

Abstract
Potent inhibitors of MenA (1,4-dihydroxy-2-naphtoate prenyltrasferase) in Mycobacterium tuberculosis are identified, and are also effective in inhibiting growth of Mycobacterium tuberculosis at low concentrations. The MenA inhibitors possess common chemical structural features of ((alkylamino)alkoxyphenyl)(phenyl)methanones. Significantly, the MenA inhibitors can be synthesized in a few steps with high overall yields. The representative MenA inhibitors are highly effective in killing nonreplicating Mycobacterium tuberculosis that is evaluated by using the Wayne low oxygen model. In addition, a series of drug resistant Mycobacterium spp. are sensitive to the MenA inhibitors. The results are expected to be of significance in terms of discovering new lead compounds that can be developed into new drugs to combat unmet diseases caused by Mycobacterium tuberculosis.
Keywords: New drug target for Mycobacterium tuberculosis, menaquinone biosynthesis, MenA, MenA inhibitors, nonreplicating Mycobacterium tuberculosis
I. INTRODUCTION
Mycobacterium tuberculosis (Mtb) causes tuberculosis (TB) and is responsible for nearly two million deaths annually [1]. Estimates indicate that one-third of the world population is infected with latent Mtb. In particular, people who are HIV-AIDS patients are susceptible to TB infection. Moreover, the emergence of multidrug-resistant (MDR) strains of Mtb seriously threatens TB control and prevention efforts [2]. MDR-Mtb is very expensive to treat; the estimates suggest that it may be ten times as expensive as drug-sensitive Mtb, especially considering the fact that patients with MDR need treatment for three years or more. One third of the 42 million people living with HIV/AIDS worldwide are co-infected with Mtb. Approximately 90% of the people living with HIV die within a few months of becoming sick with TB, if they do not receive proper TB treatment. Persons infected with both HIV and Mtb are 30 times more likely to progress to active TB disease. Recent studies have shown that infection with Mtb enhances replication of HIV and may accelerate the progression of HIV infection to AIDS [3]; for example, the risk of HIV-infected patients developing TB is 5–15% per year after an infectious contact [4]. The current recommended approach to TB treatment is the local directly observed treatment strategy (DOTS) [5]. Even where DOTS has been established, if the MDR rate is locally high, first line drugs (isoniazid, rifampicin, pyrazinamide, and ethambutol) alone give an unacceptably low cure rate. Clinical responses of MDR-TB patient to first line drug have been poor, and in some cases there is no response at all [6]. Second line drugs (amikacin, cycloserine, ethionamide, kanamycin, capreomycin, clofazimine, para-aminosalicylic acid, ciprofloxacin, and ofloxacin) are often poorly effective and tolerated [7]. There are significant problems present with respect to treatment of AIDS and TB co-infected patients. Rifampicin and isoniazid (key drugs of the DOTS therapy) interact with the cytochrome P450 3A4 enzyme pathways, one of the enzymes responsible for drug metabolism. In addition, rifampicin strongly interacts with non-nucleoside reverse transcriptase and protease inhibitors for HIV infections [8]. Thus, clinicians avoid starting Highly Active Antiretroviral Therapy (HAART), which consists of three or more highly potent anti-HIV drugs (commonly reverse transcriptase inhibitors and protease inhibitors), until the TB infection has been cleared [9].
In connection with the ongoing studies on the development of “novel” antimycobacterial agents, we discovered 1,4-dihydroxy-2-naphtoate prenyltrasferase (MenA) inhibitors which also effective in killing Mtb at low concentrations [10]. The purpose of this article is to describe these findings in full, including previously undisclosed molecules and in vitro assay data.
II. NEW TB DRUG TARGETS
There is urgent need and significant interest in developing new TB drugs, however, no new class of TB drugs has been developed in the past 40 years [11–15]. Numerous co-crystal structures of bacterial essential enzymes with their inhibitor molecules have been resolved to date. However, rational drug designs based on essential enzymes existing in Mycobacterium spp. have never been achieved successfully. It may be due in part to the lack of 1) appropriate library molecules to screen unexploited bacterial target proteins, and 2) understanding of mycobacterial physiology. On the other hand, a medium-throughput screening approach using whole cell resulted in the reinvestigations of several promising leads. As result of extensive medicinal chemistry efforts, the clinical trial drugs such as diarylquinoline (R207910, an inhibitor of F1F0 proton pump of ATP synthase), and nitroimidazoles (PA-824 and OPC-67683, their molecular targets remain undefined, but the molecules are active against cell wall lipid biosynthesis) were developed. Due, in large part, to the resurgent efforts of the TB Alliance (The Global Alliance for TB Drug Development) and its public/private partners, numerous compounds have been developed in order to improve current TB-chemotherapies. An excellent comprehensive review of new anti-tuberculosis chemotherapies including the structures, mode of actions, and pharmacokinetics and pharmacodynamics was recently reported [16]. If several of these drug leads become FDA approved anti-TB drugs, the management of drug-resistant TB would be improved. However, many TB drug leads reported are modifications of known antibacterial reagents, and thus their mode of actions remain the same. Ultimate goal of development of the treatment of TB infections is to discover novel antibacterial agents which interfere with novel (or unexploited) bacterial molecular target.
Mycobacteria are obligate aerobes. However, it has been known that tubercle bacilli encounter hypoxic environments in acute disease as well as in latent infection, and the capability of tubercular bacilli to adapt to hypoxic conditions appears to play an important role in vivo [17–20]. For example, Mtb is presumed to lie in a nonreplicating (NR) state (dormancy), particularly in the caseous nodules of the lungs where the lesions have little access to oxygen, and can survive for years in the host by entering a dormant state. About 10% of patients with latent TB are reactivated to cause the risk of fatal diseases [17]. Thus, in addition to necessity of drugs for the treatment of MDR-Mtb, the development of drugs that kill Mtb in any state is very important. However, no TB drugs are effective in killing the dormant form of Mtb in vivo. Wayne and co-workers established a link between oxygen starvation and TB drug resistance; they demonstrated that upon depletion of oxygen in culture, Mtb terminates growth and develops into a characteristic dormant form [21–22]. Importantly, it was observed that the dormant form of Mtb was found to be resistant to most of clinically utilized antimycobacterial agents [23].
The lipid-soluble electron carriers (lipoquinones) occupy a central and essential role in electron transport, and thus, ATP synthesis. The lipoquinones involved in the respiratory chains of bacteria consist of menaquinones and ubiquinones. From the taxonomic studies it is evident that majority of Gram-positive bacteria including Mycobacterium spp. utilize only menaquinone in their electron transport systems [24], and menaquinone biosynthesis is essential for survival of non-fermenting Gram-positive bacteria [25–27]. On the other hand, in the electron transport systems Gram-negative organisms such as E. coli utilize ubiquinone (CoQ) under aerobic conditions, and menaquinone under anaerobic conditions. Moreover, the electron transport chain in humans does not utilize menaquinone. Clearly, the electron transport chain is central component in the production of ATP and the subsequent growth of bacteria (Figure 1). It is speculated that although dormant bacilli are considered to have a less active metabolism and less energy reserves, ATP synthesis in oxidative phosphorylation must be active even in the dormant form in Mtb. Thus, inhibition of menaquinone synthesis could have profound effects on maintenance of dormancy in Mtb. This concept may be supported by the several reports that phenothiazines inhibited NADH:menaquinone oxidoreductase, the first enzyme in bacterial respiratory chain, were effective in killing nonreplicating Mtb [28]. We recently demonstrated that 1) inhibition of MenA (1,4-dihydroxy-2-naphthoate prenyltransferase) (highlighted in Figure 2) showed significant growth inhibitory activities against drug resistant Mycobacterium spp. [10], and 2) effectiveness of MenA inhibitors in killing dormant Mtb in vitro using the Wayne model [21–23].
Figure 1.
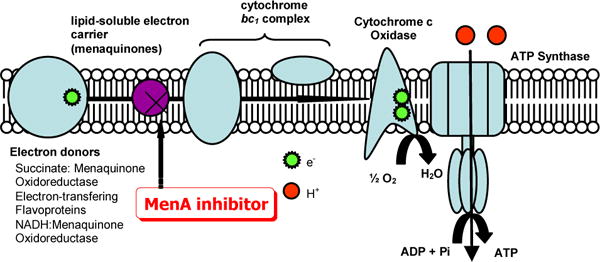
Proposed schematic electron flow in Mtb.
Figure 2.
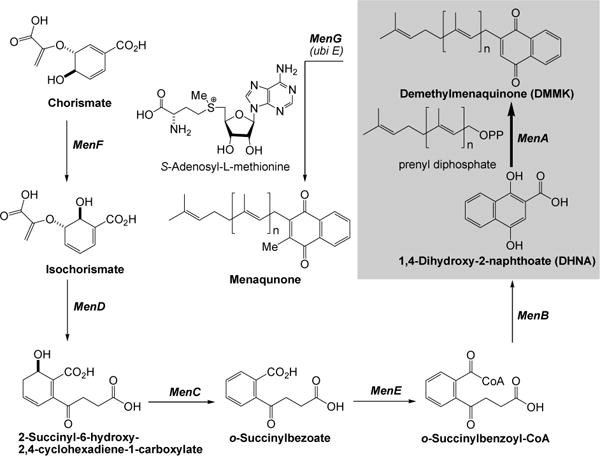
Menaquinone biosynthesis in E. coli.
III. MenA (1,4-DIHYDROXY-2-NAPTHATE PRENYLTRASFERASE)
Menaquinones are the predominant isoprenoid lipoquinones of Gram-positive bacteria, whereas bacteria such as E. coli utilize both menaquinone and ubiqinone (benzoquinone ring structure) (vide supra). The details of the biosynthesis of menaquinone in E. coli. have been reviewed [29–31]. In E. coli the synthesis of menaquinone is accomplished by seven enzymes (MenA-MenG). These enzymes are encoded by 2 clusters of genes. The men cluster consists of the menB,C,D,E,F and a separate cluster containing menA and menG [32]. Menaquinones are identified by the length and chemical structure of the prenyl side chain. The biosynthesis of menaquinone takes place via the intersection of two separate pathways. 1,4-Dihydroxy-2-naphthoate is synthesized from chorismate catalyzed by MenF, D, C, E, and B. On the other hand, prenyl diphosphate of the appropriate size (n = 8 in Mtb, Figure 2) is biosynthesized by the chain-elongation reaction. The naphthoate ring is then prenylated to form demethylmenaquinone (DMMK). MenG catalyzes methylation of DMMK to form menaquinone. Although menaquinone synthesis has been relatively extensively studied in E. coli (due in part to the availability of the men mutants), the synthesis of menaquinone in other organisms has received little attention. Menaquinone synthesis in Mtb has been ignored and virtually no information is available about the properties of these enzymes even though they are distinct from the enzymes involved in synthesizing ubiquinone in humans.
Among the menaquinone biosynthesis proteins, MenA is a membrane associated protein that catalyzes prenylation of 1,4-dihydroxy-2-naphtoate (DHNA) to form demethylmenaquinone (DMMK) (highlighted in Figure 2) [33]. Analysis of the amino acid sequence of MenA was revealed that MenA is likely to have five transmembrane segments, and there are highly conserved Asp which would be located in the inner-plasma membrane as being predicted by using a prediction program (Sosui) [34]. The activity is absolutely dependent on the presence of the divalent cations such as Mg2+. Thus, it is likely that such divalent cations form ion pairs with Asp residues existing in the catalytic site of MenA. As shown in schematic electron flow (Figure 1), MenA inhibitors are able to block the electron flow, resulting in the inhibition of ATP generation. We have obtained experimental evidences which support that “MenA is a valid therapeutic target for TB infections”; for examples, 1) all Gram-positive species tested were susceptible to the MenA inhibitors, on the contrary, all Gram-negative species tested were not susceptible to the MenA inhibitors [10], 2) bacterial growth can be rescued by supplementation with vitamin K1 or K2 at concentrations of many fold higher than the MIC of the MenA inhibitors [35], and 3) growth of drug resistant Mycobacterium spp. was inhibited by the MenA inhibitors.
IV. MenA INHIBITORS: DESIGN AND SYNTHESIS
There is no detailed biosynthetic study of a formal decarboxylative prenylation of 1,4-dihydroxy-2-naphthoate (DHNA) catalyzed by MenA. We hypothesized that the prenylation of DHNA occurs at the C3-position to produce 3-prenyl-1,4-dioxo-1,4-dihydronaphthalene-2-carboxylate (i in Figure 3) whose carboxylate would interact with cationic amino acid residues or Mg2+ ions directly or indirectly through water molecules(s) in the binding site; this hypothesis was concluded based on the observation that 1) the activity of MenA is an absolute requirement for divalent cations, and 2) highly conserved Asps exist in inner-plasma membrane (vide supra). The intermediate i would be tautomerized to form its keto-form ii which concomitantly undergoes decarboxylation to furnish demethylmenaquinone (DMMK). The intermediate i should have a stronger affinity against MenA than DMMK, thus, DMMK may readily be dissociated from the MenA catalytic site.
Figure 3.
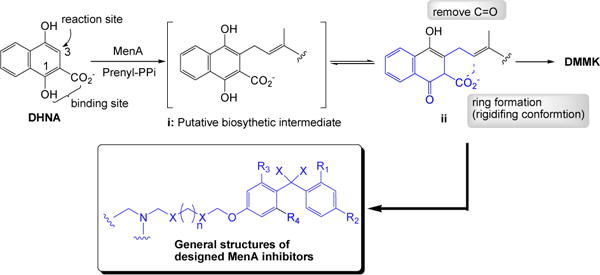
Design of MenA inhibitors.
We designed tertiary or secondary amine or hydrazine containing mimics of ii 1) in hope that the amine moiety would interact with Asp residue(s) directly or through the divalent cation(s) in the active site, and 2) in which the chemically unstable 1,4-quinone system is replaced with the hydrophobicly substituted-benzophenones (Figure 3). In this design for MenA inhibitors, the prenyl moiety of DMMK is mimicked by appropriate length of carbon chain or its heteroatom containing analogs.
As illustrated in Scheme 1, the designed DMMK mimics were synthesized in four to six steps in solution or in four to five steps on the polymer-support. The halogen-substituted (4-methoxyphenyl)(phenyl)methanone derivatives 5 were synthesized by using Friedel-Crafts acylations of 2,4-halogenated benzoyl chlorides 1a~e and anisole derivatives 3a~g with AlCl3 in PhNO2. Regioselectivities of Friedel-Crafts acylation reactions were vary depending on the structure of anisole derivatives 3a~g; however, in all cases the p-addition products were predominant, and the undesired o-addition products were readily separated by chromatography.
Scheme 1.
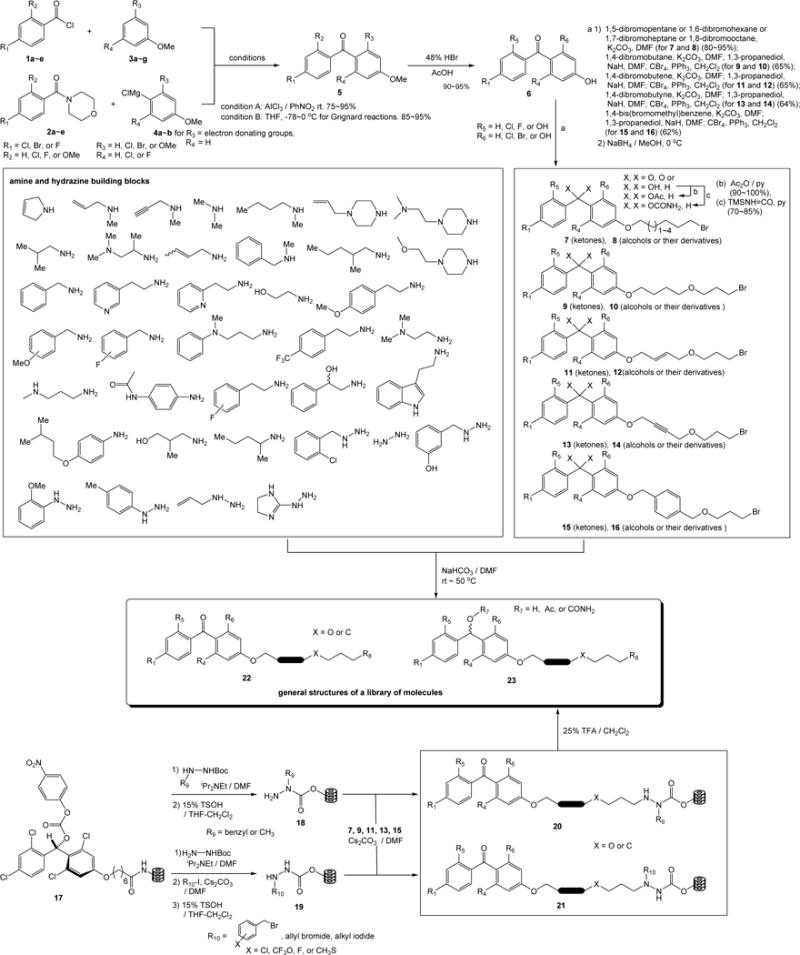
Generation of a library of molecules in solution and on polymer-support.
Alternatively, the benzophenone derivatives were synthesized via Grignard reactions of the morpholin amides 2a~e with 1-halo-4-methoxybenzene or its derivatives. Although a limited number of Grignard reagents can be prepared by the halogen-metal exchange reactions with iPrMgCl, these reactions gave rise to the desired benzophenone in over 90% yield without contamination of the over-alkylation product [36]. Demethylation(s) of the halogen-substituted (4-methoxyphenyl)(phenyl)methanone derivatives 5 by the treatment with HBr in aq. AcOH at 130 °C provided the (4-hydroxyphenyl)(phenyl)methanone derivatives 6 in 90~95% yield. The phenolic alcohol at the 4-position was selectively alkylated with the dibromo compounds to yield the desired monobromide in 85~95% yield. The molecules 9, 11, 13, and 15 were synthesized by the additional two steps of alkylation with 1,3-propanediol and subsequent bromination (CBr4, PPh3) reactions. The diphenylmethanol derivatives 8, 10, 12, 14, and 16 were synthesized by treatment of the corresponding carbonyls with NaBH4 at 0 °C. The generated alcohols could be further functionalized with acylating reagents or a wide variety of isocyanates. In generation of a library of molecules in Scheme 1, however, acetylations with Ac2O and carbamations with trimethylisocyanate were performed. Amination reactions of the bromides 7~16 with a wide variety of amines and hydrazine building blocks furnished a library of molecules in solution as pure forms after chromatographic purifications. To efficiently synthesize hydrazine containing molecules we applied the carbamate linker grafted onto Lantern™ [37] in which selective alkylation at the Nβ position of hydrazine carboxylate resins with the bromide 7, 9, 11, 13, and 15 provided the 1,2-disubstituted and 1,1-disubstituted hydrazine derivatives after cleavage from the resin 20 and 21, respectively [38]. Thus, a library of diphenylmethanone and diphenylmethanol derivatives was efficiently generated in short steps in solution or on the polymer-support.
V. IN VITRO ASSAYS AGAINST MenA AND Mycobacterium tuberculosis
The library of molecules was evaluated in an enzymatic assay in vitro (IC50) against MtbMenA. In these assays [3H]-farnesyl diphosphate was utilized in place of nonaprenylphosphate (n=8 in Figure 2). Into the assay mixtures contained 500 μM DHNA, 10 μM [3H]-farnesyl diphosphate, 5 mM MgCl2, 0.1% CHAPS in 100 mM MOPS (pH 8) and an appropriate amount of membrane protein a various concentrations of the synthesized molecules (Scheme 1) were added [28]. IC50 values were determined using GraFit version 5.0. A series of molecules exhibited IC50 of less than 10 μM against MenA and these molecules were evaluated in mycobacterial growth assay (MIC). Interestingly, in many cases the MIC values were in good agreement with the IC50 values. Representative MenA inhibitors which also exhibited the MIC value of less than 10 μg/mL were summarized in Figure 4. In these in vitro screening analyses of the library molecules it was revealed that 1) the effective substitution pattern (R1, R4, R5, and R6) in the benzophenone moiety should be hydrogen for R4 and R5; the hydrogen atom for the R4 and R5 positions showed significantly better activity than other groups such as the OH or Cl or Br group, 2) C5~C7 of sp3 carbon chain length of the linker (between the phenolic oxygen and terminal nitrogen atom) decreased mycobacterial growth inhibitory activity, on the other hand, the C8 carbon chain length of linker increased the MenA enzyme inhibitory and mycobactericidal activities, 3) the hetero atom such as oxygen can be introduced at the X-position in 22 and 23 without decreasing the activities, 4) the linker moiety can be further diversify by sp2 or sp carbons (the moiety highlighted in 22 and 23), and 5) the allylmethylamine, benzylamine, ethylenediamine, and benzylhydrazine derivatives were the effective terminal amine structures. In general, the reduced forms 23 exhibited lowered MenA inhibitory and mycobactericidal activities. However, in vitro activities of the diphenylmethanol derivatives 23 were improved by the modification of R4 and R5 groups, and carbamation of the alcohol group as seen in 23a (Figure 4). Among the potent MenA inhibitors shown in Figure 4, 22a, named allylaminomethanone-A, was resynthesized and determined MICs against a variety of strains of Mycobacterium spp. As summarized in Table 1, growth of drug resistant Mycobacterium spp. was inhibited by 22a (entries 2 and 3) at low concentrations.
Figure 4.
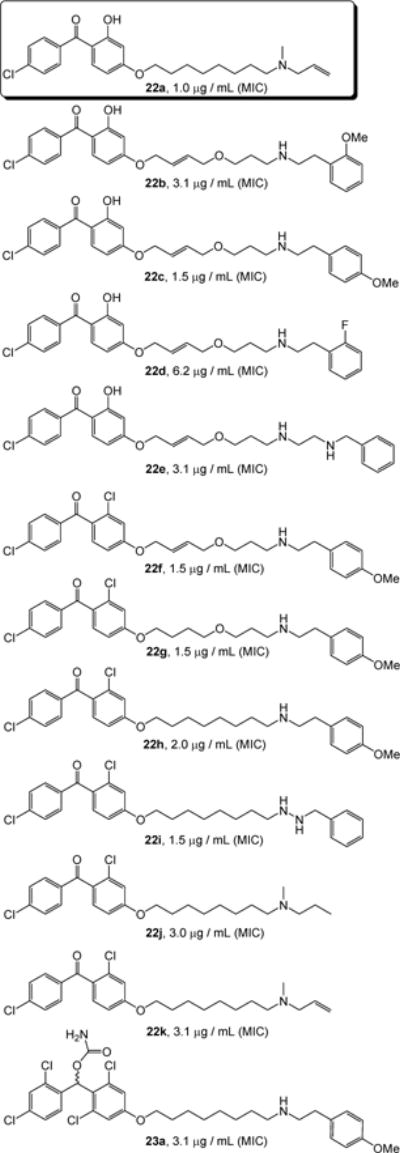
The active MenA inhibitors possessing mycobactericidal activity (MIC <10 μg/mL against H37Rv strain).
Table 1.
MICs of 22a and representative antimycobacterial agents (RFP and INH).
| Entry | Species and strain | MIC (μg/mL)a | ||
|---|---|---|---|---|
| 22a | RFPb | INHc | ||
| 1 | M.tuberculosis H37Rv | 1.0 | 0.2 | 0.1 |
| 2 | M.tuberculosis H37Rv INHr | 1.5 | 0.2 | >25 |
| 3 | M.tuberculosis H37Rv RFPr | 1.5 | >25 | 0.05 |
| 4 | M.bovis BCG Tokyo | 3.1 | 0.1 | 0.1 |
| 5 | M.avium Flamingo | 6.3 | 3.1 | >25 |
| 6 | M.intracellulare ATCC15984 | 6.3 | 3.1 | 12.5 |
| 7 | M.aurum | 6.3 | 0.78 | 6.3 |
| 8 | M.fortuitum NIHJ1615 | 12.5 | >25 | 6.3 |
| 9 | M.smegmatis Takeo | 12.5 | >25 | 12.5 |
The agar dilution method was used.
RFP: rifampicin.
INH: isoniazid.
VI. ACTIVITY of MenA INHIBITORS AGAINST LONG-TERM NONREPLICATING Mycobacterium tuberculosis
In order to study the dormancy response we evaluated the effectiveness of allylaminomethanone-A (22a) against dormant bacteria under low oxygen conditions using the Wayne model [21] with minor modification. An aerobic preculture was diluted 100-fold in tubes and tubes were closed with rubber septa to ensure the anaerobic growth of bacteria. The diluted bacterial suspension was degassed with N2 and incubated anaerobically. After 24 days (well into nonreprecating phase 2 as defined by the Wayne model), the MenA inhibitor(s) in degassed solution was introduced into dormant phase Mtb and incubated for additional 96 h, after which the bacterial suspension was diluted and plated. Culturable bacteria were then grown aerobically at 37 °C and colonies were counted [39]. As summarized in Figure 5, at the concentrations of 10 μg/mL 22a exhibited 320-, 180- and 3-fold more effective in killing nonreplicating bacteria than ethambutol, isoniazid or rifampicin. Significantly, no countable colonies were observed after treatment with 22a at 50 μg/mL concentrations. It is worth mentioning that 22a exhibited the most active in killing nonreplicating Mtb in vitro among antimycobacterial drugs tested in our laboratories.
Figure 5.
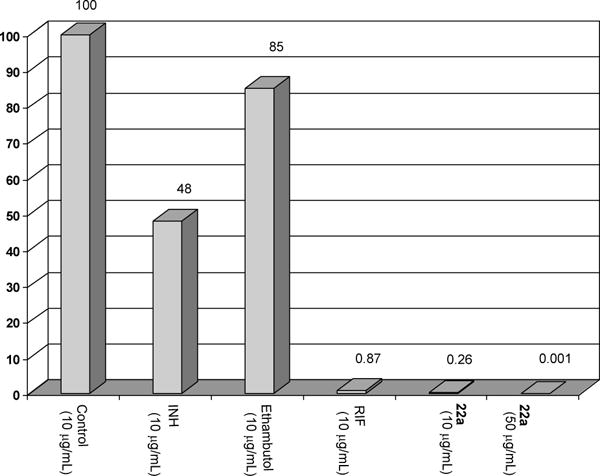
Bacterial survival after exposure to 22a and representative antimycobacterial agents.
VII. DISCUSSION AND CONCLUSION
One of ultimate goals of TB drug development is to develop a drug which has also activity against human latent tuberculosis infection. As descried above, human utilizes ubiquinone in the electron transport chain, whereas Mtb use only menaquinones. However, it has not been addressed whether menaquinone biosynthetic enzyme genes are expressed in any growth condition in Mtb. The mycobactericidal effect of the representative MenA inhibitor such as 22a against nonreplicating Mtb strongly suggested that electron transport is important in maintaining bacterial viability under conditions of restricted oxygen. Several data suggested a role for the DosR/DosS/DosT signaling system, which is required for Mtb genetic response to hypoxia and nitric oxide, in the adaptation of Mtb to conditions that trigger reversible bacterial stasis in vitro, and thus the DosR/DosS/DosT signaling system may contribute to latency in vivo [40]. MenA inhibitors seem to be able to block the electron flow without inducing a dormancy response in Mtb, suggesting that nonreplicating Mtb will behave as if they are in aerobic conditions. Clearly the electron transport chains are central component in the production of ATP and the subsequent bacterial growth (vide supra). It is conceptually very interesting that MenA inhibitors can be developed as indirect ATP synthesis inhibitors. The data descried here indicate that lipoquinones would be a unique and new antimycobacterial target.
We previously reported that the other MenA inhibitors inhibited growth of drug-resistant Gram-positive bacteria (i.e. MRSA, VRSA, and linezolid resistant MRSA) at low concentrations, and all Gram-negative bacteria tested were not susceptible to the MenA inhibitors even at high concentrations [10]. These data strongly indicate that MenA is likely to be a valid drug target in not only Mycobacterium spp. but also other Gram-positive pathogens. The MenA inhibitors described here can be synthesized cost-efficiently and structural modifications to improve the MenA and mycobacterial growth inhibitory activities can be achieved in a time efficient manner. Our recent studies on pharmacokinetics revealed that the advanced MenA inhibitors (structures not shown) possess excellent oral absorption and high distribution to lung tissue. The results are expected to be of significance in terms of discovering new lead compounds that can be developed into new drugs to combat drug-resistant and nonreplicating Mycobacterium tuberculosis. We are currently optimizing the structure of the MenA inhibitor leads to improve mycobactericidal activity in vivo. In addition, because combination therapy will remain mandatory to combat Mycobacterium tuberculosis, we will establish possible combination of MenA inhibitor with known antimycobactericidal drugs. These data will be reported elsewhere.
EXPERIMENTAL
General Procedures and Methods
IR absorptions on NaCl plates were run on a Perkin Elmer FT-IR 1600. 1H NMR spectral data were obtained using Varian 300, 400 MHz instruments. The residual solvent signal was utilized as an internal reference. 13C NMR spectral data were obtained using a Varian 100 MHz spectrometer. Chemical shifts were reported in parts per million (ppm) downfield from TMS, using the middle resonance of CDCl3 (77.0 ppm) as an internal standard. For all NMR spectra, δ values are given in ppm and J values in Hz. Mass spectra were obtained at Colorado State University’s Central Instrument Facility. Reagents and solvents are commercial grade and were used as supplied. Reaction vessels were flame-dried or oven-dried and cooled under an inert atmosphere when necessary.
Syntheses of MenA inhibitors, 22 and 23
Detailed synthesis of 22 through the Friedel-Crafts acylation reactions of 1a~e and 3a~g and additional chemical steps were described in reference 10 [10].
Synthesis of 6 via the Grignard reagents 4b and subsequent demethylation
To a stirred solution of 4b (R3=OMe, R4=H, 40 mmol), which was obtained by the halogen-metal exchange reaction of 1-iodo-2,4-dimethoxybenzene with iPrMgCl, in THF (50 mL) was added 2a (R1=Cl, R2=H, 33.9 mmol) in THF (20 mL) at −78 °C. The reaction mixture was warmed to 0 °C over 1h and quenched with aq. NH4Cl. The water phase was extracted with EtOAc and the combined organic phase was washed with brine, dried over Na2SO4, and evap. in vaccuo. This was subjected to hydrolysis with 48% HBr (50 mL) in AcOH (25 mL) at 120 °C. After 36h, all volatiles were evaporated. Purification by silica gel chromatography (5:1, hexanes:EtOAc) provided (4-chlorophenyl)(2,4-dihydroxyphenyl)methanone (6; R1, R4, R5, R6 = Cl, H, H, OH, 29.0 mmol, 86% overall) as a white powder. Data for (2-chloro-4-hydroxyphenyl)(4-chlorophenyl)methanone: 1H-NMR (CDCl3, 300 MHz): δ 7.52 (d, J = 7.8 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.34 (m, 1H), 6.37 (s, 1H), 6.30 (d, J = 8.7 Hz, 1H), 3.99 (bs, 2H); 13C-NMR (CDCl3, 100 MHz): 198.9, 166.1, 165.6, 137.9, 136.9, 135.9, 131.5, 130.5, 128.8, 112.5, 108.6, 103.4; IR (neat, cm−1): 3288, 2923, 1629, 1599, 1540; HRMS (FAB) calcd. for C13H10O3Cl (M+H+) 249.0313, and observed 249.0305.
Synthesis of 7
To a stirred solution of (4-chlorophenyl)(2,4-dihydroxyphenyl)methanone (6; R1, R4, R5, R6 = Cl, H, H, OH, 7.51 mmol) in DMF (20 mL) was added K2CO3 (5.2 g, 37.6 mmol) and 1,8-dibromooctane (5.1 g,18.8 mmol). After 12h, the reaction mixture was quenched with water and extracted with hexanes. The combined extracts were washed with brine, dried over Na2SO4, and concentrated in vaccuo. Purification by silica gel chromatography (30:1, hexanes:EtOAc) to provide (4-(8-bromooctyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (6.38 mmol, 85 %) as a liquid. Data for (4-(8-bromooctyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (7; R1, R4, R5, R6 = Cl, H, H, OH): 1H-NMR (CDCl3, 300 MHz): δ 12.6 (s, 1H), 7.73 (m, 2H), 7.52 (d, J = 8.4 Hz, 1H), 7.42 (m, 2H), 6.59 (dd, J = 2.4 & 8.7 Hz, 1H), 6.49 (d, J = 2.1 Hz, 1H), 4.06 (t, J = 6.3 Hz, 2H), 3.87 (t, J = 6.3 Hz, 2H), 1.91 (m, 4H), 1.45 (m, 8H).; 13C-NMR (CDCl3, 75 MHz): 205.6, 163.3, 159.1, 138.0, 132.3, 130.7, 128.1, 121.0, 105.4, 99.5, 68.1, 34.0, 32.7, 29.1, 28.8, 28.7, 28.1, 25.7.; LRMS (FAB) C21H24ClO3Br found = 438.
Synthesis of 22a
To a stirred solution of (4-(8-bromooctyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (7; R1, R4, R5, R6 = Cl, H, H, OH, 0.046 mmol) in THF (1 mL) were added NaHCO3 (36.2 mg, 0.46 mmol) and N-methylprop-2-en-1-amine (32.6 mg, 0.46 mmol). After 3days at r.t., the reaction mixture was filtered and all volatiles were evaporated. Purification by PTLC (10:1, hexanes:EtOAc) provided 22a (0.044 mmol, 95 %) as a white powder. Data for 22a: 1H NMR (300 MHz, CDCl3): δ 12.20 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.40 (m, 3H), 6.97 (d, J = 5.4 Hz, 1H), 6.90 (s,1H), 6.16 (m, 1H), 5.57 (d, J = 10.2 Hz, 2H), 3.82 (t, J = 6.0 Hz, 2H), 3.61 (m, 2H), 3.04 (m, 1H), 2.89 (m, 1H), 2.73 (s, 3H), 1.79 (m, 2H), 1.23 (m, 10H).; 13C-NMR (CDCl3, 100 MHz): 194.8, 158.9, 139.1, 138.3, 137.9, 130.7(3c), 128.1(2c), 126.1, 125.9(2c), 120.8, 113.8, 68.0, 58.2, 55.2, 39.3, 28.9, 28.8, 28.7, 26.5, 25.4, 23.6.; HRMS (FAB) calcd. for C25H33O3ClN (M+H+) 430.2071, and observed 430.2065. RT: 8.8 min. (Column: Supelco Discovery HS C-18, Solvent: MeOH, FR: 0.5 mL/min).
Synthesis of 11
To a stirred solution of (4-chlorophenyl)(2,4-dihydroxyphenyl)methanone (6; R1, R4, R5, R6 = Cl, H, H, OH, 4.03 mmol) in DMF (20 mL) was added K2CO3 (8.1 mmol) and 1,4-dibromobutene (10.1 mmol). After 3h at 0 °C, the reaction mixture was quenched with water and extracted with hexanes. The combined extracts were washed with brine, dried over Na2SO4, and concentrated in vaccuo. Purification by silica gel chromatography (15:1, hexanes:EtOAc) provided E-(4-(4-bromobut-2-enyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (3.62 mmol, 90 %) as a liquid. To a stirred solution of 1,3-propanediol (18.1 mmol) in DMF (10 mL) at 0 °C was added NaH (60%, 2.1g, ~54.3 mmol). After 10 min., (E)-(4-(4-bromobut-2-enyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (3.62 mmol) in DMF (4 mL) was added. The reaction mixture was stirred for 2h and quenched with 0.5 N HCl, and extracted with hexanes. The combined extracts were washed with brine, dried over Na2SO4, and concentrated in vaccuo. Purification by silica gel chromatography (20:1, hexanes:EtOAc) to provide E-(4-chlorophenyl)(2-hydroxy-4-(4-(3-hydroxypropoxy)but-2-enyloxy)phenyl)methanone (1.36 g, 3.44 mmol, 95 %) as a liquid. To a stirred solution of E-(4-chlorophenyl)(2-hydroxy-4-(4-(3-hydroxypropoxy)but-2-enyloxy)phenyl)methanone (3.44 mmol) in CH2Cl2 (10 mL) was added PPh3 (4.51 g, 17. 2 mmol) and CBr4 (5.6 g, 17.2 mmol). After 1h at r.t., the reaction mixture was evaporated in vaccuo. The crude mixture was purified by silica gel chromatography (20:1, hexanes:EtOAc) to provide (E)-(4-(4-(3-bromopropoxy)but-2-enyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (3.44 mmol, 100 %) as a liquid. Data for (E)-(4-(4-(3-bromopropoxy)but-2-enyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (11; R1, R4, R5, R6 = Cl, H, H, OH): 1H-NMR (CDCl3, 300 MHz): δ 12.53 (s, 1H), 7.60 (m, 2H), 7.47 (m, 3H), 6.52 (d, J = 2.4 Hz, 1H), 6.44 (dd, J = 2.7 & 9.0 Hz, 1H), 5.96 (m, 2H), 4.61 (m, 2H), 4.06 (m, 2H), 3.59 (t, J = 6.0 Hz, 2H), 3.54 (t, J = 6.6 Hz, 2H), 2.14 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 198.6, 166.2, 165.2, 137.8, 136.5, 134.9, 131.0. 130.3, 128.6, 126.2, 112.9, 107.9, 101.9, 70.5, 68.1, 67.8, 32.8, 30.6. IR (neat, cm−1): 3208, 2930, 1745, 1635,1590; LRMS (FAB) C20H20ClO4Br found = 438.
Synthesis of 22c
To a stirred solution of E-(4-(4-(3-bromopropoxy)but-2-enyloxy)-2-hydroxyphenyl)(4-chlorophenyl)methanone (11; R1, R4, R5, R6 = Cl, H, H, OH, 0.023 mmol) in THF (1 mL) was added NaHCO3 (0.23 mmol) and 2-(4-methoxyphenyl)ethanamine (0.12 mmol). After 24h at r.t., the reaction mixture was filtered and evaporated. Purification by PTLC (10:1, CHCl3: MeOH) to provide 22c (0.022 mmol, 96 %) as a white powder. Data for 22c: 1H-NMR (CDCl3, 300 MHz): δ 7.41 (m, 2H), 7.13 (m, 2H), 6.96 (m, 2H), 6.85 (m, 2H), 6.54 (d, J = 9.0 Hz, 1H), 6.72 (d, J = 2.7 Hz, 1H), 6.16 (dd, J = 2.4, 9.0 Hz, 1H), 5.94 (m, 2H), 4.55 (m, 2H), 4.03 (m, 2H), 3.72 (s, 3H), 3.58 (t, J = 6.0 Hz, 2H), 3.53 (m, 4H), 2.93 (t, J = 6.6 Hz, 2H), 2.13 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 198.5, 168.6, 162.8, 160.2, 132.6, 130.6, 130.4, 130.3, 130.1, 130.0, 128.9, 128.7, 128.6, 126.8, 115.3, 106.2, 102.5, 70.6, 69.0, 67.7, 55.8, 47.2, 36.3, 35.4, 29.9; IR (neat, cm−1): 3052, 2917, 1601, 1509, 1487; LRMS (FAB) C29H32ClO5N found = 497.
Synthesis of other MenA inhibitors
The other MenA inhibitors were synthesized according the procedures described above, however, different building blocks were used. The MenA inhibitors possessing hydrazine group were synthesized according to the procedures described in reference 38 [38].
Enzymatic assay against MenA
The enzymatic activity was characterized using membrane fractions prepared from M. tuberculosis essentially as previously described. M. tuberculosis (H37Rv) was grown to mid-log phase in glycerol-alanine-salts medium, washed with saline and harvested by centrifugation. The resulting pellet was irradiated for 18 h at 2,315 Rads/min using a JL Shepard instrument with a 137Cs source. This exposure was calculated to kill 100% of the bacteria but retain 90% of enzyme activity. The washed cell pellet was resuspended in homogenization buffer containing 50 mM MOPS (pH 7.9), 0.25 M sucrose, 10 mM MgCl2 and 5 mM 2-mercaptoethanol and disrupted by probe sonication on ice with a Sanyo Soniprep 150 (10 cycles of 60 sec on and 90 sec off). The resulting suspension was centrifuged at 27,000 X g for 15 min. The pellet was discarded and the supernatant was centrifuged at 100,000 X g for 1 h in a Beckman Ti70.1 rotor. The pellet (membranes) was resuspended in homogenization buffer, divided into aliquots and frozen at 70 °C. The protein concentration was estimated using a BCA protein assay kit (Pierce, Rockford, IL). MenA assays were conducted essentially as previously described. Assay mixtures contained 500 μM DHNA, 10 μM [3H]-farnesyl diphosphate (American Radiolabeled Chemicals, St. Louis, MO), 5 mM MgCl2 and 0.1% CHAPS in 100 mM MOPS (pH 8) and an appropriate amount of membrane protein. Reactions were stopped by the addition of 1 ml of 0.1 M AcOH in MeOH. The resulting mixture was extracted twice with 3 ml of hexanes and the combined extracts were evaporated to dryness under N2 and redissolved in CHCl3: MeOH (2:1). An aliquot was taken for liquid scintillation counting and the remaining material was subjected to TLC on silica gel plates, which were developed in hexanes: Et2O (95:5). Radioactive spots on the thin layer plated were located and quantitated using a Bioscan System 200 Imaging Scanner. IC50 values were calculated for the inhibitors and compared to the MIC values, in all cases tested there was a good correlation between IC50 and MIC.
MIC determinations against M. tuberculosis H37Rv
We applied colorimetric microtiter plate based method with Alamar blue/visual inspection for the MIC determination with M. tuberculosis. The MIC of all compounds synthesized was determined first against H37Rv by the colorimetric microtiter plate based method with Alamar blue/visual inspection. The compounds demonstrated good activity against H37Rv was tested against MDR-TB strains. The MDR strains are all resistant to isoniazid (INH), streptomycin, rifampicin (RIF), or ethambutol (EMB), and some are resistant to pyrazinamide (PZA) and/or additional second line drugs. Drug susceptibility test methods can vary depending on the size of the inoculum (i.e. density of the cells). We will standardize the size of the inoculum using the frozen stocks with known CFU/mL (approx. 1 ×104). Compounds were tested at 20 μM initially; each microtitre plate has both positive and negative controls and the outside wells are filled with water to help prevent dehydration. If inhibitory activity occurs at 20 μM (this is usually 10 μg/ml of a compound with approx. 500 MW) in the primary screen, serial dilutions of each compound were prepared, and MICs were determined.
Acknowledgments
We thank the national Institute of Health and Colorado State University for generous financial supports. We are grateful to Drs. Matsumoto (Otsuka Pharmaceutical) and Lenaert (Colorado State University) for conducting MIC assays, and in vitro assay using Wayne model, respectively.
REFERENCES AND NOTES
- 1.Rouhi AM. Tuberculosis: A tough adversary. Chem Eng News. 1999;77:52. [Google Scholar]
- 2.Cohen J. New TB drug promises shorter, simpler treatment. Science. 2004;306:1872. doi: 10.1126/science.306.5703.1872. [DOI] [PubMed] [Google Scholar]
- 3.Nunn P, Kochi A. A deadly duo-TB and AIDS. World Health. 1993;46:7. [Google Scholar]
- 4.Aaron L, Saadoun D, Calatroni I, Launay O, Memain N, Vincent V, Marchal G, Dupont B, Bouchaud O, Valeyre D, Lortholary O. Tuberculosis in HIV-infected patients: a comprehensive review. Cli Microbiol Infect. 2004;10:388. doi: 10.1111/j.1469-0691.2004.00758.x. [DOI] [PubMed] [Google Scholar]
- 5.Warner DF, Mizrahi V. Tuberculosis chemotherapy. Influence of stress- and damage-response pathways on drug efficacy. Cli Microbiol Rev. 2006;19:558. doi: 10.1128/CMR.00060-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Telzak EE, Chirgwin KD, Nelson ET, Matts JP, Sepkowitz KA, Benson CA, Perlman DC, El-Sadr WM. Predictors for multidrug-resistant tuberculosis among HIV-infected patients and response to specific drug regimens. Terry Beirn Community Programs for Clinical Research on AIDS (CPCRA) and the AIDS Clinical Trials Group (ACTG), National Institutes for Health. Int J Tuberclo Lung Dis. 1999;3:337. [PubMed] [Google Scholar]
- 7.Crofton J, Horne N, Miller F. Clinical tuberculosis. 2nd. Macmillan; London: 1999. [Google Scholar]
- 8.Nunn P, Brindle R, Carpenter L, Odhiambo J, Wasunna K, Newnham R, Githui W, Gathua S, Omwega M, Mcadam K. Ame Rev Respir Dis. 1992;146:849. doi: 10.1164/ajrccm/146.4.849. [DOI] [PubMed] [Google Scholar]
- 9.Lawn SD, Bangani N, Vogt M, Bekker LG, Badri M, Ntobongwana M, Dockrell HM, Wilkinson RJ, Wood R. Utility of interferon-γ ELISpot assay responses in highly tuberculosis-exposed patients with advanced HIV infection in South Africa. Bio Med Cent Infect Dis. 2007;(28):99. doi: 10.1186/1471-2334-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurosu M, Narayanasamy P, Biswas K, Dhiman R, Crick DC. Discovery of 1,4-dihydroxy-2-naphthoate prenyltransferase inhibitors: New drug leads for multidrug-resistant Gram-positive pathogens. J Med Chem. 2007;50:3973. doi: 10.1021/jm070638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIlleron H, Wash P, Burger A. Antimicrob Agents Chemother. 2006;50:1170. doi: 10.1128/AAC.50.4.1170-1177.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchison DA. The search for new sterilizing anti-tuberculosis drugs. Front Biosci. 2004;9:1059. doi: 10.2741/1293. [DOI] [PubMed] [Google Scholar]
- 13.Duncan K, Barry CE., III Prospects for new antitubercular drugs. Curr Opin Microbiol. 2004;7:460. doi: 10.1016/j.mib.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Immanuel C, Gurumurthy P, Ramachandran G, Venkatesan P, Chandrasekaran V, Prabhakar R. Bioavailability of rifampicin following concomitant administration of ethambutol or isoniazid or pyrazinamide or a combination of the three drugs. Indian, J med Res. 2003;118:109. [PubMed] [Google Scholar]
- 15.Kremer LS, Besra GS. Expert Opin Investig Drugs. 2002;11:1033. doi: 10.1517/13543784.11.8.1033. [DOI] [PubMed] [Google Scholar]
- 16.Potero JL, Rubio M. Expert Opin Ther Patents. 2007;17:617. [Google Scholar]
- 17.Iona E, Giannoni F, Pardini M, Brunori L, Orefici G, Fattorini L. Antimicrobial Agents and Chemothrapy. 2007;51:1537. doi: 10.1128/AAC.01468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boon C, Li R, Qi R, Dick T. J Bacteriol. 2001;183:2672. doi: 10.1128/JB.183.8.2672-2676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKinney JD, Hoener zu Bentrup K, Munoz-Elias EJ, Miczack A, Chen B, Chan WT, Swenson D, Sacchettini C, Jacobs WR, Russell DG. Nature. 2000;406:735. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 20.Weber I, Fritz C, Ruttkowski S, Kreft A, Bange FC. Anaerobic nitrate reductase (narGHJI) activity of Mycobacterium bovis BCG in vitro and its contribution to virulence in immunodeficient mice. Mol Microbiol. 2000;35:1017. doi: 10.1046/j.1365-2958.2000.01794.x. [DOI] [PubMed] [Google Scholar]
- 21.Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996;64:2062. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wayne LG, Sramek HA. Antigenic differences between extracts of actively replicating and synchronized resting cells of Mycobacterium tuberculosis. Infect Immun. 1979;24:363. doi: 10.1128/iai.24.2.363-370.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wayne LG, Sramek HA. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1994;13:908. doi: 10.1128/aac.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suvana KD, Stevenson R, Meganathan R, Hudspeth MES. Menaquinone (vitamin K2): biosynthesis localization and characterization of the menA gene from Escherichia coli. J Bacteriol. 1998;180:2782. doi: 10.1128/jb.180.10.2782-2787.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truglio JJ, Theis K, Feng Y, Gajda R, Machutta C, Tong PJ, Kisher C. Crystal structure of Mycobacterium tuberculosis MenB, a key enzyme in vitamin K2 biosynthesis. J Biol Chem. 2003;24:42352. doi: 10.1074/jbc.M307399200. [DOI] [PubMed] [Google Scholar]
- 26.Collins MD, Jones D. The distribution of isoprenoid quinone structure types in bacteria and their taxonoic implications. Microbiological Review. 1981;45:316. doi: 10.1128/mr.45.2.316-354.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bishop DHL, Pandya KP, King HK. Ubiquinone and vitamin K in bacteria. Biochem J. 1962;83:606. doi: 10.1042/bj0830606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weistein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. Inhibitors of type II NADH: menaquinone oxidoreductase represent a class of antitubercular drugs. PNAS. 2005;102:4548. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin JL, M McMillan F. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12:783. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 30.Bentley R, Meganathan R. Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiol Rev. 1982;46:241. doi: 10.1128/mr.46.3.241-280.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bentley R. Biosynthesis of vitamin K and other natural naphthoquinones. Pure Appl Chem. 1975;41:47. [Google Scholar]
- 32.Shineberg B, Young IG. Biosynthesis of bacterial menaquinones: the membrane-associated 1,4-dihydroxy-2-naphthoate octaprenyltransferase of Escherichia coli. Biochemistry. 1976;15:2754. doi: 10.1021/bi00658a007. [DOI] [PubMed] [Google Scholar]
- 33.Minnikin DE. Lipids: complex lipids, their chemistry, biosynthesis and roles. In: Ratledge C, Stanford J, editors. The Biology of Mycobacteria. Academic Press; London: 1982. pp. 95–184. [Google Scholar]
- 34.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 35.In vitro rescue studies in the presence of 22a (~2μM) bacterial growth was not completely rescued by the supplementation with vitamin K even at 400 μM concentrations. It was reported that total vitamin K (phylloquinone and menaquinone) concentrations are 1.19±0.16 in plasma and 28.0±4.3 nmol/L, respectively. Active vitamin K is expected to be much lower concentrations than the total vitamin K. In addition, although no study on plasma kinetics of the active vitamin K to localized Mycobacterium tuberculosis in host has been reported, it is speculated that uptake of vitamin K by bacteria is limited by low diffusion efficiency of hydrophobic characteristics of vitamin K.
- 36.Kurosu M, Kishi Y. Reaction of methylcerium reagent with tertiary amides: Synthesis of saturated and unsaturated ketones from tertiary amides. Tetrahedron Lett. 1998;39:4793. [Google Scholar]
- 37.Lantern™ (trademark of Mimotopes Pty, Ltd) is cylindrical in appearance and polystyrene grafted surface.
- 38.Kurosu M, Narayanasamy P, Crick DC. High-throughput synthesis of substituted hydrazine derivatives. Heterocycles. 2007;73:169. [Google Scholar]
- 39.Lenaerts AJ, Bitting C, Woolhiser L, Gruppo V, Marietta KS, Johnson CM, Orme IM. Evaluation of a 2-pyridone, KRQ-10018, against Mycobacterium tuberculosis in vitro and in vivo. Antimicr Agents Chemother. 2008;52:1513. doi: 10.1128/AAC.00897-07. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allylaminomethanine-A (22a) did not upregulate either DosS or DosR. Crick. D. C. unpublished data