

Abstract
Introduction
Autophagy is a cellular stress response that plays key roles in physiological processes, such as adaptation to starvation, degradation of aberrant proteins or organelles, anti-microbial defense, protein secretion, and innate and adaptive immunity. Dysfunctional autophagy is recognized as a contributing factor in many chronic inflammatory diseases, including inflammatory bowel disease (IBD). Genetic studies have identified multiple IBD-associated risk loci that include genes required for autophagy, and several lines of evidence demonstrate that autophagy is impaired in IBD patients. How dysfunctional autophagy contributes to IBD onset is currently under investigation by researchers.
Key messages
Dysfunctional autophagy has been identified to play a role IBD pathogenesis by altering processes that include: (1) intracellular bacterial killing, (2) anti-microbial peptide secretion by Paneth cells, (3) pro-inflammatory cytokine production by macrophages, (4) antigen presentation by dendritic cells, (5) goblet cell function, and (6) the endoplasmic reticulum stress response in enterocytes. The overall effect of dysregulation of these processes varies by cell type, stimulus, as well as cellular context. Manipulation of the autophagic pathway may provide a new avenue in the search for effective therapies for IBD.
Conclusion
Autophagy plays multiple roles in IBD pathogenesis. A better understanding of the role of autophagy in IBD patients may provide better subclassification of IBD phenotypes and novel approaches to disease management.
Keywords: autophagy, Crohn’s disease, genetics, inflammatory bowel disease, ulcerative colitis
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are inflammatory bowel diseases (IBD) that affect approximately 1.3 million people in the USA [1]. In the USA, the cost burden of IBD care is estimated to be between $2–3 billion dollars per year in direct costs [2]. These debilitating disorders of the gastrointestinal tract are thought to result from dysregulation of the gastrointestinal immune response due to a complex combination of genetic, microbial, and environmental factors. Both disorders are characterized by a breakdown of the intestinal epithelial barrier function leading to an inappropriate immune response to bacterial antigens and chronic inflammation [3].
The etiology of IBD is complex; genetic studies suggest the presence of susceptibility genes, which when combined with other factors, alter the immune response of the innate and acquired immune systems to commensal flora to result in excessive, chronic inflammation. This chronic inflammation has been attributed to either an inability to remove low-level, chronic inflammatory stimuli, an enhanced and inappropriate inflammatory response, or impaired resolution of inflammatory events [4]. Carriage of IBD susceptibility alleles is not sufficient for disease development and studies link environmental and microbial factors for disease onset. This is evidenced by a low concordance of IBD prevalence in monozygotic twins for CD (20–55%) and UC (6.3–17%)[5]. Other studies reveal an increase in IBD development in countries with historically low IBD prevalence, such as India and China, as these nations have become more industrialized [6]. Additionally, population-based studies in second-generation immigrants to industrialized countries indicate that there is a strong environmental influence on the incidence of IBD, particularly in those who immigrate at a younger age [7, 8]. Furthermore, microbial profiling studies have linked alterations in the composition and location of the intestinal microbiota to the pathogenesis of IBD [9]. Although no causative microbe has yet been identified, increasing evidence supports the expansion of opportunistic pathogens (“pathobionts”), such as adherent-invasive Escherichia coli strains (AIEC), as contributors to the chronic inflammation of IBD[10]. These findings highlight the complexity and interplay of genetic susceptibility, microbes, and environmental factors in the pathogenesis of IBD.
Autophagy-related genes linked to IBD susceptibility
One key breakthrough in understanding the underlying mechanisms of IBD pathogenesis came from large scale genetic analyses, which uncovered key molecular pathways of IBD susceptibility [11]. Currently, there are there are over 200 recognized IBD-associated genetic loci [12, 13]. Many of these loci encode or regulate the function of key receptors and signaling proteins in an array of immune-related responses, including the interleukin-23 receptor and Janus-activated kinase (JAK) signaling, genes involved in innate mucosal defense, cytokine production, lymphocyte activation, epithelial barrier integrity, and multiple proteins involved in autophagy [12]. These findings opened up a new paradigm in the study of IBD pathogenesis that focused on dysregulation of key molecular pathways, rather than analysis of single risk genes or isolated cell types.
The first links between autophagy and Crohn’s disease were identified in genome-wide association studies (GWAS) that uncovered several new susceptibility loci for CD, including polymorphisms in the autophagy genes, autophagy-related gene 16 like 1 (ATG16L1) and immunity-related, GTPase family M (IRGM) [11]. Since then, additional genetic studies have uncovered multiple genetic variants in key components of all steps of the autophagic pathway in IBD patients (Figure 1) [14–16]. There is some overlap in these autophagy-related genetic variants in both CD and UC; however, the majority of the identified variants are more strongly associated with ileal CD. Despite this strong association, the positive predictive value for disease development in individuals carrying autophagy variants is low. A notable example is the ATG16L1 T300A polymorphism, which is linked with CD susceptibility, but is also present in a large proportion of healthy individuals who do not develop IBD [17]. Complete knockout of Atg3, Atg5, Atg7 or Atg16L1 is lethal in mice, and impairment of either Atg7 or Atg16L1 results in severe Crohn’s-disease-like transmural ileitis[18]. These findings are significant because they point towards autophagy as a key pathway in IBD, and disruption of any of the steps results in a similar phenotype.
Figure 1. IBD risk alleles are found in multiple components of the autophagy pathway.
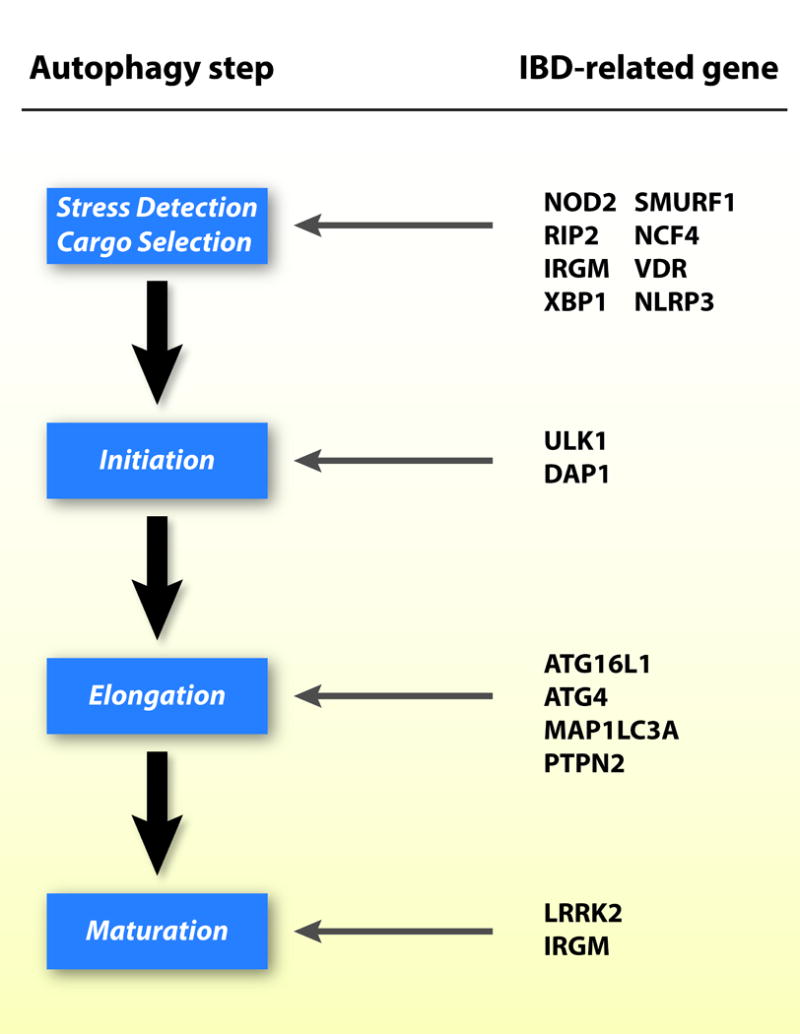
NOD 2, nucleotide-binding oligomerization domain-containing protein 2; SMURF1, Smad ubiquitination regulatory factor 1; RIP2, receptor-interacting protein 2; NCF4, neutrophil cytosolic factor 4; IRGM, immunity-related GTPase family M; VDR, vitamin D receptor; XBP1, X-box-binding protein 1; NLRP3, NOD-like receptor pyrin domain-containing protein 3; ULK1, unc-51-like kinase complex 1; DAP1, death-associated protein 1; ATG16L1, autophagy-related gene 16 like 1; ATG4, autophagy-related protein 4; MAP1LC3A, microtubule-associated protein 1 light chain 3 alpha; PTPN2, protein tyrosine phosphatase non-receptor type 2; LRRK2, leucine rich repeat kinase 2.
Overview of the process of autophagy
Autophagy plays a major role in regulation of metabolic and inflammatory pathways. Impaired autophagy is implicated as a cause or contributing factor for many autoimmune and chronic inflammatory conditions, including IBD. The word autophagy means “self-eating”. It is a stepwise, lysosome-mediated catabolic process by which eukaryotic cells identify and encapsulate cytosolic components for degradation and recycling as an energy source for the cell (Figure 2). Autophagy is a cellular stress response that serves as a protective mechanism by which cells degrade damaged organelles, clear protein aggregates, and survive periods of nutrient starvation, as well as regulates intracellular pathogen clearance and cytokine secretion. It is essential to the functioning of both the innate and acquired immune system, and it has a major role in the cell survival. Although there are different types of autophagy, the focus of this review is macroautophagy, and it will hereafter be referred to as autophagy [15].
Figure 2. The stepwise process of autophagy and sites of dysfunction in IBD. (A).
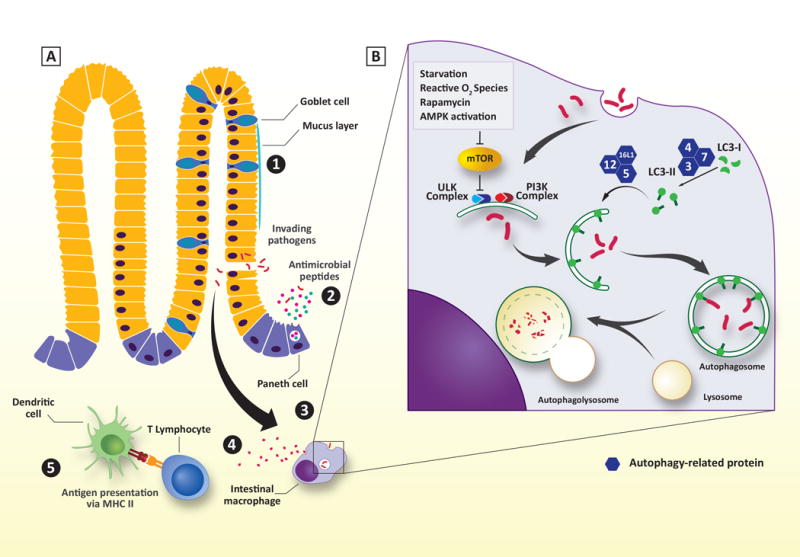
Schematic representation of the intestinal border. Numbers indicate affected sites due to autophagic dysfunction in IBD. (1) Goblet cell function and mucus layer. (2) Paneth cell function. (3) bacterial clearance, ER stress response (not shown). (4) Cytokine production. (5) Antigen presentation by intestinal dendritic cells. (B) Overview of the stepwise process of autophagy. See text for detailed explanation.
Although the classical role of autophagy is to detect, encapsulate, and degrade intracellular materials or pathogens, it is now known that autophagy has additional roles that include secretion of cytokines, antimicrobial peptides (AMPs) and mucins, antigen presentation, and apoptosis. Autophagy is a stepwise process that includes stressor detection, initiation of a multi-lamellar membrane structure, cargo selection, elongation of the autophagosomal membrane, and finally fusion of the autophagosome with the lysosome for degradation (Figure 2).
Autophagy begins when sensors detect specific cues and convert them into signals that are ultimately relayed to the autophagic machinery [19, 20]. Multiple signals lead to the stimulation of autophagy (i.e. nutrient starvation, reactive oxygen species, protein aggregates, etc.) and the majority of these signals converge on two regulators of autophagy, the mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK). Under physiologic conditions, mTOR is activated by growth factors and AMPK activity is inhibited by high intracellular levels of ATP [19, 21]. Cellular stressors which increase intracellular AMP production activate AMPK, which in turn blocks mTOR activity. Loss of mTOR activity is a central signal to activate an autophagic response and may represent a potential therapeutic target to correct defective stressor detection in IBD patients.
Once a stressor is detected, two kinase complexes are activated to recruit autophagosome nucleating proteins and stimulate the elongation of the multi-lamellar autophagosomal membrane. One direct target of mTOR is unc-51-like kinase complex (ULK), propagates the stimulatory signal to a phosphatidylinositol-3-kinase (PI3K) class III lipid kinase complex. The PI3K complex is essential for modifying the forming lipid membrane to create docking sites for additional Atg proteins necessary for cargo selection and membrane elongation.
Two different ubiquitin-like conjugation systems are involved in elongation and closure of the autophagosomal membrane. The Atg7/10 system functions to covalently link Atg12 to Atg5, creating a nucleating partner for a larger Atg16L1 complex that associates with autophagosomal membranes. The other conjugation system (Atg4/7/3) targets the cytosolic microtubule-associated protein 1A/1B-light chain 3 (LC3), called LC3-I. LC3-I is cleaved by the protease Atg4 and then conjugated to phosphatidylethanolamine by the ligases Atg3 and Atg7, forming LC3-II. LC3-II is then inserted into the forming autophagosomal membranes through the actions of the Atg16L1 complex to provide docking sites for cargo selection adaptor proteins, such as sequestosome 1 (SQSTM1/p62), neighbor of BRCA1 (NBR1), nuclear dot protein 52kDa (NDP52), and optineurin [15, 22, 23]. The autophagosome then closes and fuses with lysosomes to form autophagolysosomes and degrade the enclosed cargo.
Processes affected by dysfunctional autophagy in IBD
Variants in autophagy-related genes affect multiple aspects of intestinal innate and acquired inflammatory responses (figure 2). These include abnormalities in bacterial, fungal, and viral clearance, as well as antimicrobial peptide production by Paneth cells, cytokine production, antigen presentation, and response to endoplasmic reticulum (ER) stress [24]. Recent studies show that autophagic defects also decreases goblet cell function, production of mucus membrane defenses, and the absorptive function of microvilli [25].
Goblet cell mucus secretion
Goblet cells are modified columnar epithelial cells in the intestines that synthesize and secrete high levels of mucin glycoproteins (i.e. mucin 2/MUC2), antimicrobial factors (i.e. trefoil factor), and mucus cross-linking proteins, such as Fc-gamma binding protein. This creates the mucus layer which is the first line of defense for the epithelial barrier and maintains a critical gap between the normal gut microbiota and the epithelial surface. In IBD patients, this mucus layer has been demonstrated to be defective, allowing the commensal microbiota to form dense biofilms in close proximity to the epithelium that drive chronic inflammation[26]. Disruption of autophagy by genetic knockout or RNA interfence (RNAi)-mediated knockdown of Atg5, Atg7, or LC3 in mice or cell culture models results in altered goblet cell morphology and markedly decreased mucus secretion [27, 28]. Similar findings were observed in a knock-in mouse model expressing the Atg16L1 T300A variant [29]. These findings suggest that defective autophagy in goblet cells may contribute to IBD onset through increasing the interaction between the microbiota and the epithelium that may drive chronic inflammation or the development of immune responses against the normal microbiota.
Paneth cell crinophagy
Paneth cells are highly specialized epithelial cells of the small intestine that coordinate many physiological functions, including antimicrobial peptide (AMP) production and secretion via crinophagy. They produce high levels of lysozyme, regenerating islet derived protein 3 gamma (RegIIIγ), and human defensin 5 and 6 (HD-5 and HD-6), which play major roles in maintaining the mucus barrier and shaping the microbial composition of the gut [25, 30]. In CD patients carrying risk variants in ATG16L1 or nucleotide-binding oligomerization domain-containing protein 2 (NOD2), the Paneth cell morphology is abnormal with fewer and more disorganized secretory granules present [31]. In more mechanistic studies utilizing mutant mice, decreased expression of Atg5, Atg7, or Atg4B also resulted in abnormal Paneth cell function, including fewer and disorganized granules, and increased production of inflammatory mediators [32]. Recent studies demonstrated that additional IBD risk genes, NOD2 and leucine rich repeat kinase 2 (LRRK2), play roles in Paneth cell lysozyme packaging and secretion as well, suggesting that this autophagic response is regulated by multiple IBD risk genes [31]. A link more direct link to human IBD was identified through studies of transgenic mice with reduced Atg16L1 expression that demonstrated a requirement for an additional environmental insult for the development of altered Paneth cell morphology and CD-like ileitis [14]. These findings suggest that dysfunctional autophagy in Paneth cells may contribute to IBD onset through induction of intestinal dysbiosis that sensitizes these individuals to environmental triggers of IBD.
Xenophagy – a mechanism of microbial clearance
Xenophagy is a term given to autophagy-mediated pathogen clearance [33]. Defective xenophagy is seen in cells carrying ATG16L1 T300A or IRGM disease risk variants, resulting in enhanced AIEC and S. typhimurium intracellular survival [34, 35]. Additionally, CD-associated AIEC stimulates target cells to produce microRNAs against transcripts for multiple, essential autophagy components to decrease expression of these components and blockade of xenophagy [36]. Other disease-associated genetic variants that are associated with defective xenophagy include NOD2, receptor-interacting protein 2 (RIP2), protein tyrosine phosphatase non-receptor type 2 (PTPN2) and sequestosome-1 (SQSTM1/p62). NOD2 is an intracellular protein, which is involved in recognition bacterial wall components, and activation of an anti-microbial response through multiple but inter-related inflammatory pathways. NOD2 is thought to interact with Atg16L1 to guide it to sites of bacterial entry, and cells expressing CD-associated NOD2 variant proteins are unable to localize autophagosomes to the correct cellular location for bacterial capture [15]. SQSTM1/p62 is an adaptor protein that targets cargo to the forming autophagosome, and disease-associated variants also result in AIEC intracellular survival and viable bacteria within the cytoplasm of epithelial cells [37]. Finally, a polymorphism in the coding region for leucine-rich repeat kinase 2 (LRRK2), which is important for autophagosomal maturation, is also linked to defective xenophagy [14]. These findings highlight how multiple steps in the autophagy pathway are impaired by disease-associated variants and pathway dysfunction enhances intracellular bacterial survival in IBD.
Inflammatory cytokine regulation
The human intestine is host to a large number of microorganisms including bacteria, fungi, and viruses. This presents a challenge to intestinal mucosal immune cells, which need to be able to respond to pathogens, but not be activated by the resident microbiota. Therefore, the gut macrophages are programed to preferentially respond with antimicrobial, and not inflammatory, responses within the mucosa [38]. However, in IBD patients the macrophages respond conversely, with an exaggerated inflammatory response to bacteria and ineffective bacterial clearance [15]. In mouse models, deficiencies in proteins involved in the autophagy pathway (i.e. Atg7 and Atg16L1) result in significantly higher secretion of IL-1β and IL-18 in response to inflammatory triggers uniquely in macrophages, both in vitro and in vivo [15, 39] [16]. In contrast to this, inflammatory cytokine release is not enhanced from epithelial cells obtained from patients with either ATG16L1, IRGM or MAP1LC3B risk alleles[40]. This highlights the important role cellular context plays in determining the functional effect of CD risk allele carriage. It also suggests that autophagy plays an important role not only in mounting an immune response against invading pathogens, but also in balancing the inflammatory responses within the cell to reduce intestinal inflammation when appropriate.
Antigen presentation by dendritic cells
Dendritic cells are an integral part of the adaptive immune system. They are found in lymphoid tissue or immune organs, as well as at interfaces between the body and the environment, such as the intestinal mucosa. The main function of dendritic cells is antigen processing and presentation to lymphocytes through both major histocompatibility complexes class I (MHC I) and class II (MHC II). Dysfunctional autophagy interferes with these processes, as demonstrated in animal models with depleted Atg5 expression [15, 41]. In vitro, mouse dendritic cells that are Atg7-deficient are unable to stimulate CD4+ T cell activation when exposed to T. gondii antigens [42]. Further evidence of the role of autophagy in dendritic cell function comes from studies that showed that impaired autophagy resulted in lack of peptide citrullination and presentation of these peptides by DCs. In addition, deletion of Atg16l1 in a mouse model resulted in increased T-cell stimulation by dendritic cells [25]. NOD2-mediated autophagy is required for both bacterial handling and generation of MHC-II restricted CD4+ T cell responses in DCs. A strong link between defects in antigen presentation and IBD in humans is apparent. DCs isolated from CD patients carrying risk variants in NOD2 or ATG16L1 showed impaired MHC II antigen presentation [43]. The findings of these studies suggest that autophagy dysfunction not only affects the efficiency of antigen presentation, but also the repertoire of peptides presented to T cells, the strength of activation, and phenotype of the adaptive immune response.
Endoplasmic reticulum (ER) stress response
Endoplasmic reticulum stress has been suggested to be a contributor to IBD pathogenesis and an early marker of autophagy dysfunction. Increased levels of multiple proteins activated by ER stress were found in the uninflamed intestinal mucosa of IBD patients, as well as in the mucosa of healthy individuals with an ATG16L1 T300A risk genotype [25]. Unfolded proteins trigger ER stress. The unfolded protein response (UPR) mediates cellular response through multiple pathways. Inositol-requiring enzyme 1 (IRE1) is one of those pathways and is associated with both IBD and autophagy. X-box binding protein 1 (XBP1) is a key component in IRE1 pathway. Decreased expression of XBP1 as a result of a genetic variant is associated with IBD [15]. Also, interference with the IRE1 pathway in epithelial cells results in loss of goblet cells and decreases in the intestinal epithelial barrier [44]. These findings suggest that through ER stress response pathways, autophagy has a role in multiple interrelated aspects of the immune response including xenophagy, goblet cell function, and Paneth cell function.
Avenues for IBD therapy through manipulation of autophagy
Multiple studies link defects in autophagy with increased risk of IBD development, suggesting that strategies to target this essential cellular response therapeutically may provide clinical benefit to certain IBD patients. These links come from both genetic studies of IBD risk alleles, as well as emerging studies demonstrating that multiple environmental factors (i.e. smoking, diet, pharmaceuticals, toxins, etc.) also decrease autophagic flux.
Loss of mTOR activity is a central signal for activation of an autophagic response and may represent a potential therapeutic target to correct defective stressor detection in IBD patients. Two mTOR inhibitors (sirolumus and everolimus) showed efficacy in small clinical studies as a treatment for refractory CD. Sirolimus (a.k.a. rapamycin) is an immunosuppressant that has many clinical applications, including prevention of rejection in transplanted organs, and it also stimulates autophagy by inhibiting mTOR [45]. Sirolimus was also shown to be therapeutically effective in a small retrospective cohort study in children who suffered from refractory CD [46]. Everolimus is a similar compound that showed marked and sustained improvement of disease activity in treatment of refractory CD patients in a randomized control trial, but it was not more efficacious than azathioprine.[47]. Furthermore, in animal studies, the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) reduced disease activity in a mouse colitis model by modulating inflammatory responses, potentially through autophagy activation [48]. Although much more research needs to be done, these findings suggest that the use of autophagy stimulating drugs may have promise as a novel treatment for IBD.
Other evidence suggests that autophagy modulation can be controlled through natural products or compounds synthesized naturally. Recent data suggest that melatonin has autophagy modulating properties through its effects on sirtuin activity. [49]. Vitamin D is another naturally synthesized compound that controls autophagic responses through regulation of ATG16L1 and NOD2 expression. Low vitamin D receptor levels in the intestine have been linked to abnormal Paneth cell morphology, impaired xenophagy, and intestinal microbiome dysbiosis [50]. These findings indicate that vitamin D supplementation may be beneficial for IBD patients through stimulating autophagy-mediated pathways and needs further investigation. A natural product, feijoa (also known as pineapple guava), is a fruit native to South America. Hydrophilic feijoa fraction (F3) stimulated autophagy and reduced NFκB-driven inflammation in murine intestinal epithelial cells [51]. Finally, other natural compounds, such as the mushroom Ganoderma lucidum used in traditional Chinese medicine, were identified to modulate autophagic pathways in colon cancer cells, and are potential candidates to test in the context of IBD [52].
Conclusion
The pathogenesis of inflammatory bowel disease is complex and involves multiple risk factors that lead to chronic intestinal inflammation in a genetically susceptible host. Many of the genetic variants that are associated with CD and UC affect regulatory proteins that influence the immune response of the intestines, including proteins that control the autophagic machinery within cells. The autophagic response pathway intersects both the adaptive and the innate immunity pathways, and IBD-associated genetic polymorphisms affect all steps of the autophagy pathway. Autophagy dysfunction can affect bacterial clearance, maintenance of a strong mucus membrane defense through dysfunction of goblet cells and Paneth cells, appropriate pro-inflammatory cytokine secretion, shaping an effective adaptive immune response through diverse and efficient antigen presentation to T cells, and control of the ER stress response. The specific functional effects of autophagy defects is dependent on cell type and stimulus and the synergistic effects of these defects drives disease onset. Mapping the function of key regulators in the autophagic pathway opens the door to identification of potential natural or synthetic compounds that may be beneficial in IBD therapy. The relationship between autophagy and IBD provides insights into new avenues of translational research, and improved diagnostic and therapeutic solutions for IBD patients.
Acknowledgments
Research in our laboratory is supported by grants from the Department of Defense Peer Reviewed Medical Research Program (PR110887 to C.M.), the National Institutes of Health (T32DK083251 and UL1TR000439), and sponsored research from the STERIS Corporation and Metagenics, Inc. We are also grateful for philanthropic support by Gerald and Nancy Goldberg to the CCF IBD Group. The study sponsors did not play any role in the design, collection, analysis, or interpretation of the data and the views presented are solely those of the authors.
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Kappelman MD, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol. 2007;5(12):1424–9. doi: 10.1016/j.cgh.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Park MD, Bhattacharya J, Park K. Differences in healthcare expenditures for inflammatory bowel disease by insurance status, income, and clinical care setting. PeerJ. 2014;2:e587. doi: 10.7717/peerj.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spalinger MR, Rogler G, Scharl M. Crohn’s disease: loss of tolerance or a disorder of autophagy? Dig Dis. 2014;32(4):370–7. doi: 10.1159/000358140. [DOI] [PubMed] [Google Scholar]
- 4.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 5.Gordon H, et al. Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflamm Bowel Dis. 2015;21(6):1428–34. doi: 10.1097/MIB.0000000000000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loftus EV, Jr, Sandborn WJ. Epidemiology of inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31(1):1–20. doi: 10.1016/s0889-8553(01)00002-4. [DOI] [PubMed] [Google Scholar]
- 7.Benchimol EI, et al. Inflammatory bowel disease in immigrants to Canada and their children: a population-based cohort study. Am J Gastroenterol. 2015;110(4):553–63. doi: 10.1038/ajg.2015.52. [DOI] [PubMed] [Google Scholar]
- 8.Li X, et al. Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: a nationwide follow-up study. Inflamm Bowel Dis. 2011;17(8):1784–91. doi: 10.1002/ibd.21535. [DOI] [PubMed] [Google Scholar]
- 9.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146(6):1489–99. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Medina M, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15(6):872–82. doi: 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 11.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lange KM, Barrett JC. Understanding inflammatory bowel disease via immunogenetics. J Autoimmun. 2015 doi: 10.1016/j.jaut.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 13.Liu JZ, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015 doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kernbauer E, Cadwell K. Autophagy, viruses, and intestinal immunity. Curr Opin Gastroenterol. 2014;30(6):539–46. doi: 10.1097/MOG.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian C, Kabi A, McDonald C. Role of Autophagy-Related Genes in the Pathology of Inflammatory Bowel Disease. 2013 [Google Scholar]
- 16.Randall-Demllo S, Chieppa M, Eri R. Intestinal epithelium and autophagy: partners in gut homeostasis. Front Immunol. 2013;4:301. doi: 10.3389/fimmu.2013.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collazo CM, et al. Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med. 2001;194(2):181–8. doi: 10.1084/jem.194.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adolph TE, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503(7475):272–6. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sica V, et al. Organelle-Specific Initiation of Autophagy. Mol Cell. 2015;59(4):522–39. doi: 10.1016/j.molcel.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 20.Brest P, et al. Autophagy and Crohn’s disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med. 2010;10(5):486–502. doi: 10.2174/156652410791608252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 23.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9(9):859–64. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scharl M, Rogler G. Inflammatory bowel disease: dysfunction of autophagy? Dig Dis. 2012;30(Suppl 3):12–9. doi: 10.1159/000342588. [DOI] [PubMed] [Google Scholar]
- 25.Baxt LA, Xavier RJ. Role of Autophagy in the Maintenance of Intestinal Homeostasis. Gastroenterology. 2015 doi: 10.1053/j.gastro.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swidsinski A, Loening-Baucke V, Herber A. Mucosal flora in Crohn’s disease and ulcerative colitis - an overview. J Physiol Pharmacol. 2009;60(Suppl 6):61–71. [PubMed] [Google Scholar]
- 27.Chen GY, Stappenbeck TS. Mucus, it is not just a static barrier. Sci Signal. 2014;7(323):pe11. doi: 10.1126/scisignal.2005357. [DOI] [PubMed] [Google Scholar]
- 28.Patel KK, et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013;32(24):3130–44. doi: 10.1038/emboj.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lassen KG, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111(21):7741–6. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaishnava S, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–8. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha JD, Schlossmacher MG, Philpott DJ. LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat Immunol. 2015;16(9):898–900. doi: 10.1038/ni.3255. [DOI] [PubMed] [Google Scholar]
- 32.Cadwell K, et al. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy. 2009;5(2):250–2. doi: 10.4161/auto.5.2.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardet A, Xavier RJ. Common alleles that influence autophagy and the risk for inflammatory bowel disease. Curr Opin Immunol. 2012;24(5):522–9. doi: 10.1016/j.coi.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Elliott TR, et al. Defective macrophage handling of Escherichia coli in Crohn’s disease. J Gastroenterol Hepatol. 2015;30(8):1265–74. doi: 10.1111/jgh.12955. [DOI] [PubMed] [Google Scholar]
- 35.Fujita N, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol. 2013;203(1):115–28. doi: 10.1083/jcb.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen HT, et al. Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology. 2014;146(2):508–19. doi: 10.1053/j.gastro.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 37.Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012;14(6):791–807. doi: 10.1111/j.1462-5822.2012.01768.x. [DOI] [PubMed] [Google Scholar]
- 38.Smythies LE, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem. 2010;285(25):19593–604. doi: 10.1074/jbc.M109.069955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 40.Scharl M, et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm Bowel Dis. 2012;18(7):1287–302. doi: 10.1002/ibd.21891. [DOI] [PubMed] [Google Scholar]
- 41.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32(2):227–39. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu E, Van Grol J, Subauste CS. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-gamma production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015;17(4):275–84. doi: 10.1016/j.micinf.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–7. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 44.Zhang HS, et al. The Endoplasmic Reticulum Stress Sensor IRE1alpha in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J Biol Chem. 2015;290(24):15327–36. doi: 10.1074/jbc.M114.633560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Massey DC, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut. 2008;57(9):1294–6. doi: 10.1136/gut.2008.157297. [DOI] [PubMed] [Google Scholar]
- 46.Mutalib M, et al. The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J Crohns Colitis. 2014;8(12):1730–4. doi: 10.1016/j.crohns.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 47.Reinisch W, et al. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn’s disease. Am J Gastroenterol. 2008;103(9):2284–92. doi: 10.1111/j.1572-0241.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 48.Bai A, et al. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther. 2010;333(3):717–25. doi: 10.1124/jpet.109.164954. [DOI] [PubMed] [Google Scholar]
- 49.Motilva V, et al. New paradigms in chronic intestinal inflammation and colon cancer: role of melatonin. J Pineal Res. 2011;51(1):44–60. doi: 10.1111/j.1600-079X.2011.00915.x. [DOI] [PubMed] [Google Scholar]
- 50.Sun J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy. 2015:0. doi: 10.1080/15548627.2015.1072670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nasef NA, et al. Extracts of Feijoa Inhibit Toll-Like Receptor 2 Signaling and Activate Autophagy Implicating a Role in Dietary Control of IBD. PLoS One. 2015;10(6):e0130910. doi: 10.1371/journal.pone.0130910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oliveira M, et al. A methanolic extract of Ganoderma lucidum fruiting body inhibits the growth of a gastric cancer cell line and affects cellular autophagy and cell cycle. Food Funct. 2014;5(7):1389–94. doi: 10.1039/c4fo00258j. [DOI] [PubMed] [Google Scholar]