

Abstract
Patterns of gene expression in tumors can arise as a consequence of or result in genomic instability, characterized by the accumulation of somatic copy number alterations (SCNAs) and point mutations (PMs). Expression signatures have been widely used as markers for genomic instability, and both SCNAs and PMs could be thought to associate with distinct signatures given their different formation mechanisms. Here we test this notion by systematically investigating SCNA, PM, and transcriptome data from 2660 cancer patients representing 11 tumor types. Notably, our data indicate that similar expression signatures can be derived from correlating gene expression with either SCNA or PM load. Gene sets related to cell growth and proliferation generally associated positively, and immunoregulatory gene sets negatively, with variant burden. In-depth analyses revealed several genes whose de-regulation correlates with SCNA but not with PM burden, yielding downstream effectors of TP53 and MYC signaling unique to high-SCNA tumors. We compared our findings to expression changes observed in two different cancer mouse models with persistent mitotic chromosomal instability, observing a decrease in proliferative expression signatures. Our results suggest that overexpression of cell-cycle–related genes are a characteristic of proliferation, and likely tumor evolution, rather than ongoing genomic instability.
Genomic instability, defined as an elevated rate in the accumulation of somatic genetic variants, is one of the most distinguishing characteristics between tumor and normal tissue. By facilitating clonal diversification, genomic instability is considered an enabling trait impacting other hallmarks of cancer (Hanahan and Weinberg 2011). Somatic variants comprise point mutations (PMs) and somatic copy number alterations (SCNAs), the latter of which encompass segmental or whole-chromosome aneuploidies (Negrini et al. 2010). PMs and SCNAs arise via different molecular processes, impose distinct stresses upon the cell, and, depending on the context, can complementarily contribute to cancer development by activating or inactivating cancer-associated genes (Kandoth et al. 2013; Zack et al. 2013). PMs can emerge from errors during the procession of DNA polymerases during DNA replication in S phase and from DNA mismatch repair defects (Alexandrov et al. 2013; Helleday et al. 2014). On the other hand, diverse events can lead to SCNAs, including inappropriately repaired double-strand breaks, replication fork stalling, and template switching, as well as errors in chromosome segregation (Branzei and Foiani 2010; Schvartzman et al. 2010; Janssen et al. 2011; Burrell et al. 2013; Yang et al. 2013; Bakhoum et al. 2014). Chromosome instability (CIN) is a term describing a high rate of SCNA formation. While chromosomal gains can by themselves destabilize the genome (Sheltzer et al. 2011; Passerini et al. 2016), it has been proposed that highly aneuploid tumors can also reacquire a high degree of genomic stability (Sheltzer 2013).
SCNAs exert specific as well as global effects on gene expression (Sheltzer et al. 2012; Stingele et al. 2012; Durrbaum et al. 2014). For instance, oncogenes such as MYC or PIK3CA are often focally amplified in cancer (Zack et al. 2013). On the other hand, single chromosome gains mediate widespread effects on expression, which depending on context may lead to fitness disadvantages or advantages (Williams et al. 2008; Sheltzer et al. 2012; Stingele et al. 2012; Durrbaum et al. 2014). Modeling whole chromosome aneuploidies in yeast, plant, and mammalian cells has shown that stoichiometric imbalances of proteins emanating from additional chromosomal material can result in proteotoxic stress and decreased proliferation (Torres et al. 2010; Tang et al. 2011; Oromendia et al. 2012; Stingele et al. 2012; Donnelly et al. 2014; Santaguida and Amon 2015; Santaguida et al. 2015). Lines of research across multiple species (both multicellular and microbial) have underscored that aneuploidy, though immediately disadvantageous, can be beneficial under circumstances of selection (Selmecki et al. 2006, 2015; Rancati et al. 2008; Rowald et al. 2016). Thus, while CIN can promote cancer evolution, too much instability may compromise cell population maintenance or expansion (Sotillo et al. 2010; Lee et al. 2011; Silk et al. 2013; Kuznetsova et al. 2015; Selmecki et al. 2015; Rowald et al. 2016).
Given its relevance for cancer development, ample research has focused on identifying key determinants of genomic instability. Defects in DNA mismatch repair genes as well as POLE mutations, for example, exacerbate errors during DNA replication and dramatically heighten PM burdens (Liu et al. 1995; The Cancer Genome Atlas Network 2012; The Cancer Genome Atlas Research Network et al. 2013; Palles et al. 2013; Shlien et al. 2015). The causative factors behind CIN and the origin of SCNAs, however, are not as clearly defined yet. To identify genes that may result in or cooperate with CIN, several studies have investigated correlations between gene expression and the degree to which a cancer genome is affected by SCNAs (Carter et al. 2006; Endesfelder et al. 2014; Fehrmann et al. 2015). Originally proposed as a signature up-regulated in CIN tumors, the CIN70 signature comprises numerous cell cycle genes (Carter et al. 2006). It is tempting to assume that their appearance in the CIN70 implicates these genes as directly involved in CIN. Indeed, when de-regulated individually in experimental settings, many of these genes have been reported to result in mitotic abnormalities (Sotillo et al. 2007; Neumann et al. 2010; Nam and van Deursen 2014). However, experimental models have indicated that cell cycle genes can also be down-regulated in the context of CIN and aneuploidy (Torres et al. 2007; Williams et al. 2008; Sheltzer et al. 2012; Stingele et al. 2012; Sheltzer 2013; Durrbaum et al. 2014). A more recent study also adopted a correlative approach using a karyotypic index of numerical heterogeneity, defined as the degree of cell-to-cell heterogeneity with regard to chromosome content, as a predictor for CIN in human cell lines (Sheltzer 2013). This study showed that cell cycle gene expression negatively correlates with increased karyotype heterogeneity in cell lines. Based on the hypothesis that the expression changes associated with primary CIN are more akin to the stress response of single chromosome aneuploidies, an alternative signature, the HET70, was hence proposed to reflect clonal diversity and CIN (Sheltzer 2013).
While providing valuable initial insights into associations between signatures reflecting CIN, karyotype heterogeneity, and gene expression, these studies did not investigate changes in gene expression occurring in association with PM accumulation, which in some cancers play a much more predominant role in tumorigenesis than SCNAs (The Cancer Genome Atlas Network 2012; The Cancer Genome Atlas Research Network et al. 2013; Kim et al. 2013). Here, we investigated, across multiple cancer types, gene expression signatures associated with the accumulation of PMs and SCNAs and compared them to those previously reported for CIN.
Results
Expression signatures associated with increasing SCNAs also associate with PM accumulation
We investigated data from The Cancer Genome Atlas (TCGA) encompassing exonic PM, SCNA, and expression data from 11 tumor types representing 2660 specimens (with matched SCNA and exonic PM data) (Supplemental Fig. S1; summarized in Table 1; The Cancer Genome Atlas Research Network et al. 2013; Zack et al. 2013). We implemented two filtering steps. First, with the aim of identifying expression changes corresponding to the global state of a cancer genome, we corrected for dosage effects which might bias the analysis toward genes affected by recurrent SCNAs (see Methods; Supplemental Fig. S2A–C; Supplemental Data 1). Second, given the varying numbers of samples for each tumor type, we employed independent filtering, discarding the genes with the least variance in their expression to minimize multiple testing penalties (Supplemental Fig. S2D). We then computed Spearman's correlation between each individual gene's expression and the total number of PMs and SCNAs per sample, respectively, across all genes (Fig. 1A). A negative correlation for a gene indicates that samples with high variant burden have lower expression of that gene than samples with low variant burden. Conversely, positively correlating genes are more highly expressed in variant-high tumors as compared to variant-low tumors.
Table 1.
Summary of tumor-specific SCNA and exonic PM counts of samples used in this study
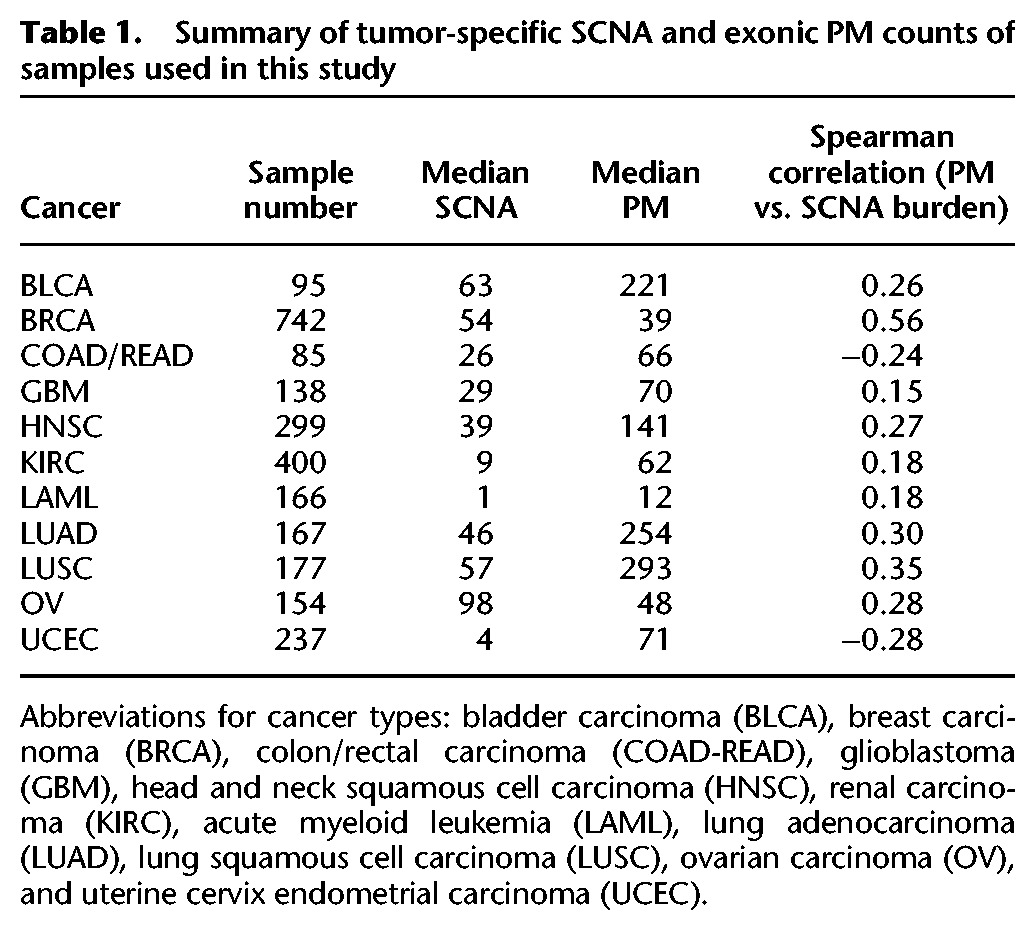
Figure 1.

Comparative correlative analysis between gene expression, exonic PM burden, and SCNA burden. (A) Summary of correlation analysis workflow. (B) Gene-wise comparisons between SCNA burden (x-axis) and PM burden (y-axis). Shown are Spearman correlations across 11 tumor types from TCGA. Tumor type: bladder carcinoma (BLCA), breast carcinoma (BRCA), colon/rectal carcinoma (COAD-READ), glioblastoma (GBM), head and neck squamous cell carcinoma (HNSC), renal carcinoma (KIRC), acute myeloid leukemia (LAML), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian carcinoma (OV), and uterine cervix endometrial carcinoma (UCEC). Gray: FDRPM > 5% and FDRSCNA > 5%; blue: FDRPM > 5% and FDRSCNA < 5%; red: FDRPM < 5% and FDRSCNA > 5%; black: FDRPM < 5% and FDRSCNA < 5%. Representative correlation plots between variant burdens and expression shown in side panels. Colors represent specified FDR cutoffs (see legend). Representative examples of genes whose expression (RSEM, RNA-seq by expectation maximization values) are positively (CDCA8) or negatively (CDKN1A) correlating with SCNA or PM burden are shown in the side panel.
When comparing the resulting correlation coefficients, a striking similarity for PMs and SCNAs became apparent (Fig. 1B; Supplemental Data 1). In seven out of 11 cancer types, the expression of most genes correlated in the same direction with either variant burden, with the most strongly correlating genes often being associated significantly both with PM and SCNA burden (false discovery rate [FDR] < 5%). This included head and neck cancer (HNSC), breast (BRCA) cancer, kidney clear cell carcinoma (KIRC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), acute myeloid leukemia (LAML), and bladder cancer (BLCA)—cancer types that accumulate both SCNAs and PMs. For colorectal carcinoma (COAD-READ), this pattern appeared reversed, with the expression of most genes correlating in the opposite direction with either variant burden, whereas for endometrial cancer (UCEC) there was no clear relationship between the patterns seen for PM and SCNA burden (Fig. 1B). The remaining two tumor types, glioblastoma (GBM) and ovarian cancer (OV), showed only a few significantly correlating genes. These correlation patterns were typically preserved even when we separated SCNAs into two classes, as either focal or whole-chromosome copy number alterations, and analyzed them separately (Supplemental Fig. S2E,F).
A common proliferation signature correlates with somatic variant burden
Next, we sought to further our analyses by identifying biological pathways enriched for genes correlating positively or negatively with SCNA or PM accumulation. To do so, we used two independent approaches. First, we employed Gene Set Enrichment Analysis (GSEA) (Subramanian et al. 2005) to determine which biological pathways were composed of genes the expression of which was positively or negatively correlated with increased variant burden (Fig. 2A,B). Notably, pathways involved in cell cycle regulation and transcription were positively associated both with SCNA and PM burden across cancer types, with cell-cycle–related pathways being the most enriched pathway both for SCNAs and PMs (Fig. 2B; Supplemental Data 2). Other pathways, though comparably affected by SCNA or PM burden, were differentially enriched depending on cancer type. For instance, translational regulation tended to be negatively enriched in AML, OV, and UCEC, yet positively enriched in HNSC, KIRC, and LUSC, possibly reflecting alternative priorities in protein homeostasis depending on the malignancy. Interferon γ signaling and other immune-related pathways negatively correlated with SCNA as well as PM burden with the exception of COAD-READ and BLCA, where these were positively associated with PM burden (normalized enrichment score, NESCOAD-READ = 2.04, FDRGSEA < 10−6 and NESBLCA = 1.68, FDRGSEA = 0.0081) but negatively associated with SCNA burden (NESCOAD-READ = −2.03, FDRGSEA < 10−6 and NESBLCA = −2.03, FDRGSEA = 0.0002), respectively (Fig. 2B).
Figure 2.

Pathway and transcriptional regulation prediction analyses of genes associated with PM and SCNA burden. (A) Examples of 2D gene set enrichment analyses (GSEA). Genes from sample Reactome gene sets are superimposed on HNSC and LUSC correlation plots. (B) GSEA across 11 tumor types. −log10-transformed FDRs are displayed on the y-axis with directionality denoting the direction of the enrichment. Colors denote tumor types described in the legend. (Left panel) SCNA correlation enrichments. (Right panel) PM correlation enrichments. Dotted lines denote cutoffs corresponding to FDRGSEA < 0.05. (C) iRegulon transcription factor prediction of top 200 genes correlating with SCNA (left) or PM (right) burden. Per tumor type (rows), transcription factors (columns) predicted to regulate a significant number (number within tile) are shown. In cases where a transcription factor is predicted, the correlation coefficient of that factor's gene itself is denoted by the color (see legend).
Second, we attempted to predict if specific transcriptional programs were responsible for the observed correlation patterns. Exploring the genes that were correlating highly with increased SCNA or PM burdens, we noted that in many tumor types, master transcriptional regulators responsible for the progression through the cell cycle tended to be among the highest correlating genes (e.g., E2F1 in BRCA, ρSCNA = 0.61) (Ren et al. 2002; Laoukili et al. 2005; Bieda et al. 2006). To complement the previous analysis, we thus employed iRegulon (Janky et al. 2014), a framework used to mine available transcription factor (TF) motif and ChIP-seq data to infer potential transcriptional regulators of an input set of genes. We selected the top 200 genes most highly correlating with SCNA or PM burden and predicted transcription factors potentially regulating them. We found that, in both cases, the majority of genes are predicted to be regulated by TFs, including E2F1, E2F4, TFDP1, FOXM1, and MYBL2, which are themselves highly correlated with somatic variant burden in several tumor types (Fig. 2C; Supplemental Data 3). These TFs are well known to cooperate in regulating various phases of the cell cycle (Laoukili et al. 2005; Sadasivam and DeCaprio 2013). This pattern was not observed in all tumors and was found predominantly among gynecological and lung cancers. In contrast, few recurrently predicted TFs were found to regulate the 200 lowest correlating genes (Supplemental Fig. S3), though these included EP300 and GATA3, both genes with previously described roles in tumor suppression (Gayther et al. 2000; Kandoth et al. 2013). These results indicate that cell-cycle–related pathways are typically up-regulated in the context of either high PM and SCNA accumulation, respectively.
Both SCNA and PM burden converge independently on a common proliferation signature
Most tumor types analyzed here accumulate both PMs and SCNAs throughout their evolution (Table 1). It is thus possible that similarities between transcriptional signatures associated with PMs and SCNAs are caused by their mutual co-occurrence. To exclude this possibility, we focused on UCEC and COAD-READ tumors, where PM and SCNA accumulation are nearly mutually exclusive (Muzny et al. 2012; The Cancer Genome Atlas Network 2012; The Cancer Genome Atlas Research Network 2013). For UCEC, we first classified tumors as either PM-low (<100 PMs) or SCNA-low (<15 SCNAs) (see Methods). Our stratification left us with two groups of ∼100 samples each, within which tumors were relatively invariable for one variant type but highly variable for the other (Fig. 3A). After separation of the tumor samples into two groups, we analyzed the correlation between expression and SCNA burden in PM-low tumors as well as PM burden in SCNA-low tumors. We noted a remarkable similarity between both resulting correlation patterns, with many genes correlating to a significant extent with both SCNA as well as PM accumulation and similarities in gene correlation becoming more apparent with stratification (for both UCEC and COAD-READ) than without stratification (Fig. 3B; Supplemental Fig. S3B; Supplemental Data 1). Again, the highest and most mutually correlating genes were those involved in the cell cycle, for example, E2F1 (ρSCNA = 0.63, ρPM = 0.57). We observed similar results for COAD-READ (Supplemental Fig. S4A,B). Reassuringly, GSEA analysis confirmed the results we obtained in an unstratified setting for both tumor types, yielding a down-regulation in translational regulatory genes and up-regulation of cell cycle/proliferation gene sets (Supplemental Fig. S4C; Supplemental Data 2). These results support the notion that two independent trajectories for tumorigenesis, namely PM or SCNA accumulation, converge on the same gene expression signatures associated with increased proliferation.
Figure 3.

Comparative correlative analysis of gene expression in tumors stratified by variant type. (A) Endometrial cancers (UCEC) segregated based on low-SCNA (red) or low-PM (blue) burden. Insets show sample composition of low-SCNA (left) and low-PM (right) tumors. (B) Comparison between SCNA burden correlations in low-PM tumors (x-axis) and PM burden correlations in low-SCNA tumors (y-axis). Gray: FDRPM > 5% and FDRSCNA > 5%; blue: FDRPM > 5% and FDRSCNA < 5%; red: FDRPM < 5% and FDRSCNA > 5%; black: FDRPM < 5% and FDRSCNA < 5%. Lower right inset displays distribution of the unstratified UCEC correlation distribution.
Next, we related our data to signatures of chromosome instability from two previous studies. Notably, the expression of CIN70 genes (Carter et al. 2006), comprising mostly cell-cycle–related genes, positively correlated with both PM and SCNA burden in stratified as well as unstratified COAD-READ and UCEC settings (Supplemental Fig. S4D,E). The HET70 proposed to describe the response associated with clonal heterogeneity and CIN in cell lines (Sheltzer 2013) was not found to associate with SCNA burden (NESCOAD-READ = −1.33, FDRGSEA = 0.343 and NESUCEC = −1.24, FDRGSEA = 0.233). Collectively, these results indicate that proliferation-like signatures are not exclusive to tumors accumulating SCNAs.
Genes uniquely correlating with SCNA burden reflect preceding genetic events in cancer evolution
Having characterized the commonalities between SCNA and PM burden correlations, we sought to use our approach to identify genes correlating uniquely with SCNA burden. To this end, we again took advantage of the tumor-type–specific analyses in UCEC and COAD-READ, since in these tumor types there is no positive relationship between PM and SCNA burden. We intersected genes that either negatively or positively correlated only with SCNAs, defining a consensus “down” (N = 89) and “up” (N = 70) gene set (Fig. 4A; Supplemental Data 3). Though no specific pathway enrichments were noted by GSEA, iRegulon analysis predicted the “down” gene set to be enriched for targets of a few transcription factors, most notably TP53 (e.g., CDKN1A) and RFX5 (e.g., HLA-DMB and CCND1) (Fig. 4B; Supplemental Data 3). The “up” gene set was enriched especially for targets of MYC but also NFKB1, the latter having been previously implicated in the regulation of senescence in response to DNA damage (Mercurio and Manning 1999; Piret et al. 1999; Chien et al. 2011; Kang et al. 2015; Voce et al. 2015). Among genes predicted to be targeted by both TFs was HSF1, a key regulator of protein homeostasis and previously shown to help trisomic and tetrasomic cell lines cope with aneuploidy (Santagata et al. 2013; Donnelly et al. 2014). These results are consistent with TP53 mutation and loss and MYC amplification, these being common events seen in tumors with complex karyotypes including UCEC and COAD-READ (The Cancer Genome Atlas Network 2012; The Cancer Genome Atlas Research Network 2013; Zack et al. 2013; Lawrence et al. 2014).
Figure 4.

Comparison of gene signatures present in UCEC and COAD-READ. (A) Correlation distributions of COAD-READ and UCEC. Black dots represent FDRSCNA < 5% and FDRPM > 5%. Colored dots represent regulated genes predicted by iRegulon (corresponding transcription factors in legend). Text describes overlap between negative (down consensus) and positive (up consensus) correlating sets. (B) Predicted transcriptional networks based on iRegulon. Circular nodes represent target genes, with predicted regulators denoted by diamonds.
Cancer mouse models of CIN reveal a decrease in pro-proliferative transcription
Interpretation of CIN in cancer genomes is complicated by the fact that patient samples are usually collected many rounds of cell division after SCNA generation. We therefore sought to analyze the consequences on gene expression during forced CIN in solid tumor model systems. To this end, we first compared two previously published tetracycline-inducible breast cancer mouse models initiated with mutant KrasG12D with or without coexpression of Mad2l1 (Sotillo et al. 2010; Rowald et al. 2016), a regulatory component of the spindle assembly checkpoint, under the control of the MMTV promoter (hereafter abbreviated as K, for Kras, and KM, for Kras+Mad2l1, respectively) (Sotillo et al. 2007, 2010). Although both models show the same spectra of recurrent SCNAs (e.g., loss of Chromosome 4 and gain of Chromosome 2), tumor development and mitotic phenotypes differ greatly between K and KM. Whereas K tumors develop rapidly and display few mitotic defects ex vivo, tumors initiated under a KM background lag in their progression owing to mitotic defects early on and demonstrate persistently high rates of mitotic errors and hence CIN ex vivo (Rowald et al. 2016). Accordingly, KM tumors in general exhibit a higher burden of whole or partial chromosome aneuploidies when compared to the K tumor model (Rowald et al. 2016).
To investigate expression signatures, we performed RNA sequencing on K and KM tumors and employed DESeq2 to identify genes differentially expressed between both tumor models. Only a few genes were found to be differentially expressed between K and KM tumors (FDR = 10%, twofold expression change) (see Methods; Supplemental Fig. S5A; Supplemental Table 1). Notably however, ranking fold changes of genes and performing GSEA revealed a down-regulation of CIN70 signature genes (NES = −3.17, FDRGSEA < 10−6) and other cell cycle-associated gene sets, as well as gene sets associated with growth, including transcription and translation (Fig. 5A–C). The HET70 (NES = −1.40, FDRGSEA = 0.0283) and several cell stress-associated gene sets were not found to be differentially enriched in KM versus K tumors (or if so, were only weakly or negatively enriched) (Fig. 5B; Supplemental Fig. S5B; Supplemental Data 2).
Figure 5.

Gene set enrichment analysis of transcriptional changes between Kras-Mad2l1 and Kras breast tumors. (A) Descriptive image of mouse model. (B) GSEA plots of select gene sets. (Left) Running-sum plots with running sum on y-axis and gene rank based on fold change on x-axis. (Right) Ranked fold changes with gene-set members denoted by shaded lines. Normalized enrichment scores (NESs) and FDRs are displayed in the plot. (C) Overview of GSEA results for breast KM versus K models. −log10-transformed FDRs are displayed on the y-axis with directionality denoting the direction of the enrichment. (D) Representative images of cultured tumor cells taken ex vivo for analyses for LC3 staining. Red arrows mark positive and yellow arrows mark negative cells. (E) Comparison of average percentages of cells staining positive for LC3 in K versus KM cells. P-value calculated using Student's t-test. (F) Comparison of LC3 staining intensities in positive staining cells between K and KM cells. P-value calculated using a linear mixed model ANOVA for the effect of genotype on observed staining intensity. At least 18 cells per tumor were used.
We performed immunofluorescence staining of six genes from the HET70 signature in ex vivo cultures, which revealed subpopulations of HET70 positive cells in both K and KM tumor explants (ranging from 0% to 30% per tumor). There were, on average, less positively stained cells in explants from KM tumors as compared to K tumors (P = 0.0164) (Supplemental Fig. S5C,D), suggesting a down-regulation of these six HET70 genes in this CIN breast tumor model and in agreement with our GSEA results. To investigate nontranscriptional responses to stress in these tumors, we further performed immunofluorescence staining of LC3, an autophagy marker known to increase in expression in response to aneuploidy (Santaguida et al. 2015). Specifically, LC3-labeled structures are reported to accumulate in aneuploid cells due to the proteotoxic stress and aggregates elicited by aneuploidy. We observed a slight trend (albeit not nominally significant) toward an increased percentage of cells staining positively for LC3 in KM versus K tumors (P = 0.0712) (Fig. 5D,E). Among positively stained cells, total LC3 intensity in KM explants was higher (P = 0.0434) (Fig. 5F), suggesting a higher proportion of cells experiencing autophagic burden in KM versus K tumors. Despite the slight increase in LC3 intensity between KM and K tumor explants, this change was not reflected in transcriptional changes of genes belonging to a previously reported TFEB-responsive signature (Settembre and Ballabio 2011; Settembre et al. 2011; Santaguida et al. 2015) associated with lysosomal stress and autophagy defects in aneuploid cells (Supplemental Table 2).
Furthermore, interferon γ signaling (NES = 2.96, FDRGSEA < 10−6) and other immune response sets (Supplemental Data 2) were found to be positively enriched in KM tumors. Additionally, some genes included in these gene sets were identified as significantly differentially expressed when analyzed individually (e.g., Cxcl10, log2 fold change = 1.24, Padjust = 0.045), hinting at an immunoregulatory response to CIN in vivo.
We additionally explored the transcriptional consequences of CIN in vivo by reanalyzing previously published microarray data obtained from K and KM tumors in the context of a mouse model of lung cancer (Sotillo et al. 2010). In notable agreement with our results in breast tumors, KM lung tumors expressed less pro-proliferative and pro-growth mRNAs than K tumors (CIN70, NES = −2.43, FDRGSEA < 10−6) (Supplemental Fig. S5E,F) with key CIN70 signature genes, including Ccnb2, showing significant down-regulation (log2 fold change = −1.02, Padjust = 0.0179) (Supplemental Data 4). Thus, tumor models of persistent CIN reveal a decrease in transcription of proliferative gene sets, an increase in immune response gene sets, and an absence of a transcriptional stress signature when compared to an isogenic, genomically more stable tumor model.
Discussion
Our understanding of genomic instability and the dynamics through which this process can fuel tumorigenesis is presently incomplete, and genomic instability hence remains an active area of research (Negrini et al. 2010; Birkbak et al. 2011; Santaguida and Amon 2015). Here, we present results highlighting an up-regulation of proliferative gene expression signatures in tumors with either a high PM or SCNA load. This is predominantly associated with the E2F family and other cell division transcription factors in human tumors. Since this proliferative signature is not specific to the accumulation of a single somatic variant class (e.g., PM vs. SCNA accumulation), it may reflect, rather than continuing instability, an “end point” which is presumably reached through previous periods of genomic instability mediated by alternative variant formation processes. We notably observed up-regulation of cell cycle genes as a common denominator between high SCNA or high PM tumors even when analyzing tumors with a dichotomy of PM and SCNA formation (UCEC and COAD-READ), which enabled us to analyze correlations of one variant class in the absence of the other. We also found that this type of signature is present in several diverse tumor types and is most prominent in gynecological and lung cancers.
The immediate consequences of PM and SCNA formation differ in that the former is less likely to collaterally harm cellular fitness than the latter (Thompson and Compton 2010; Sheltzer et al. 2012; Stingele et al. 2012; Sheltzer 2013). Tumors can take advantage of both variant classes, with some tumor types known to exhibit more recurrent SCNA-mediated driver variants, whereas others exhibit largely recurrent PM-mediated drivers (Ciriello et al. 2013). Nonetheless, the generation of a beneficial variant can occur via either mechanism and enhance clonal fitness, the ultimate manifestation of which is cell growth and division. Subsequent clonal sweeps can act to select cells harboring these driver variants, along with typically numerous passenger variants. The total burden of any variant type may therefore, at least in part, reflect the degree to which the tumor has evolved toward a pro-proliferative phenotype, and these transcriptional changes in cell division genes are likely to be a product of selection rather than reflecting mutational mechanism. The in vivo data presented in our study can help support this notion, with persistent CIN not associating with overexpression but with down-regulation of proliferative gene sets in both breast and lung Kras-driven tumors. It should also be noted in this regard that Mad2l1 overexpression has previously been shown to expedite lung tumor development in mice, a finding that warrants further investigation in the light of our novel results. Overall, our results point toward the transcriptional situation of human tumors with high SCNA burden differing markedly from controlled models where CIN is enforced in vivo. Selection for highly fit, cell cycle gene-expressing clones over the extended developmental time of a full-fledged human tumor may explain these observations.
An important additional implication of our analyses is the potential of an immune response against CIN/aneuploid tumors (Figs. 2B, 5D). This correlation is intriguing, especially considering that a link between immune surveillance and polyploidy has previously been suggested (Senovilla et al. 2012) and that cancer treatments specifically targeting immune checkpoints are currently in several clinical trials (Mellman et al. 2011; Drake et al. 2014). In contrast to our mouse model of CIN tumors, these gene sets were down-regulated with respect to SCNA burden in most clinical tumor types, perhaps reflecting an immuno-regulatory barrier to be overcome by the evolving cancer.
The HET70 and other cell-stress transcriptional signatures showed no or negative enrichment in both mouse models and the human tumors (Fig. 5B; Supplemental Fig. S4C–E), implying that this transcriptional state of cellular distress, which may be the most immediate response to somatic changes in karyotype, may be selected against during clonal sweeps in tumorigenesis. This does not exclude the possibility that cells expressing these signatures were not present or contributing earlier during tumor evolution. Evidence, both from simulation and mouse model studies, suggests that an optimal rate of SCNA generation exists, whereby a population of cancer cells can sample a fitness landscape while maintaining an actively dividing majority (Silk et al. 2013; Laughney et al. 2015). It is possible that in many cancers the primary tumor establishes a dynamic whereby the cells giving rise to novel variation are in a relative minority compared to most other cells, which divide efficiently. The slight but observable increase in LC3-positive cells in KM versus K tumors, as well as the higher LC3-intensity measurements, support this notion. The difference in the observed levels of LC3-staining in KM versus K tumors is, while significant, less pronounced than observed in the context of induced chromosome missegregation (using drugs or MAD2 siRNA knockdown) in cell culture (Santaguida et al. 2015) and may reflect the existence of a less dramatic level of instability in our mouse models of CIN. This is further supported by the lack of evidence of autophagy and lysosome stress signatures in our mouse models (Settembre and Ballabio 2011; Settembre et al. 2011; Santaguida et al. 2015). One possibility may be that taken, ex vivo, the CIN population fraction may be skewed in the context of long-term cell culture, which could be more permissive to chromosome segregation errors. Another possibility could be that responses to SCNA generation vary depending on tissue type and cannot be summarized in a single signature, which is conceivable given that variation exists between tumor types even with regard to the specific genes that correlate with a fixed SCNA burden.
With regard to a signature that reflects CIN in human tumors, we propose that, given that the cancer transcriptome (like the genome) is an average outcome of the entire population when tumor cells are sequenced in bulk, measured expression changes largely associate with fixed SCNAs caused by previously acting CIN. To this effect, we have observed that commonly deleted and amplified genes (e.g., TP53 and MYC) leave some transcriptional footprint unique to SCNA-, but not PM-, high tumors (Fig. 4). A recent study performed with fibroblasts suggests that these aberrations alone are insufficient to rescue the growth defect experienced by aneuploid cells (Sheltzer et al. 2016), in spite of the fact that TP53 disruption can permit the development of CIN tumors and positively influences E2F target transcription and cell division (Barboza et al. 2006; Schvartzman et al. 2011; Parikh et al. 2014). Intriguingly, our association between HSF1 and SCNA accumulation adds support to the hypothesis that de-regulation of protein homeostasis may serve as an adaptive mechanism in highly aneuploid tumors (Donnelly et al. 2014). It will be interesting to see in future studies to what extent the negative effects of CIN are dealt with simply via clonal selection of beneficial SCNAs or by fixed genetic changes (such as TP53 de-activation and HSF1 overexpression) that allow more proficient buffering of cellular stresses associated with CIN.
Methods
TCGA data
The filtered pan-cancer mutation (ID syn1729383) and annotation files as well as RNA-seq (ID syn1695373) and gene-specific SCNA data (IDs syn1695366 and syn169536 for normal/tumor, respectively) for 11 cancer types was retrieved from the Synapse database (www.synapse.org). Additionally, genome-wide SCNA calls from a previous TCGA study were kindly provided by R. Beroukhim (ID syn8076349) (Zack et al. 2013). Data from rectal and colon carcinomas were combined as previously described (The Cancer Genome Atlas Network 2012).
Statistical analyses
All statistical analyses were performed in the R (version 3.2.3) programming language (R Core Team 2008).
Correlation analysis
All analyses were performed per tumor type unless otherwise specified. Total SCNAs were counted from a previously published study, using a minimum size cutoff of 40,000 bp. Total somatic PMs were also counted per tumor. Previous studies did not take into account the fact that genes located on recurrent SCNAs will typically correlate with increasing SCNA burden without actually being a general consequence/cause of aneuploidy itself (Fehrmann et al. 2015). To correct for this, we conservatively omitted sample entries from a correlation calculation for a gene if for that sample the gene was itself within an SCNA, eliminating for the most part this relationship and allowing the study of global changes on the transcriptome caused by increasing SCNA burden rather than the dosage-specific ones. Specifically, this step does not omit any gene entirely from the analysis but omits samples from that gene's correlation calculation when the gene is deleted or amplified in that sample. To increase sensitivity, we then employed independent filtering, now omitting genes from the analysis with the bottom 30% of expression variation, thereby lessening the multiple testing burden. Spearman rank coefficients between a gene's expression and the total burden of either somatic variant were then calculated, and estimated P-values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method (Padjust). For the stratification analysis, cutoffs for SCNA-low and PM-low tumors were chosen to optimally minimize the variation of one variant type in the test set. For the whole chromosome versus focal SCNA analysis, we categorized SCNA events as whole/arm-level if they encompassed 50%–100% of the chromosome size and as focal if they were smaller.
Gene set enrichment analysis
Gene set enrichment analysis was performed as previously described using gene sets from the Reactome database (Subramanian et al. 2005). Briefly, for a given analysis, gene correlations were ranked in descending order, and the running sum was calculated, with the enrichment score (ES) being the highest or lowest value of the running sum. Normalized enrichment scores, accounting for variability in gene set size, as well as false discovery rates (FDRGSEA), taking into account multiple hypothesis testing, were calculated as previously described (Subramanian et al. 2005), and FDR calculations amounting to 0 are annotated as <10−6. We limited gene set sizes to between 50 and 1000 genes. An identical analysis was performed for the expression analysis, using log2 fold change ranks instead of correlation coefficients. We define an enrichment as significant when the absolute value of the NES > 2 and the FDRGSEA < 0.05.
Transcription factor prediction
The top (and bottom) 200 correlating genes of each tumor type were analyzed with the iRegulon Cytoscape plugin on default settings (Janky et al. 2014). Specifically, we chose to focus solely on transcription factor predictions with a minimum normalized enrichment score of three and controlled at an FDR of <0.05 and included only those with ChIP-seq support, which are represented in Figure 2C and Supplemental Figures S2 and S3. Clustering of tumor types was performed based on similarity of TF predictions.
Generation of mouse models
All manipulations regarding the generation of the K and KM mouse models are described in Rowald et al. (2016).
Cell imaging
For immunofluorescence, tumor cells were obtained as described in Rowald et al. (2016). Cells were cultured on coverslips coated with Collagen I (Cultrex, 344.020-01). After 24 h, cells were fixed with 4% PFA for 10 min at room temperature or with a methanol/acetone mix for 10 min at −20°C and permeabilized with 0.2% Triton X-100 PBS for 10 min. Cells were treated with blocking buffer containing 5% donkey serum, 1% BSA, and 0.2% Triton X-100 in PBS for 1 h at RT. The following primary antibodies were used: rabbit anti-LC3 (1:100, Sigma, L7543), rabbit anti-Fibronectin (1:40, Abcam, ab23750), mouse anti-Ataxin1 (1:50, Santa Cruz, sc-365343), mouse anti-Myosin X (1:50, Santa Cruz, sc-166720), mouse anti-RhoA/RhoC (1:100, Thermo Scientific, 1B3-4A10), mouse anti-Vimentin (1:50, Sigma, V2258), and rabbit anti-AKT3 (1:800, Cell Signaling, E1Z3W). Donkey anti-mouse Alexa 488 and anti-rabbit Alexa 568 secondary antibodies were used (1:500, Invitrogen), and the DNA was stained with DAPI. Images were acquired with the Leica LAS 4.5 software on a Leica SP5 confocal microscope. Quantification of LC3 fluorescence intensity was performed with ImageJ software.
Cell imaging analysis
To test for average decreases in HET70 signature genes in KM versus K tumors, we performed 10,000 permutations, scrambling, per gene, the average % positive values among K and KM tumors and calculating the amount of times all six tested genes yielded average % positive cells lower for KM tumors versus K tumors, and from this determined the empirical P-value. To test for significance between K and KM genotypes with regard to average LC3 intensity per cell, we took into account the fact that each tumor alone cannot be considered a true replicate for their respective genotype since prior, unknown tumor evolution has occurred before data acquisition. Thus, we used a linear mixed effect model, with genotype (K or KM) being the fixed effect and sample ID being a random effect, thus taking into consideration variation between tumors of the same genotype. Specifically, we log10-transformed integrated LC3 intensities to normalize the data. An ANOVA was then performed comparing a model using only sample ID and one including sample ID and genotype information to test whether the addition of genotype information significantly contributes to explaining the variation in average LC3 intensity.
RNA libraries and sequencing
Total RNA was extracted from cells from six mice (3× K, 3× KM) using the RNeasy mini kit (Qiagen). RNA quality control was performed using the 2100 Bioanalyzer platform (Agilent). A total of 500 ng total RNA was used as a starting material for the RNA sequencing libraries, which were prepared using the TruSeq strand-unspecific protocol with Ribo-Zero Gold (Illumina) and sequenced on the Illumina HiSeq 2000 platform with 2 × 51 cycles according to the manufacturer's instructions. In order to minimize batch effects, samples were processed on a Beckman Biomek FX robot following the manufacturer's instructions.
RNA sequencing analyses
RNA-seq reads were aligned to the reference built by Bowtie (Langmead et al. 2009) based on Ensembl v62 exons (genome build NCBI37/mm9) (Hubbard et al. 2002). Gene-specific read counts were obtained with the Genomic Alignments package in R, using a strand-unspecific model together with the Ensembl transcript database. Differential expression was assessed with the R package DESeq2 (Love et al. 2014). We defined significant differential expression as a gene with an FDR < 10% and an average twofold gene expression change.
Analysis of microarray data
Microarray data for 22 tumors from Sotillo et al. (2010) were downloaded from the Gene Expression Omnibus (GEO), accession number GSE19753. One microarray was excluded due to quality metrics. The limma package from Bioconductor was used to determine significantly differentiated genes between KM and K backgrounds. P-values were corrected using the Benjamini-Hochberg method. We defined significant differential expression as a gene with an FDR < 10% and an average twofold gene expression change.
Data access
RNA-based sequencing data from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession number PRJEB13611.
Supplementary Material
Acknowledgments
Principal funding for this study came from an ERC starting grant to J.O.K. (336045). In part, this work was funded through a Network of Excellence funding by the European Commission (260791 to J.O.K.). B.R.M. received funding through a Marie Curie IEF postdoctoral fellowship (328621). Work in R.S.’s laboratory is supported by an ERC starting grant (281614), the Howard Hughes Medical Institute, and the German Center for Lung Research. We thank Joachim Weischenfeldt, Alexandros Drainas, Theocharis Efthymiopolous, and Adrian Stütz for thoughtful discussions and Anton Khmelinskii for insightful comments on the manuscript. We also thank the GeneCore, the Center for Statistical Data Analysis, and IT core facilities at EMBL for excellent assistance, particularly R. Hercog, D. Pavlinic, J. Zimmermann, V. Benes, and Bernd Klaus for assistance with sequencing and analysis. The results reported here are in part based upon data generated by the TCGA Research Network, and we acknowledge the specimen donors as well as the research groups involved in the sampling, sequencing, and processing of these data.
Author contributions: C.B., B.R.M., and J.O.K. conceived of the project. C.B. performed data analyses. B.R.M. and J.O.K. supervised the project. K.R. and R.S. generated the mouse models and samples for gene expression analyses, and L.S. performed immunofluorescence analyses. C.B. wrote the paper together with B.R.M. and J.O.K. All authors contributed to discussions concerning the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.212225.116.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Silkworth WT, Nardi IK, Nicholson JM, Compton DA, Cimini D. 2014. The mitotic origin of chromosomal instability. Curr Biol 24: R148–R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G. 2006. p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci 103: 19842–19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieda M, Xu X, Singer MA, Green R, Farnham PJ. 2006. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 16: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, Tan P, Tan IB, Richardson AL, Szallasi Z, Swanton C. 2011. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 71: 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. 2010. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Bio 11: 208–219. [DOI] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. 2013. Replication stress links structural and numerical cancer chromosomal instability. Nature 494: 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Integrated genomic characterization of endometrial carcinoma. Nature 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. 2006. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38: 1043–1048. [DOI] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al. 2011. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev 25: 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. 2013. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 45: 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly N, Passerini V, Durrbaum M, Stingele S, Storchova Z. 2014. HSF1 deficiency and impaired HSP90-dependent protein folding are hallmarks of aneuploid human cells. EMBO J 33: 2374–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CG, Lipson EJ, Brahmer JR. 2014. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol 11: 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrbaum M, Kuznetsova AY, Passerini V, Stingele S, Stoehr G, Storchova Z. 2014. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics 15: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endesfelder D, Burrell RA, Kanu N, McGranahan N, Howell M, Parker PJ, Downward J, Swanton C, Kschischo M. 2014. Chromosomal instability selects gene copy-number variants encoding core regulators of proliferation in ER+ breast cancer. Cancer Res 74: 4853–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann RS, Karjalainen JM, Krajewska M, Westra HJ, Maloney D, Simeonov A, Pers TH, Hirschhorn JN, Jansen RC, Schultes EA, et al. 2015. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat Genet 47: 115–125. [DOI] [PubMed] [Google Scholar]
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, et al. 2000. Mutations truncating the EP300 acetylase in human cancers. Nat Genet 24: 300–303. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. 2014. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 15: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. 2002. The Ensembl genome database project. Nucleic Acids Res 30: 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, et al. 2014. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 10: e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. 2011. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333: 1895–1898. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. 2015. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Laird PW, Park PJ. 2013. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 155: 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova AY, Seget K, Moeller GK, de Pagter MS, de Roos JADM, Durrbaum M, Kuffer C, Muller S, Zaman GJR, Kloosterman WP, et al. 2015. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14: 2810–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. 2005. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nature Cell Biol 7: 126–136. [DOI] [PubMed] [Google Scholar]
- Laughney AM, Elizalde S, Genovese G, Bakhoum SF. 2015. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep 12: 809–820. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, et al. 2011. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 71: 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, Papadopolous N, Peltomaki P, de la Chapelle A, Hamilton SR, et al. 1995. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 9: 48–55. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Coukos G, Dranoff G. 2011. Cancer immunotherapy comes of age. Nature 480: 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F, Manning AM. 1999. NF-κB as a primary regulator of the stress response. Oncogene 18: 6163–6171. [DOI] [PubMed] [Google Scholar]
- Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, et al. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, van Deursen JM. 2014. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol 16: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. 2010. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11: 220–228. [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. 2010. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464: 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. 2012. Aneuploidy causes proteotoxic stress in yeast. Genes Dev 26: 2696–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, et al. 2013. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh N, Hilsenbeck S, Creighton CJ, Dayaram T, Shuck R, Shinbrot E, Xi L, Gibbs RA, Wheeler DA, Donehower LA. 2014. Effects of TP53 mutational status on gene expression patterns across 10 human cancer types. J Pathol 232: 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerini V, Ozeri-Galai E, de Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, Kerem B, Storchova Z. 2016. The presence of extra chromosomes leads to genomic instability. Nat Commun 7: 10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret B, Schoonbroodt S, Piette J. 1999. The ATM protein is required for sustained activation of NF-κB following DNA damage. Oncogene 18: 2261–2271. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. 2008. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135: 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev 16: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowald K, Mantovan M, Passos J, Buccitelli C, Mardin BR, Korbel JO, Jechlinger M, Sotillo R. 2016. Negative selection and chromosome instability induced by Mad2 overexpression delay breast cancer but facilitate oncogene independent outgrowth. Cell Rep 15: 2679–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, DeCaprio JA. 2013. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 13: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Mendillo ML, Tang YC, Subramanian A, Perley CC, Roche SP, Wong B, Narayan R, Kwon H, Koeva M, et al. 2013. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341: 1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Amon A. 2015. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol 16: 473–485. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Vasile E, White E, Amon A. 2015. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev 29: 2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Sotillo R, Benezra R. 2010. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. 2011. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 19: 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, et al. 2015. Polyploidy can drive rapid adaptation in yeast. Nature 519: 349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al. 2012. An immunosurveillance mechanism controls cancer cell ploidy. Science 337: 1678–1684. [DOI] [PubMed] [Google Scholar]
- Settembre C, Ballabio A. 2011. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 7: 1379–1381. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM. 2013. A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res 73: 6401–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. 2011. Aneuploidy drives genomic instability in yeast. Science 333: 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. 2012. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci 109: 12644–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, Amon A. 2016. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell 31: 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, Van Loo P, Tarpey PS, Coupland P, Behjati S, et al. 2015. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet 47: 257–262. [DOI] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. 2013. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci 110: E4134–E4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. 2007. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman JM, Socci ND, Benezra R. 2010. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464: 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. 2012. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol 8: 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. 2011. Identification of aneuploidy-selective antiproliferation compounds. Cell 144: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. 2010. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 188: 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924. [DOI] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. 2010. Identification of aneuploidy-tolerating mutations. Cell 143: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voce DJ, Schmitt AM, Uppal A, McNerney ME, Bernal GM, Cahill KE, Wahlstrom JS, Nassiri A, Yu X, Crawley CD, et al. 2015. Nfkb1 is a haploinsufficient DNA damage-specific tumor suppressor. Oncogene 34: 2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. 2008. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, Hsieh CH, Zhang C, Ren X, Protopopov A, Chin L, et al. 2013. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153: 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al. 2013. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.