse | Human | ||||||
| IND126255 | O | H | 0.25 ± 0.05 | 3.6 ± 0.9 | 14.2 ± 0.6 | 9.2 | 50.2 |
| IND126471 | S | H | 0.36 ± 0.03 | 3.0 ± 0.5 | 14.4 ± 0.2 | 17.3 | 34.3 |
| IND126463 | O | F | 0.38 ± 0.03 | 0.4 ± 0.1 | 1.3 ± 0.2 | 7.3 | 43.6 |
| IND126466 | O | OMe | 1.11 ± 0.10 | 1.6 ± 0.8 | 3.0 ± 0.1 | 25.2 | >60 |
Aryl Amides
To build a pharmacophore model of PrPSc leads, we started with 440 compounds for which EC50 values had been determined in ScN2a-cl3 cells. These were divided into 274 actives (EC50 < 10 µM) and 166 inactives (EC50 ≥ 10 µM), and pharmacophore modeling was performed using PHASE software (Dixon et al. 2006). More than half the active compounds fell within a four-site pharmacophore model of two aromatic rings linked by a hydrogen bond acceptor and a hydrogen bond donor (Lu et al. 2013). This pharmacophore structure is consistent with a biaryl amide.
Because initial 2-AMT leads had poor aqueous solubility, we focused on improving this and other drug-like physicochemical characteristics for the aryl amides. One screening hit, IND28484, had an N-benzylpiperazine substituent at the para position of the aniline moiety (Fig. 5). The piperazine group has a pKa of ∼8 and is therefore protonated at physiological pH conferring greater solubility. From other aryl amide hits, we observed that compounds with at least one biaryl in a linear or fused fashion were more potent. Introducing a biaryl group on the carbonyl side of the screening hit and varying the N-linked substituent on the piperazine led to IND116133 (Fig. 5), with an EC50 value of 22 nM in the ScN2a-cl3 cell assay (Li et al. 2013b).
Figure. 5.

Chemical structures and pharmacokinetic parameters of aryl amides, including brain Cmax and AUClast following a 10 mg/kg oral gavage.
Although CmpdB showed good efficacy in extending survival of prion-infected mice, the hydrazone functionality is a potential liability, because hydrolysis yields an aryl hydrazine, a toxic metabolite, and carcinogen (Powell and Gannett 2002). We therefore explored the SAR around the aryl amide analog of CmpdB. Introduction of electron-donating groups onto the carbonyl aryl ring improved potency; however, these compounds led to poor brain exposure, which was likely due to low metabolic stability as determined by incubation in mouse and human liver microsomes. To improve metabolic stability and pharmacokinetics, we introduced a cyclopropyl group at the 2-position of the oxazole to prevent oxidation at this site, leading to compounds such as IND114378 with an AUClast of 16.8 µM·h (Fig. 5) (Lu et al. 2013).
In addition to the N-aryl piperazine and N-aryl oxazoles, we also explored SARs for the N-aryl heterocycles and N-linked cyano benzyl series (Giles et al. 2016). Based on N-aryl benzothiazole and N-aryl benzoxazole hits, we first tested a range of commercially available analogs. In the N-aryl benzothiazole series, a 6-methyl substituent on the benzothiazole ring in combination with a furyl amide was determined to be important for potency (Giles et al. 2016). Single-dose pharmacokinetics by oral gavage showed that IND114431 (Fig. 5) had the highest brain exposure for this scaffold. For the N-aryl benzoxazole series, a methylpiperazyl nicotinamide analog, IND116135 (Fig. 5), had the best combination of potency and exposure. Various carbonyl linked aryl substituents tested on an N-(4-cyanophenyl) backbone had sub-100-nM potency, and a derivatized 3-phenylpyridine, IND126256 (Fig. 5), was identified to have the best brain exposure of the series (Giles et al. 2016).
EFFICACY
The log-rank (Mantel–Cox) test is typically chosen to compare survival of two groups (e.g., control and treated); however, its interpretation for prion survival studies can be problematic. A group of prion-inoculated mice typically shows onset of neurological disease within a few days of one another, giving a very sharp drop-off in the survival curve. Small differences between survival curves can be highly statistically significant by log-rank but experimentally meaningless if this difference could be accounted for by interexperiment variability. Such analyses are also insensitive to the magnitude of any difference: Nonoverlapping survival curves can have the same level of significance whether the gap between survival curves is a few days or a year. To enable comparison of treatments between different prion strains and mouse models that are characterized by different incubation periods, we introduced the metric of “survival index” as the ratio of vehicle- to drug-treated incubation periods (all other variables being equal), multiplied by 100 (Berry et al. 2013). A treatment that increases survival by 50% therefore has a survival index of 150.
Monitoring Disease Progression In Vivo
Although extension of survival is an unambiguous metric of efficacy, WT mice typically take ∼4 mo or more to develop clinical signs of disease. We previously showed that by utilizing bioluminescence imaging (BLI) we could monitor disease progression in Tg mice expressing a luciferase (luc) reporter driven by the mouse GFAP promoter (Tamgüney et al. 2009). In Tg(Gfap-luc) mice, astrocytic gliosis associated with incipient prion disease drives luciferase expression (Zhu et al. 2004). The substrate luciferin, delivered by intraperitoneal injection, is oxidized by luciferase to oxyluciferin in an electronically excited state, which upon relaxation to the ground state releases a photon. Because of their ∼500-nm wavelength, these photons pass through tissue and bone and are detected directly with a sensitive charge-coupled device (CCD) camera. Using this technology, we could detect disease onset in approximately half the time required to observe the onset of clinical signs (Tamgüney et al. 2009). We validated BLI in a therapeutic paradigm, dosing with CmpdB. Tg(Gfap-luc) mice expressing the WT level of PrP and bigenic Tg(MoPrP:Gfap-luc) mice overexpressing mouse PrP were inoculated with RML prions, dosed with vehicle or CmpdB, and BLI was performed weekly. In both lines, BLI in CmpdB-treated mice showed suppression of luminescence signal upregulation observed in vehicle-treated controls, indicating that compound efficacy could be determined long before extension in survival was observed (Lu et al. 2013).
2-Aminothiazoles
Compounds that showed potency in the cell assay and good brain penetration by pharmacokinetic studies were advanced to long-term dosing experiments. Consumption varied by mouse and on a daily basis over the duration of the experiment; therefore, dosing level was calculated as an average per cage. 2-AMT compounds were typically dosed at ∼200 mg/kg per day unless otherwise stated. The first two 2-AMTs tested in efficacy studies, IND24 and IND81, extended survival of Tg(Gfap-luc) mice inoculated with the RML prion strain to ∼200 d and delayed upregulation of BLI signal (Berry et al. 2013). Immunoblot analysis of brains from treated mice showed PK-resistant PrP, suggesting the mice ultimately died of prion disease. However, the characteristic three-band pattern of unglycosylated, monoglycosylated, and diglycosylated PrP was altered, leading us to suspect that strain properties were altered following efficacious treatment. To differentiate strains generated by treatment, we refer to them by appending the compound name in brackets following the inoculating strain—RML[IND24] and RML[IND81]—compared to prions in vehicle-treated mice as RML[V]. For RML[IND24] and RML[IND81], the ratio of mono- to diglycosylated PrP was significantly lower than for RML[V] (Berry et al. 2013). We then measured the conformational stability of the strains by incubating them with increasing concentrations of GdnHCl (Peretz et al. 2001). The half maximal concentrations, or [GdnHCl]1/2 values for RML[IND24] and RML[IND81], were higher than that of RML[V], but only the concentration for RML[IND24] was significantly different (Berry et al. 2013). In addition, we quantified the neuropathological changes in the brains of treated mice and determined that the distribution and intensity of vacuolation and PrP deposition was significantly different between RML[IND24] and RML[V] for multiple brain regions (Berry et al. 2013).
We then tested the infectivity properties of RML[V] and RML[IND24] in cell and animal models. When CAD5 cells were infected with RML[V], they replicated the properties of the original RML strain; however, CAD5 cells infected with RML[IND24] propagated prions resistant to concentrations of IND24 up to 20 µM, suggesting that treatment had led to a drug-resistant prion strain (Berry et al. 2013). Serial passage of the RML[IND24] strain in CAD5 cells in the presence or absence of IND24 retained the IND24-resistant properties. When RML[IND24] was serially passaged in mice further treated with IND24, incubation times were reduced to ∼165 d, and the brains of the resulting mice contained the same IND24-resistant strain propagated in the CAD5 cells, seemingly maintaining the new strain. However, when RML[IND24] prions were serially passaged in mice treated with vehicle, incubation times were ∼190 d, but infection of CAD5 cells propagated an IND24-sensitive strain. A second serial passage with vehicle had an incubation time of ∼115 d, the same as the original RML inoculum, suggesting that in vivo, the RML[IND24] strain reverts in the absence of IND24 (Berry et al. 2013).
Efficacy of IND24 was tested against two additional strains of mouse-passaged sheep scrapie: 22L and ME7, the former derived from the same original sheep scrapie brain pool as RML but with a different passaging history (Dickinson 1976), and the latter derived from an independent transmission to mice (Zlotnik and Rennie 1963). All three strains can be propagated in CAD5 cells. Whereas IND24 showed good potency against RML-infected CAD5 cells, it was lower against ME7 prions, and 22L prions appeared resistant to IND24 in culture at concentrations up to 10 µM (Giles et al. 2015). When IND24 dosing was started the day after inoculating WT mice, the survival indices for ME7- and 22L-infected WT mice were 170 and 148, respectively, compared to 173 for RML (Table 2) (Berry et al. 2013; Giles et al. 2015). Interestingly, IND24 treatment of ME7-infected mice did not appear to alter the strain type; ME7[IND24] was indistinguishable from ME7[V] by biochemical and cell infectivity metrics.
Table 2.
Survival indices following continuous dosing of IND24 in multiple prion-infected mouse models
| Prion strain | Mouse model | Dosing initiated (days postinoculation) | Survival indexa |
|---|---|---|---|
| RML | WT | 1 | 173 |
| RML | WT | 60 | 179 |
| RML | WT | 90 | 100 |
| RML | WT | −14 | 383 |
| ME7 | WT | 1 | 170 |
| ME7 | WT | 60 | 125 |
| 22L | WT | 1 | 148 |
| 22L | WT | 60 | 114 |
| RML | Tg(MoPrP) | 1 | 220 |
| SSBP/1 | Tg(OvPrP)/Prnp0/0 | 1 | 109 |
| CH1641 | Tg(OvPrP)/Prnp0/0 | 1 | 101 |
| CWD1 | Tg(ElkPrP)/Prnp0/0 | 1 | 220 |
| CWD2 | Tg(ElkPrP)/Prnp0/0 | 1 | 158 |
| sCJD(MM1) | Tg(HuPrP,M129)/Prnp0/0 | 1 | 101 |
| sCJD(VV2) | Tg(HuPrP,V129)/Prnp0/0 | 1 | 96 |
aSurvival index is the ratio of vehicle- to drug-treated incubation periods (all other variables equal), multiplied by 100 (Berry et al. 2013).
We also tested IND24 against a number of naturally occurring prion strains. Tg mice lacking endogenous expression of mouse PrP (Prnp0/0) but expressing the PrP of the host species (i.e., ovine [Ov], human [Hu], or elk) are susceptible to scrapie, CJD, and CWD, respectively. IND24 treatment did not extend survival in Tg(OvPrP)/Prnp0/0 mice inoculated with SSBP/1 or CH1641 isolates, or in Tg(HuPrP)/Prnp0/0 mice inoculated with either of the two major sCJD strains (Table 2). However, Tg(ElkPrP)/Prnp0/0 mice inoculated with two natural CWD isolates had survival indices of 158 and 220 upon IND24 treatment (Berry et al. 2013, 2015). As with the ME7 strain, IND24 treatment of CWD-infected mice did not appear to alter the strain characteristics, suggesting that the development of resistance may be strain dependent. To show that the differential effects observed were prion strain-specific rather than dependent on the Tg model used, we inoculated mice expressing a chimeric mouse/human PrP transgene, which are susceptible to both RML and CJD prions (Giles et al. 2010). IND24 treatment of Tg(Mo/HuPrP)/Prnp0/0 mice inoculated with RML prions doubled survival, but had no effect on the same mice inoculated with the CJD prions.
Dosing with CmpdB was shown to be less effective the later treatment was started (Kawasaki et al. 2007). Surprisingly, when IND24 treatment was started approximately halfway through the normal incubation period for RML-inoculated mice, it produced a similar extension in survival compared to initiating dosing the day after inoculation (Table 2). However, this relationship was strain-specific, as IND24 showed greatly reduced efficacy in ME7- and 22L-infected mice when dosing was initiated halfway through the respective incubation periods (Table 2; Fig. 6A–C) (Berry et al. 2013; Giles et al. 2015). Further experiments in RML-infected mice showed that starting dosing any later in disease progression had rapidly diminishing returns. When IND24 dosing was started at ∼75% of the untreated incubation period, no survival extension was observed. Intermittent dosing of IND24 in RML-infected mice further extended survival, but the most dramatic effect was when dosing was initiated two weeks before inoculation, which extended survival almost fourfold (Table 2; Fig. 6A) (Giles et al. 2015). Prophylactic dosing is highly relevant to the genetic prion diseases in which carriers have the mutation from embryogenesis but typically do not manifest disease until their fourth or fifth decade.
Figure 6.

Survival curves of wild-type (A–C) and Tg(MoPrP) mice overexpressing PrP (D) inoculated with RML (A and D), ME7 (B), and 22L (C) prion strains. Mice were dosed with IND24 (green) or IND125 (blue) at 200 mg/kg/d, or vehicle (red), with dosing initiated 1 (solid lines) or 60 (dashed lines) days postinoculation (dpi), or 14 d before inoculation (dotted line). (A) Starting dosing with IND24 at 1 or 60 dpi in RML-inoculated mice produced a similar extension in survival; however, prophylactic dosing greatly increased survival. (B) ME7-inoculated mice showed a smaller survival extension when dosing was started at 60 dpi than at 1 dpi. (C) 22L-inoculated mice showed limited efficacy of IND24 when dosing was started at 60 dpi. (D) In mice overexpressing PrP, continuous dosing with IND24 approximately doubles and IND125 approximately triples the untreated incubation period of RML-inoculated mice.
The second-generation 2-AMT compounds with improved potency in the cell assay were also tested in long-term efficacy studies. Despite similar values for brain exposure for IND125 and IND126 when dosing by oral gavage, efficacy in RML-infected Tg(MoPrP) mice overexpressing mouse PrP was vastly different, with IND125 generating a survival index of 302 (Fig. 6D) and IND126 generating a survival index of just 120 (Table 3). This difference may be explained by low bioavailability from the liquid diet for IND126 or induction of cytochrome p450s increasing the rate of metabolism.
Table 3.
Survival indices of RML-infected wild-type (WT) and Tg(MoPrP) mice treated with 2-AMT compounds
| IND24 | IND81 | IND125 | IND126 | IND114338 | IND126461 | |
|---|---|---|---|---|---|---|
| wt | 173 | 164 | 159 | 197 | ||
| Tg(MoPrP) | 220 | 302 | 120 | 133 |
IND114338, the pyridine-3-yl analog of IND24, had an almost 20-fold higher cellular potency in the ScN2a-cl3 cell assay, but long-term dosing of WT mice resulted in a survival index of 197, only slightly higher than that of IND24. Another potent 2-AMT derivative, IND126461, has a cyclopropylamide C-ring substitution. This compound had a similar EC50 value to IND114338 but a ∼50-fold lower brain exposure following oral gavage. In the Tg(MoPrP) line, IND126461 had a survival index of just 133 (Table 3).
Interestingly, the neuropathological distribution of PrPSc in the brains of treated mice differed with each of the compounds. IND125-treated mice, despite surviving three times longer than vehicle-treated controls, had lower levels of PrPSc than controls in multiple brain regions, with little or no deposition throughout the thalamus and much of the cortex. Despite only moderately extending survival, IND126- and IND126461-treated mice also had lower levels of PrPSc deposition throughout the brain than terminal control mice (Giles et al. 2015). In combination with the data on IND24, these changes in neuropathological phenotype suggest that each of the compounds may differentially impact the strain that is propagating, although larger time-course studies and dose response data with various treatments will be needed to uncover these details.
As with IND24, none of the improved 2-AMTs extended survival in Tg mice inoculated with CJD prions, emphasizing the strain specificity of compounds identified with the RML-infected ScN2a-cl3 assay.
Aryl Amides
The aryl amides were the most highly represented chemical scaffold from HTS, accounting for ∼15% of the hits (Silber et al. 2013a). Medicinal chemistry optimization of various subseries led to N-linked aryl piperazine (IND116133), benzothiazole (IND114431), benzoxazole (IND116135), and cyanobenzyl (IND126256) leads (Fig. 5), which were progressed to long-term efficacy studies.
Dosing IND116133 at ∼200 mg/kg per day resulted in brain levels of ∼75 µM after 3 d, and continued dosing led to toxicity after about one month. However, dosing at 25 mg/kg per day resulted in a survival index of 181 (Table 4) (Giles et al. 2016). Surprisingly, each of the other three lead compounds led to similar extension in survival despite differing potency in the cell assay and pharmacokinetics: IND114431, IND116135, and IND126256 had survival indices of 220, 233, and 227, respectively, in Tg(MoPrP) mice (Table 4) (Giles et al. 2016). As with the 2-AMT treatments, each compound appeared to produce a unique neuropathological signature. However, in contrast to IND24 and IND81, treatment with any of the four aryl amide compounds did not significantly change the glycoform ratio. Importantly, when CAD5 cells were infected with brains of mice treated with aryl amides, they propagated strains susceptible to the respective aryl amide, suggesting that drug resistance is not an inevitable outcome of effective antiprion therapeutics (Giles et al. 2016).
Table 4.
Survival indices of RML-infected wild-type (WT) and Tg(MoPrP) mice treated with aryl amide compounds
| IND116133 | IND114431 | IND116135 | IND126256 | |
|---|---|---|---|---|
| wt | 181 | |||
| Tg(MoPrP) | 220 | 233 | 227 |
ALTERNATIVE STRATEGIES TO IDENTIFY ANTIPRION COMPOUNDS
Complementary High-Throughput Screens
Using a combination of an antiprion cell culture assay and one that detects binding of recombinant PrP to brain-derived PrPSc (Bertsch et al. 2005), a library of approximately 20,000 compounds was screened. Following identification of a cluster of hits with the 3,5-diphenyl-pyrazone (DPP) structure, a focused library of DPP analogs was studied with the above assays, together with a cell-free propagation assay based on protein misfolding cyclic amplification (PMCA) (Wagner et al. 2013). Further testing of a range of analogs in vivo led to the identification of anle138b (Fig. 7A) as the lead compound. In WT mice infected with the RML prion strain, anle138b treatment resulted in a survival index of 198. Although PMCA using human PrP as substrate suggested that anle138b would be effective against human prions (Wagner et al. 2013), it did not extend survival of Tg mice infected with CJD brain homogenate (Giles et al. 2015).
Figure 7.

Chemical structures of additional compounds that extend survival of prion-infected mice. (A) Anle138b, (B) GN8, (C) LIN5044, and (D) BX912.
A screen to measure PrPC reduction was developed using a fluorescence resonance energy transfer (FRET)-based methodology. The FRET pair was attached to anti-PrP antibodies with different epitopes recognizing distinct domains on the same PrP molecule. Screening a collection of approximately 1300 drugs approved for human use identified nine hits. Using an orthogonal screen based on cell-surface immunofluorescence reduced this to two: tacrolimus and astemizole. Although both compounds reduced PrPSc in cell culture, only astemizole was reported to have an effect on survival of RML-infected mice in which treatment from ∼13%–34% of the normal incubation period (20–50 dpi) resulted in a survival index of just 104 (Karapetyan et al. 2013).
Oligonucleotides
In an attempt to stimulate innate immunity, CpG oligodeoxynucleotides were dosed repeatedly following prion inoculation (Sethi et al. 2002). As with the porphyrins, a modest extension of survival was achieved, but only in peripherally inoculated mice when dosing was started within hours of inoculation. Phosphorothioate oligonucleotides (PS-ON), in which the backbone oxygen is replaced by a sulfur atom, are resistant to nucleases and therefore have greater residence times. PS-ON reduced both PrPC and PrPSc levels in cell culture in a sequence-independent manner. PS-ON were effective when dosed prophylactically in peripherally inoculated mice or when mixed directly with the inoculum before intracerebral inoculation (Kocisko et al. 2006; Karpuj et al. 2007).
PS-ON were identified that reduced PrPC and PrPSc levels in cells. Intraventricular delivery of PS-ON targeting Prnp also reduced PrPC levels in the brain, and when intraventricular treatment was initiated the day after intracerebral inoculation, a survival index of 142 was obtained (Nazor Friberg et al. 2012).
In Silico Screen
Based on the concept that stabilizing the PrPC structure would stop the formation of PrPSc, an in silico screen of 320,000 compounds was undertaken to identify those that bind to a pocket in the PrPC structure. There were 59 hits identified that had at least one hydrogen bond to PrPC and also drug-like characteristics. Of these, 44 were tested in a GT1 cell assay propagating the mouse-passaged Fukuoka-1 strain, and one molecule, GN8 (Fig. 7B), had an EC50 value of 1–2 µM. GN8 was dosed subcutaneously starting at ∼50% of the untreated incubation period or intraventricularly starting at ∼35% of the untreated incubation period, resulting in survival indices of 114 and 115, respectively (Kuwata et al. 2007).
Polythiophenes
Certain polythiophene molecules show conformation-dependent fluorescent spectra and have been used to characterize prion strains in histological sections (Sigurdson et al. 2007). Ex vivo experiments showed that these and related polythiophenes also reduced prion infectivity by stabilizing PrPSc (Margalith et al. 2012). Because polythiophenes are unlikely to cross the BBB, mice were dosed by intraventricular delivery; one compound, LIN5001, generated a modest survival index of 136 when given prophylactically and an index of 120 when dosing was started at ∼30% of the normal incubation period of RML-infected WT mice (Herrmann et al. 2015). Replacement of the sulfur in the central thiophene ring with selenium increased the prophylactic survival index to 150. Interestingly, molecular modeling based on an unrelated β-sheet amyloid led to the design of LIN5044 (Fig. 7C) with a survival index of 188 for prophylactic treatment (Herrmann et al. 2015).
Targeting Toxic Cascades
The precise mechanism by which prion disease leads to neurotoxicity is poorly understood. Synaptic failure and neuronal loss in prion disease is associated with phosphorylation of PERK, which in turn phosphorylates eIF2α causing translational repression. It has, therefore, been posited that promoting translational recovery should be neuroprotective. Expression of GADD34, a specific eIF2α phosphatase, reduced eIF2α-P and slowed neurodegeneration, whereas an inhibitor of eIF2α-P phosphorylation enhanced it (Moreno et al. 2012). However, these treatments resulted in survival indices of 103 and 95, respectively. Dosing with the PERK inhibitor GSK2606414 replicated the neuropathological and behavioral recovery but failed to extend survival because of toxicity (Moreno et al. 2013). The small molecule ISRIB restores translation downstream of eIF2α. ISRIB administration in prion-infected mice was initiated at ∼60% of the untreated incubation period and was neuroprotective, but it led to a survival index of only 105 because of excessive body weight loss (Halliday et al. 2015).
Another study showed that prion infection activated 3-phosphoinositide-dependent kinase-1 (PDK1). PDK1 reduces the sheddase activity that cleaves PrPC, which in turn increases PrPSc. Conversely, inhibition of PDK1 should increase PrPC shedding and potentially extend survival of prion-infected mice. As with GSK2606414 and ISRIB, long-term dosing with the specific PDK1 inhibitor BX912 (Fig. 7D) is known to be toxic. When 22L-infected WT mice were treated with BX912 at ∼75% of the untreated incubation period, it produced a survival index of 117 (Pietri et al. 2013). Interestingly, BX912 shares a structural similarity to GN8. Whether the antiprion effect of BX912 acts through the stabilization of PrP, or GN8 acts as a PDK1 inhibitor, remains to be determined.
The Notch-1 intracellular domain (NICD) is generated from the γ-secretase catalyzed cleavage of Notch-1. NICD is increased in the brains of prion-infected mice preceding dendritic atrophy and loss, and is decreased by the γ-secretase inhibitor LY411575. Dosing RML-infected mice with a combination of LY411575 and quinacrine starting at ∼40% of the incubation period reduced PrPSc and prevented dendritic atrophy and loss; however, the toxicity of LY411575 meant no extension in survival was observed (Spilman et al. 2008).
CONCLUSION
Although none of the antiprion therapies reported to date have shown efficacy in a CJD-infected mouse model, the screening paradigm and medicinal chemistry efforts outlined above describe the most effective antiprion therapeutics reported to date. Developing a cell line for the propagation of human prions remains a top priority, on which the same drug discovery paradigm will be repeated. The mechanisms of action for many of the compounds reported above are not yet determined, and, ultimately, a therapeutic strategy for prion diseases may require multiple complementary methods of intervention.
With the understanding that more prevalent neurodegenerative diseases, such as Alzheimer's and Parkinson's, progress via conformational templating of misfolded proteins, the HTS studies outlined above (and potentially even some of the compounds themselves) may provide a paradigm to accelerate the discovery of drugs to address the growing societal challenges caused by these devastating diseases.
ACKNOWLEDGMENTS
The authors acknowledge support from the National Institutes of Health (AG002132 and AG031220), Daiichi Sankyo, Henry M. Jackson Foundation, CurePSP, Dana Foundation, Glenn Foundation, Mary Jane Brinton Fund, Sherman Fairchild Foundation, and a gift from the Rainwater Charitable Foundation.
Footnotes
Editor: Stanley B. Prusiner
Additional Perspectives on Prion Diseases available at www.perspectivesinmedicine.org
REFERENCES
- Ahn M, Ghaemmaghami S, Huang Y, Phuan P-W, May BCH, Giles K, DeArmond SJ, Prusiner SB. 2012. Pharmacokinetics of quinacrine efflux from mouse brain via the P-glycoprotein efflux transporter. PLoS ONE 7: e39112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret A, Tagliavini F, Forloni G, Bate C, Salmona M, Colombo L, De Luigi A, Limido L, Suardi S, Rossi G, et al. 2003. Evaluation of quinacrine treatment for prion diseases. J Virol 77: 8462–8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D, Giles K, Oehler A, Bhardwaj S, DeArmond SJ, Prusiner SB. 2015. Use of a 2-aminothiazole to treat chronic wasting disease in transgenic mice. J Infect Dis 212: S17–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DB, Lu D, Geva M, Watts JC, Bhardwaj S, Oehler A, Renslo AR, DeArmond SJ, Prusiner SB, Giles K. 2013. Drug resistance confounding prion therapeutics. Proc Natl Acad Sci 110: E4160–E4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertsch U, Winklhofer KF, Hirschberger T, Bieschke J, Weber P, Hartl FU, Tavan P, Tatzelt J, Kretzschmar HA, Giese A. 2005. Systematic identification of antiprion drugs by high-throughput screening based on scanning for intensely fluorescent targets. J Virol 79: 7785–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. 1994. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68: 7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone I, Belton L, Walker AS, Darbyshire J. 2008. Intraventricular pentosan polysulphate in human prion diseases: An observational study in the UK. Eur J Neurol 15: 458–464. [DOI] [PubMed] [Google Scholar]
- Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. 1994. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1: 19–30. [PMC free article] [PubMed] [Google Scholar]
- Butler DA, Scott MRD, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. 1988. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 62: 1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Race RE. 1992. Potent inhibition of scrapie-associated PrP accumulation by Congo red. J Neurochem 59: 768–771. [DOI] [PubMed] [Google Scholar]
- Caughey WS, Raymond LD, Horiuchi M, Caughey B. 1998. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc Natl Acad Sci 95: 12117–12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL. 1961. Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 277: 1378–1379. [DOI] [PubMed] [Google Scholar]
- Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. 2006. Kuru in the 21st century—An acquired human prion disease with very long incubation periods. Lancet 367: 2068–2074. [DOI] [PubMed] [Google Scholar]
- Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, et al. 2009. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): A patient-preference trial. Lancet Neurol 8: 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SJ, Lewis V, Brazier M, Hill AF, Fletcher A, Masters CL. 2002. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann Neurol 52: 503–506. [DOI] [PubMed] [Google Scholar]
- Cuillé J, Chelle PL. 1939. Transmission experimentale de la tremblante a la chevre. C R Acad Sci 208: 1058–1060. [Google Scholar]
- Diack AB, Head MW, McCutcheon S, Boyle A, Knight R, Ironside JW, Manson JC, Will RG. 2014. Variant CJD: 18 years of research and surveillance. Prion 8: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson AG. 1976. Scrapie in sheep and goats In Slow virus diseases of animals and man (ed. Kimberlin RH.), pp. 209–241. North-Holland, Amsterdam. [Google Scholar]
- Diringer H, Ehlers B. 1991. Chemoprophylaxis of scrapie in mice. J Gen Virol 72: 457–460. [DOI] [PubMed] [Google Scholar]
- Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. 2006. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 20: 647–671. [DOI] [PubMed] [Google Scholar]
- Doh-ura K, Iwaki T, Caughey B. 2000. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 74: 4894–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. 2004. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol 78: 4999–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohgu S, Yamauchi A, Takata F, Sawada Y, Higuchi S, Naito M, Tsuruo T, Shirabe S, Niwa M, Katamine S, et al. 2004. Uptake and efflux of quinacrine, a candidate for the treatment of prion diseases, at the blood–brain barrier. Cell Mol Neurobiol 24: 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers B, Diringer H. 1984. Dextran sulphate 500 delays and prevents mouse scrapie by impairment of agent replication in spleen. J Gen Virol 65: 1325–1330. [DOI] [PubMed] [Google Scholar]
- Farquhar C, Dickinson A, Bruce M. 1999. Prophylactic potential of pentosan polysulphate in transmissible spongiform encephalopathies. Lancet 353: 117. [DOI] [PubMed] [Google Scholar]
- Gajdusek DC, Gibbs CJ Jr, Alpers M. 1966. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 209: 794–796. [DOI] [PubMed] [Google Scholar]
- Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR. 2011. 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem 54: 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind MD, Kuo AL, Wong KS, Haman A, Devereaux G, Raudabaugh BJ, Johnson DY, Torres-Chae CC, Finley R, Garcia P, et al. 2013. Quinacrine treatment trial for sporadic Creutzfeldt–Jakob disease. Neurology 81: 2015–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB. 2009. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog 5: e1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, May BCH, Renslo AR, Prusiner SB. 2010a. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol 84: 3408–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Ullman J, Ahn M, St. Martin S, Prusiner SB. 2010b. Chemical induction of misfolded prion protein conformers in cell culture. J Biol Chem 285: 10415–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CJ Jr., Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB. 1968. Creutzfeldt–Jakob disease (spongiform encephalopathy): Transmission to the chimpanzee. Science 161: 388–389. [DOI] [PubMed] [Google Scholar]
- Giles K, Glidden DV, Patel S, Korth C, Groth D, Lemus A, DeArmond SJ, Prusiner SB. 2010. Human prion strain selection in transgenic mice. Ann Neurol 68: 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, De Nicola GF, Patel S, Glidden DV, Korth C, Oehler A, DeArmond SJ, Prusiner SB. 2012. Identification of I137M and other mutations that modulate incubation periods for two human prion strains. J Virol 86: 6033–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Berry DB, Condello C, Hawley RC, Gallardo-Godoy A, Bryant C, Oehler A, Elepano M, Bhardwaj S, Patel S, et al. 2015. Different 2-aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. J Pharmacol Exp Ther 355: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Berry DB, Condello C, Dugger BN, Li Z, Oehler A, Bhardwaj S, Elepano M, Guan S, Silber BM, et al. 2016. Optimization of aryl amides that extend survival in prion-infected mice. J Pharmacol Exp Ther 358: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Woerman AL, Berry DB, Prusiner SB 2017. Bioassays and inactivation of prions. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a023499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, et al. 2013. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: Large scale survey. BMJ 347: f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlow WJ. 1959. Scrapie and kuru. Lancet 274: 289–290. [Google Scholar]
- Haïk S, Brandel JP. 2014. Infectious prion diseases in humans: Cannibalism, iatrogenicity and zoonoses. Infect Genet Evol 26: 303–312. [DOI] [PubMed] [Google Scholar]
- Haïk S, Brandel JP, Salomon D, Sazdovitch V, Delasnerie-Lauprêtre N, Laplanche JL, Faucheux BA, Soubrié C, Boher E, Belorgey C, et al. 2004. Compassionate use of quinacrine in Creutzfeldt–Jakob disease fails to show significant effects. Neurology 63: 2413–2415. [DOI] [PubMed] [Google Scholar]
- Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM, et al. 2015. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6: e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann US, Schutz AK, Shirani H, Huang D, Saban D, Nuvolone M, Li B, Ballmer B, Åslund AKO, Mason JJ, et al. 2015. Structure-based drug design identifies polythiophenes as antiprion compounds. Sci Transl Med 7: 299ra123. [DOI] [PubMed] [Google Scholar]
- Hitchcock SA, Pennington LD. 2006. Structure–brain exposure relationships. J Med Chem 49: 7559–7583. [DOI] [PubMed] [Google Scholar]
- Huang Y, Okochi H, May BC, Legname G, Prusiner SB, Benet LZ, Guglielmo BJ, Lin ET. 2006. Quinacrine is mainly metabolized to mono-desethyl quinacrine by CYP3A4/5 and its brain accumulation is limited by P-glycoprotein. Drug Metab Dispos 34: 1136–1144. [DOI] [PubMed] [Google Scholar]
- Ingrosso L, Ladogana A, Pocchiari M. 1995. Congo red prolongs the incubation period in scrapie-infected hamsters. J Virol 69: 506–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P, Fallahi M, Hodder P, Weissmann C, Lasmezas CI. 2013. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc Natl Acad Sci 110: 7044–7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuj MV, Giles K, Gelibter-Niv S, Scott MR, Lingappa VR, Szoka FC, Peretz D, Denetclaw W, Prusiner SB. 2007. Phosphorothioate oligonucleotides reduce PrPSc levels and prion infectivity in cultured cells. Mol Med 13: 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. 2007. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol 81: 12889–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin RH, Walker CA. 1983. The antiviral compound HPA-23 can prevent scrapie when administered at the time of infection. Arch Virol 78: 9–18. [DOI] [PubMed] [Google Scholar]
- Klatzo I, Gajdusek DC, Zigas V. 1959. Pathology of kuru. Lab Invest 8: 799–847. [PubMed] [Google Scholar]
- Kocisko DA, Vaillant A, Lee KS, Arnold KM, Bertholet N, Race RE, Olsen EA, Juteau JM, Caughey B. 2006. Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob Agents Chemother 50: 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, May BCH, Cohen FE, Prusiner SB. 2001. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci 98: 9836–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, et al. 2003. Abbreviated incubation times for human prions in mice expressing a chimeric mouse–human prion protein transgene. Proc Natl Acad Sci 100: 4784–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, Nakamura HK, Kimura K, Kawasaki M, Takakura Y, et al. 2007. Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci 104: 11921–11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Gever JR, Rao S, Widjaja K, Prusiner SB, Silber BM. 2013a. Discovery and preliminary structure–activity relationship of arylpiperazines as novel, brain-penetrant antiprion compounds. ACS Med Chem Lett 4: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Rao S, Gever JR, Widjaja K, Prusiner SB, Silber BM. 2013b. Towards optimization of arylamides as novel, potent, and brain-penetrant antiprion lead compounds. ACS Med Chem Lett 4: 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Silber BM, Rao S, Gever JR, Bryant C, Gallardo-Godoy A, Dolghih E, Widjaja K, Elepano M, Jacobson MP, et al. 2013c. 2-Aminothiazoles with improved pharmacotherapeutic properties for treatment of prion disease. Chem Med Chem 8: 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Giles K, Li Z, Rao S, Dolghih E, Gever JR, Geva M, Elepano ML, Oehler A, Bryant C, et al. 2013. Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J Pharmacol Exp Ther 347: 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. 2007. Prion strain discrimination in cell culture: The cell panel assay. Proc Natl Acad Sci 104: 20908–20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalith I, Suter C, Ballmer B, Schwarz P, Tiberi C, Sonati T, Falsig J, Nyström S, Hammarström P, Åslund A, et al. 2012. Polythiophenes inhibit prion propagation by stabilizing prion protein (PrP) aggregates. J Biol Chem 287: 18872–18887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, McLean CY, Tung JY, Yu LPC, Gambetti P, et al. 2016. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 8: 322ra329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al. 2012. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485: 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, et al. 2013. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5: 206ra138. [DOI] [PubMed] [Google Scholar]
- Nazor Friberg K, Hung G, Wancewicz E, Giles K, Black C, Freier S, Bennett F, DeArmond SJ, Freyman Y, Lessard P, et al. 2012. Intracerebral infusion of antisense oligonucleotides into prion-infected mice. Mol Ther Nucleic Acids 1: e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman PK, Todd NV, Scoones D, Mead S, Knight RSG, Will RG, Ironside JW. 2014. Postmortem findings in a case of variant Creutzfeldt–Jakob disease treated with intraventricular pentosan polysulfate. J Neurol Neurosurg Psychiatry 85: 921–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, et al. 1999. Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46: 224–233. [PubMed] [Google Scholar]
- Peretz D, Scott M, Groth D, Williamson A, Burton D, Cohen FE, Prusiner SB. 2001. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10: 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietri M, Dakowski C, Hannaoui S, Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A, Mouillet-Richard S, Haik S, Bailly Y, Peyrin JM, et al. 2013. PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer's diseases. Nat Med 19: 1124–1131. [DOI] [PubMed] [Google Scholar]
- Powell JH, Gannett PM. 2002. Mechanisms of carcinogenicity of aryl hydrazines, aryl hydrazides, and arenediazonium ions. J Environ Pathol Toxicol Oncol 21: 1–31. [PubMed] [Google Scholar]
- Priola SA, Raines A, Caughey WS. 2000. Porphyrin and phthalocyanine antiscrapie compounds. Science 287: 1503–1506. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. 2012. A unifying role for prions in neurodegenerative diseases. Science 336: 1511–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. 1983. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 35: 349–358. [DOI] [PubMed] [Google Scholar]
- Race RE, Fadness LH, Chesebro B. 1987. Characterization of scrapie infection in mouse neuroblastoma cells. J Gen Virol 68: 1391–1399. [DOI] [PubMed] [Google Scholar]
- Race RE, Caughey B, Graham K, Ernst D, Chesebro B. 1988. Analyses of frequency of infection, specific infectivity, and prion protein biosynthesis in scrapie-infected neuroblastoma cell clones. J Virol 62: 2845–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D. 2005. Search for a prion-specific nucleic acid. J Virol 79: 10796–10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schätzl HM, Laszlo L, Holtzman DM, Tatzelt J, DeArmond SJ, Weiner RI, Mobley WC, Prusiner SB. 1997. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J Virol 71: 8821–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Lipford G, Wagner H, Kretzschmar H. 2002. Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet 360: 229–230. [DOI] [PubMed] [Google Scholar]
- Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Polymenidou M, Schwarz P, Leclerc M, Hammarstrom P, Wüthrich K, Aguzzi A. 2007. Prion strain discrimination using luminescent conjugated polymers. Nat Methods 4: 1023–1030. [DOI] [PubMed] [Google Scholar]
- Silber BM, Gever JR, Li Z, Gallardo-Godoy A, Renslo AR, Widjaja K, Irwin JJ, Rao S, Jacobson MP, Ghaemmaghami S, et al. 2013a. Antiprion compounds that reduce PrPSc levels in dividing and stationary-phase cells. Bioorg Med Chem 21: 7999–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber BM, Rao S, Fife KL, Gallardo-Godoy A, Renslo AR, Dalvie DK, Giles K, Freyman Y, Elepano M, Gever JR, et al. 2013b. Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice. Pharm Res 30: 932–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber BM, Gever JR, Rao S, Li Z, Renslo AR, Widjaja W, Wong C, Giles K, Freyman Y, Elepano M, et al. 2014. Novel compounds lowering the cellular isoform of the human prion protein in cultured human cells. Bioorg Med Chem 22: 1960–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Lessard P, Sattavat M, Bush C, Tousseyn T, Huang EJ, Giles K, Golde T, Das P, Fauq A, et al. 2008. A gamma-secretase inhibitor and quinacrine reduce prions and prevent dendritic degeneration in murine brains. Proc Natl Acad Sci 105: 10595–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamgüney G, Francis KP, Giles K, Lemus A, DeArmond SJ, Prusiner SB. 2009. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci 106: 15002–15006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, Sidle KCL, Collinge J, DeArmond SJ, Prusiner SB. 1994. Transmission of Creutzfeldt–Jakob disease from humans to transgenic mice expressing chimeric human–mouse prion protein. Proc Natl Acad Sci 91: 9936–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83: 79–90. [DOI] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. 1996. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274: 2079–2082. [DOI] [PubMed] [Google Scholar]
- Tsuboi Y, Doh-ura K, Yamada T. 2009. Continuous intraventricular infusion of pentosan polysulfate: Clinical trial against prion diseases. Neuropathology 29: 632–636. [DOI] [PubMed] [Google Scholar]
- Vorberg I, Raines A, Story B, Priola SA. 2004. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J Infect Dis 189: 431–439. [DOI] [PubMed] [Google Scholar]
- Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, et al. 2013. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease. Acta Neuropathol 125: 795–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Jucker M. 2015. Neurodegenerative diseases: Expanding the prion concept. Annu Rev Neurosci 38: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung L, Huang Y, Lessard P, Legname G, Lin ET, Baldwin M, Prusiner SB, Ryou C, Guglielmo BJ. 2004. Pharmacokinetics of quinacrine in the treatment of prion disease. BMC Infect Dis 4: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Ramboz S, Hewitt D, Boring L, Grass DS, Purchio AF. 2004. Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci Lett 367: 210–212. [DOI] [PubMed] [Google Scholar]
- Zlotnik I, Rennie JC. 1963. Further observations on the experimental transmission of scrapie from sheep and goats to laboratory mice. J Comp Pathol 73: 150–162. [DOI] [PubMed] [Google Scholar]