

Abstract
There are two members of the vasohibin (VASH) family, VASH1 and VASH2. VASH1 is expressed mainly in endothelial cells to inhibit angiogenesis, whereas VASH2 is expressed mainly in cancer cells to stimulate tumor growth. The aim of the present study was to establish neutralizing monoclonal antibody (mAb) against human VASH2 and apply it as an anti‐cancer treatment. We previously raised mAb against several synthetic peptides of hVASH1, and found that one of them exhibited neutralizing activity against hVASH1. Because of the similarity in the amino acid sequences between VASH1 and VASH2, we hypothesized that they shared the bioactive center. When we mutated four amino acids within the region, the mutant VASH2 lost its pro‐angiogenic activity. Therefore, we raised mAb against a synthetic peptide overlapping the mutated amino acids of hVASH2, and isolated one clone (1760) that almost completely inhibited the stimulatory effect of hVASH2 on the migration of and tube formation by endothelial cells. When we used this clone 1760 antibody for cancer treatment, the peritoneal injection of it inhibited both tumor growth and angiogenesis in a mouse xenograft model of human cancer cells. In terms of anti‐tumor activity, 25 mg/kg of clone 1760 was equivalent to 5 mg/kg of bevacizmab. From these results, we propose the targeting of human VASH2 with neutralizing mAb as a new strategy for cancer treatment.
Keywords: Angiogenesis, monoclonal antibody, mutant protein, synthetic peptide, vasohibin‐2
Angiogenesis (i.e. the formation of new capillaries) is an essential event in various developmental or remodeling processes that take place under physiological and pathologic settings, including cancers. Indeed, angiogenesis is recognized as one of the principal hallmarks of cancers, and, thus, has become a target for the treatment of various cancers.1 Cancer cells express angiogenesis stimulators to develop tumor neo‐vessels. Among those stimulators, vascular endothelial growth factor (VEGF) is thought to play the principal role, as it stimulates the migration and proliferation of, and tube formation by, endothelial cells (EC) Based on these notions, anti‐angiogenic drugs that block the VEGF signaling pathway have been developed; and they are now in clinical use for the treatment of various cancers.2 The benefits of such drugs are well‐proved, but because of problems such as drug resistance and/or side effects, the development of novel anti‐angiogenic drugs with different modalities is anticipated.2
Previously we searched for and isolated novel angiogenesis regulators that we designated as vasohibin‐1 (VASH1) and vasohibin‐2 (VASH2).3, 4 VASH1 is an intrinsic angiogenesis inhibitor preferentially expressed in EC,3 whereas VASH2 is a homolog of VASH1 contradictorily exhibiting pro‐angiogenic activity.4, 5 These two regulators lack the classical signal sequence for secretion, but are efficiently secreted when they bind to small vasohibin‐binding protein (SVBP).6 Importantly, VASH2 is expressed in various cancers and promotes tumor growth.7, 8, 9, 10, 11, 12, 13 Thus, VASH2 can be a target for anti‐angiogenesis therapy in cancers. Indeed, a previous study of ours revealed that the knockdown of VASH2 in cancer cells by the local injection of its siRNA results in notable inhibition of tumor growth and tumor angiogenesis in a murine xenograft model of ovarian cancer.14
In the present study we developed anti‐human VASH2 neutralizing monoclonal antibodies for such therapeutic use. To do so, we identified the bioactive center of human VASH2 responsible for its pro‐angiogenic activity, made a synthetic peptide of that domain, and immunized mice with the synthetic peptide. Thereby, we developed a neutralizing anti‐human VASH2 monoclonal antibody and tested it as an anti‐cancer treatment.
Materials and Methods
Cell culture and reagents
The murine Leydig tumor cell line MLTC‐1 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and was chosen because we had previously confirmed that endogenous VASH2 is not expressed in this cell line.9 Human ovarian serous adenocarcinoma cell line SKOV‐3 was used as described previously.9 Cells were cultured in RPMI1640 medium (Wako Pure Chemical Industries, Osaka, Japan) supplemented with 10% heat‐inactivated FBS (BioWest, S.A.S, Nuaille, France). HUVEC were obtained from Kurabo Industries (Osaka, Japan) and were cultured in type I collagen‐coated dishes (Iwaki, Chiba, Japan) containing endothelial basal medium (EBM)‐2 (Lonza, Walkersville, MD, USA) supplemented with EGM‐2‐MV‐SingleQuots (Lonza) containing VEGF, FGF‐2, insulin‐like growth factor‐1, epidermal growth factor and 5% FBS. We used more than three different lots of HUVEC and confirmed the reproducible responses. All cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Recombinant human VASH2 protein was described previously.4 Bevacizumab was obtained from Chugai Pharmaceutical (Tokyo, Japan).
Establishment of wild type or mutated human. VASH2‐expressing murine tumor cell clones
Human VASH2 (hVASH2) cDNA was cloned into the pCALL2‐pcDNA3.1/Hygro vector as described previously.9 A mutated hVASH2 cDNA fragment was obtained by replacing Lys281, Glu282, Leu283 and Glu284 with four consecutive alanine residues by PCR using primers 5′‐TGCAGCCCTTATGTCAGCCCTCAG‐3′ and 5′‐GCCGCGAAATATGCCAGGGACATG‐3′. This fragment cDNA was cloned into the pCALL2‐pcDNA3.1/Hygro vector as was the hVASH2 cDNA. MLTC‐1 cells were transfected with wild‐type (WT) or mutated VASH2 expression vector or control vector with Effectene transfection reagent (QIAGEN, Venlo, Netherlands) according to the manufacturer's instructions. After the transfection, the cells were selected in hygromycin‐containing medium (Invitrogen Life Technologies, Carlsbad, CA, USA). Finally, WT or mutated VASH2‐expressing clones and control clones were established.
RT‐PCR
Total RNA was extracted from cell cultures with ISOGEN‐II (Nippon Gene, Toyama, Japan) according to the manufacturer's instructions. The concentration of extracted total RNA was determined by using a Nanodrop 2000c spectrophotometer (Thermo Scientific, Wilmington, DE, USA). First‐strand cDNA was generated with ReverTra Ace (Toyobo, Osaka, Japan). The RT‐PCR procedure was performed in a DNA thermal cycler (Takara Bio, Tokyo, Japan). PCR conditions consisted of an initial denaturation step at 94°C for 5 min followed by 35 cycles comprising a 15‐s phase at 94°C (denaturation), a 30‐s phase at 56°C (annealing) and a 30‐s phase at 72°C (extension). PCR products were separated on a 2% agarose gel and visualized under ultraviolet rays by ethidium bromide staining. The primer pairs used were as follows: human glyceraldehyde‐3‐phosphate dehydrogenase gene forward primer, 5′‐ACCACAGTCCATGCCATCAC‐3′, and reverse primer, 5′‐TCCACCACCCTGTTGCTGTA‐3′; human VASH2 forward primer, 5′‐ACGTCTCAAAGATGCTGAGG‐3′, and reverse primer, 5′‐CTCTCCGACCCAAGTGAGAA‐3′; and mutated human VASH2‐specific forward primer, 5′‐GCTGACATAAGGGCTGCAGC ‐3′, and reverse primer, 5′‐AGCCCACTTCATTCAGAGTG ‐3′.
Cell proliferation
Proliferation of tumor cells was measured by carrying out the Tetra COLOR One cell proliferation assay (Seikagaku, Tokyo, Japan). Briefly, wild‐type or mutated hVASH2‐expressing clones of MLTC‐1 and the mock transfectants were seeded at a density of 2 × 103 cells/well in a 96‐well plate and then incubated at 37°C. After 72 h, 5 μL of Tetra COLOR One was added to each well. The mixture was subsequently incubated for an additional 3 h, after which the absorbance at 450 nm was measured.
Cell migration
Wild‐type or mutated hVASH2‐expressing clones of MLTC‐1 (1 × 106 cells) and mock transfectants were seeded into the lower chambers of Boyden chambers (Corning, Lowell, MA, USA) and cultured in serum‐free medium for 24 h for the secretion of wild‐type or mutated hVASH2 protein. Then HUVEC were plated at 5 × 104 cells/well in the upper chambers (8.0‐μm pore size) of the Boyden chambers. After incubation at 37°C for 4 h, the cells that had migrated across the membrane were stained with Giemsa, and those in five random fields were counted at the magnification of ×200. To assess the neutralizing activity of hVASH2 mAb, we added several kinds of Abs to the lower chamber of a Boyden chamber, on the bottom of which WT hVASH2‐expressing cells had been seeded, as mentioned above.
Tube formation
Wild‐type or mutated hVASH2‐expressing clones of MLTC‐1 cells and the mock transfectants were cultured in serum‐free medium for 24 h. The conditioned medium (CM) was then obtained from each cell culture dish. Next, cellular components were removed from the CM by using a Millex GP filter (0.22 μm; PES, 33mm; Millipore, Billerica, MA, USA). The bottoms of 24‐well plates were coated with 500 μL of growth factor‐reduced Matrigel matrix (BD Biosciences, San Diego, CA, USA). Then 1 × 105 serum‐starved HUVEC in 500 μL of M199 medium containing 0.5% FBS were added to the coated plates, and 250 μL of CM obtained as described above were added at the time of plating. After a 6‐h incubation, the tube formation was visualized under a phase‐contrast microscope, and the branching points were counted in four random fields at ×40 magnification. To assess the neutralizing activity of hVASH2 mAb, we added various kinds of Abs (5 μg/mL) to wells containing 1 × 105 serum‐starved HUVEC in M199 medium.
Monoclonal antibody
The mouse monoclonal antibodies against synthetic human VASH2 peptide were established by Bio Matrix Research (Chiba, Japan). Briefly, after mice had been immunized six times with PEGylated Cys‐VASH2 peptide (Leu269‐Arg288 of human VASH2 protein), spleen cells from the immunized mice were fused with myeloma cells to generate hybridomas.
Dot blot analysis
Aliquots of 50 ng of purified recombinant human VASH2 protein were spotted onto PVDF membranes. The blot was blocked for 1 h at room temperature with 5% nonfat dry milk in PBS supplemented with 0.1% Tween 20 (PBST) and then incubated at 4°C with conditioned culture medium collected from hybridoma clones that had been cultured overnight. Anti‐human VASH2 mAb (5E3 clone)4 and mouse normal IgG (Santa Cruz) were used as positive and negative controls, respectively. Next, the membrane was washed three times with PBST followed by incubation with HRP‐conjugated secondary antibody in 5% nonfat dry milk‐PBST for 1 h at room temperature. After three more washes with PBST, immunoreactive protein spots were detected by using Immobilon Western HRP Substrate (Millipore) and an LAS‐4000 (Fuji Photo Film, Tokyo, Japan).
ELISA
Recombinant His6‐tagged hVASH2 protein was added to each well in His Grab Copper Coated High Binding Capacity plates (Thermo Scientific) and incubated for 1 h at room temperature. The plates were then washed three times with PBST and blocked with PBS containing 2% Block Ace (Dainippon Pharmaceutical, Osaka, Japan), 10% sucrose, and 0.1% ProClin150 (Sigma‐Aldrich, St. Louis, MO, USA) for 30 min at room temperature. The monoclonal antibodies against hVASH2 (1 μg/mL in PBS containing 1% Block Ace and 0.1% ProClin150) were added to the plates, and the samples was incubated for 1 h at room temperature. Next, the plates were washed three times with PBST and then incubated with HRP‐conjugated secondary antibody in PBS containing 1% Block Ace and 0.1% ProClin150 for 1 h at room temperature. After three additional washes with PBST, the immunoreactivity was visualized by incubation with TMB substrate solution (Cell Signaling Technology). Subsequently, the reaction was terminated by the addition of STOP solution (Cell Signaling Technology); and the absorbance at 450 nm was then measured with an iMark Microplate Reader (Bio‐Rad).
Western blot analysis
Purified recombinant human VASH2 protein was separated by SDS‐polyacrylamide gel electrophoresis and electrotransferred to PVDF membranes. Detection of protein bands immunoreactive with anti‐human VASH2 mAb (1760 clone) was performed according to the standard procedure as described under the subheading ‘Dot blot analysis’.
Animal studies
This study was approved by Tohoku University Center for Gene Research and carried out under the guidelines for animal experimentation of Tohoku University. Vash2 lacZ/lacZ mice are described elsewhere.5 Female 6–8‐week‐old BALB/c nude mice were obtained from Charles River Laboratories Japan (Yokohama, Japan). The mice were kept in a specific pathogen‐free environment at the animal facility of Tohoku University.
Inoculation of mice with WT‐VASH2 and Mut‐hVASH2 transfectants
A total of 3 × 106 wild‐type or mutated hVASH2‐expressing clonal MLTC‐1 cells or mock transfectants were inoculated s.c. into the dorsal flank of 6‐to‐8‐week‐old Vash2 lacZ/lacZ mice. Tumor size was measured with digital calipers twice per week, and the tumor volume was estimated by using the following formula: volume = 1/2 × (long diameter) × (short diameter)2. At day 26, the mice were killed and subcutaneous tumors were harvested for subsequent analyses.
Treatment of mice with anti‐hVASH2 neutralizing mAb
SKOV‐3 cells were injected s.c. in the dorsal flank of BALB/c nude mice at 1 × 106 cells/mouse. When the tumors became measurable (approximately 5 mm in diameter), we initiated the administration of each mAb. Based on the results of in vitro analysis, we chose clone 1760, which had the highest neutralizing activity, and applied it to an in vivo experimental model.
To assess the anti‐tumor effect of clone 1760, we administered the agent i.p. at various doses: 10, 15, 25 or 50 mg/kg twice per week for 3 weeks. For comparison with the existent anti‐angiogenic agent bevacizumab, mice were treated i.p. with bevacizumab (5 mg/kg) or clone 1760 (25 mg/kg) twice per week for 3 weeks. Control mice were treated with 5 mg/kg of mouse IgG (ab37355; Abcam Japan, Tokyo, Japan) twice weekly for 3 weeks. Then, 22 days after initiation of treatment, the mice were killed; and their tumors were harvested, weighed and snap frozen for future studies. Each experimental group consisted of eight animals. Serial tumor growth was monitored before and after each injection of antibody, and the tumor volume was calculated according to the following formula: volume = 1/2 × (long diameter) × (short diameter)2.
Immunohistochemical analysis
For the immunohistochemical analysis of tumor angiogenesis, the tumors were frozen in optimal cutting temperature (OCT) compound (Sakura, Tokyo, Japan), cut into 7‐μm sections, fixed in methanol for 20 min at −20°C, and blocked with 1% BSA in PBS for 45 min at room temperature. Primary antibody reactions were performed overnight at 4°C with rat monoclonal antibody against mouse CD31 (BD Biosciences) at a dilution of 1:500, and with mouse monoclonal antibody against mouse α smooth muscle actin (αSMA; Sigma‐Aldrich) at a dilution of 1:800. Secondary antibody reactions were performed for 1 h at room temperature with Alexa Fluor 488‐conjugated donkey anti‐rat IgG or Alexa Fluor 594‐conjugated goat anti‐mouse IgG (Molecular Probes, Eugene, OR, USA) at a dilution of 1:500. After the sections had been washed three times with PBS, they were covered with fluorescence mounting medium (Dako, Carpintera, CA, USA). All samples were analyzed with a BZ‐9000 fluorescence microscope (Keyence, Osaka, Japan) at room temperature. For evaluation of tumor angiogenesis, the vascular luminal area and the number of sprouts were calculated in five different fields of each tumor section by using BZ‐H1C software (Keyence).
Statistical analysis
Student's t‐test was used to test for a significant difference between two groups. Dunnet's test was used for multiple comparisons. P < 0.05 was considered to indicate a statistically significant difference.
Results
Prediction of the bioactive center for hVASH2
We previously raised mAb against five different synthetic peptides of hVASH1 (Fig. S1). Among them, a mAb against hVASH1286–299 (4E12) exhibited neutralizing activity.15 Because of the similarity in amino acid sequence between hVASH1 and hVASH2 in this 286–299 region, we predicted that the bioactive center of hVASH2 might be analogous to that of hVASH1. To test this hypothesis, we developed mutant hVASH2 cDNA by replacing four amino acids, KELE, to AAAA (Fig. S1). We then stably transfected MLTC‐1 cells, in which endogenous VASH2 is not expressed, with WT or mutant (Mut) hVASH2 cDNA (Fig. 1a). WT‐hVASH2 exhibited the retarded cell growth in vitro, but Mut‐hVASH2 did not (Fig. 1b). When those cells were inoculated in VASH2 lacZ/LacZ mice, the tumor growth of WT‐hVASH was augmented, but that of Mut‐hVASH2 was not (Fig. 1c). The difference of tumor growth between parental and VASH2‐transfected MLTC‐1 cells was bigger when inoculated in VASH2 lacZ/LacZ mice. This is probably because host cells of WT mice infiltrated in the tumor also produced VASH2. Therefore, we used VASH2 lacZ/LacZ mice to eliminate the possible influence of host derived VASH2. When tumor tissue was evaluated, tumor angiogenesis was augmented in WT‐hVASH2, but not in Mut‐hVASH2 (Fig. 1d).
Figure 1.
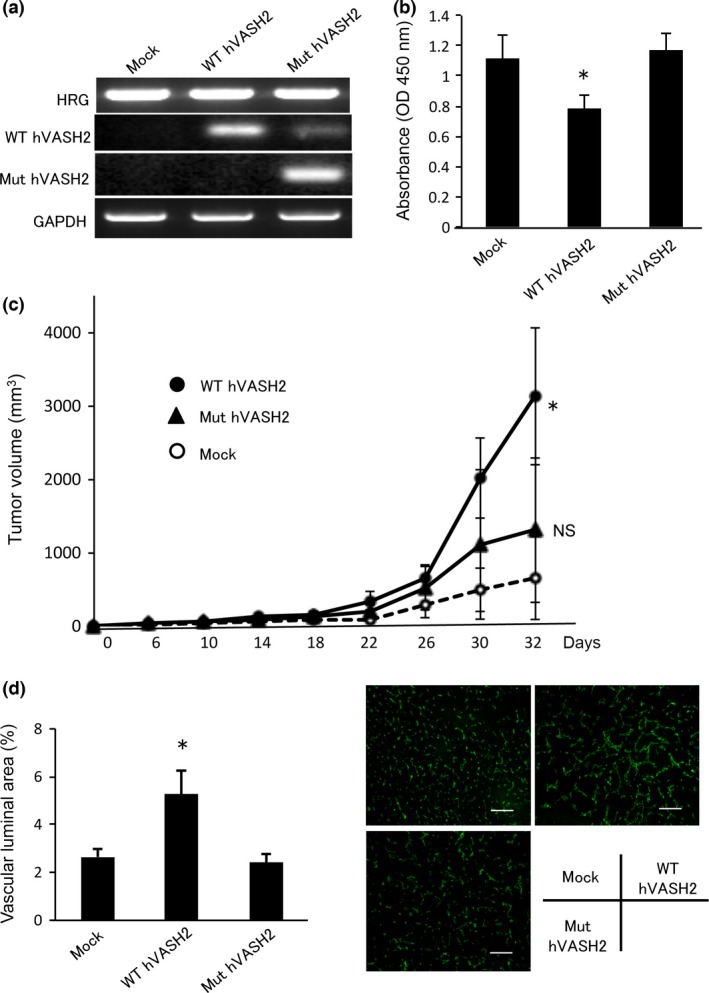
Establishment of human VASH2 mutant. (a) MLTC‐1 cells, in which VASH2 is not endogenously expressed, were stably transfected with WT‐hVASH2 or Mut‐hVASH2 cDNA. Stable transfection of these genes was confirmed by RT‐PCR analysis. (b) Cell proliferation was determined as described in the “Materials and Methods”. We repeated this experiment three times and confirmed the reproducibility. Representative data are presented as the mean and SD (n = 4 wells). *P < 0.05 (vs mock). (c) Transfectants were inoculated into Vash2 lacZ/lacZ mice, and tumor growth was monitored. Data are presented as the mean and SD (n = 9). *P < 0.05 (vs mock), NS (vs mock). (d) Tumor vessels were examined immunohistochemically with anti‐CD31 Ab (green), and the vascular luminal area was quantified. Data are presented as the mean and SD (n = 4). *P < 0.05 (vs mock). HRG, hygromycin‐resistance gene; NS, not significant.
We further examined the pro‐angiogenic activities of VASH2 on HUVEC by using those transfectants or their CM. WT‐hVASH2 on the lower chamber stimulated the migration of HUVEC across the filter (Fig. 2a), and CM from WT‐hVASH2 stimulated the tube formation by HUVEC on the Matrigel (Fig. 2b). In contrast, Mut‐hVASH2 or CM from Mut‐hVASH2 lacked such stimulatory activities (Fig. 2a,b). We could not determine hVASH2 and Mut‐hVASH2 protein concentration, as the ELISA system we had was not sensitive enough to determine their concentration in the medium. It is, therefore, possible that there was a large difference in quantities between VASH2 and Mut‐hVASH2, and that might explain the difference of their effects. However, we did not think that was likely from the results shown in Figure 1, and concluded that the bioactive center of hVASH2 for its pro‐angiogenic activity resided in the predicted amino acid region.
Figure 2.
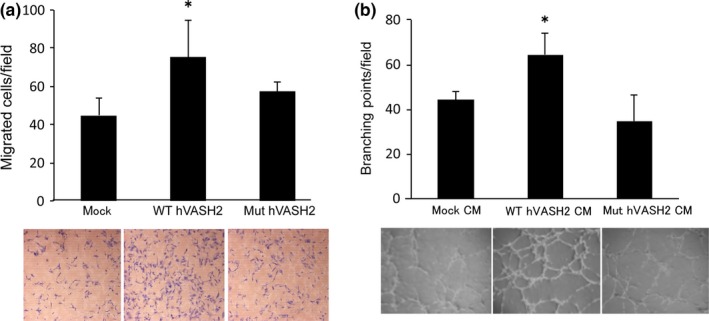
Mut hVASH2 lacked stimulatory effect on the migration of and tube formation by HUVEC. (a) Migration of HUVEC toward WT‐hVASH2 or Mut‐hVASH2 transfectants was determined by Transwell migration assay. The HUVEC that migrated across the membrane were Giemsa‐stained and counted in five random fields at a magnification of ×200. We repeated this experiment six times and confirmed the reproducibility. Representative data are presented as the mean and SD (n = 6 fields). *P < 0.05 (vs mock). (b) Tube formation by HUVEC on the Matrigel was determined. The branching points were counted in four random fields at a magnification of ×40. We repeated this experiment three times and confirmed the reproducibility. Representative data are presented as the mean and SD (n = 4 wells). *P < 0.05 (vs mock).
Establishment of a neutralizing anti‐hVASH2 monoclonal antibody
Based on the results regarding the hVASH2 bioactive center as described above, we synthesized peptide hVASH2269–289 (Fig. S1, box) and developed mAb against it. Because the mAb were raised against the synthetic peptide, we screened mAb by ELISA using recombinant hVASH2 protein and picked up six positive clones (71, 671, 1632, 1696, 1760 and 2356) that reacted with recombinant hVASH2 protein. We then examined the effect of these clones, as well as the antiserum from the mouse initially immunized with the synthetic peptide, on the HUVEC migration assay. When Wt‐hVASH2 cells were plated on the lower chamber, HUVEC on the upper chamber migrated across the filter. When antibodies were added to the lower chamber, either the antiserum, clone 71 or clone 1760 inhibited the migration of HUVEC across the filter (Fig. 3a). We confirmed by dot blot analysis that clones 71 and 1760 recognized recombinant hVASH2 protein, with clone 1760 being more efficient (Fig. 3b). Moreover, western blot analysis showed the recognition of recombinant hVASH2 protein by clone 1760 (Fig. 3c).
Figure 3.
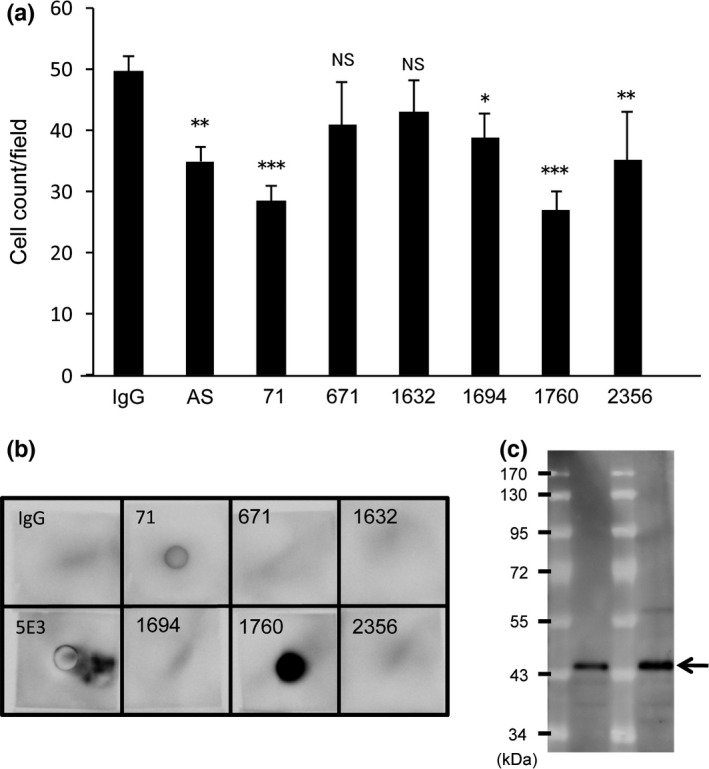
Establishment of anti‐hVASH2 monoclonal antibody (mAb). (a) Migration of HUVEC toward WT‐hVASH2 transfectants was examined by Transwell migration assay. Mouse normal IgG (5 μg/mL), antisera or mAb from the indicated clone (5 μg/mL) was added to the lower chamber. The HUVEC that migrated across the membrane were Giemsa‐stained and counted in five random fields at a magnification of ×200. Data are presented as mean and SD (n = 6). Dunnet's test was used for multiple comparisons. *P < 0.05, **P < 0.01, ***P < <0.001 (vs IgG). (b) Dot blot analysis was performed as described in the “Materials and Methods”. (c) Western blot analysis with clone 1760 was performed as described in the “Materials and Methods”. The arrow indicates VASH2 protein.
Next, we compared the neutralizing activities of clones 71 and 1760. As our recombinant hVASH2 protein retained little bioactivity, we used the CM from Wt‐hVASH2 cultures to stimulate the migration of and tube formation by HUVEC. The results revealed that clone 1760 was more potent than clone 71 in inhibiting the migration of and tube formation by HUVEC stimulated with CM from Wt‐hVASH2 cells (Fig. 4a,b).
Figure 4.
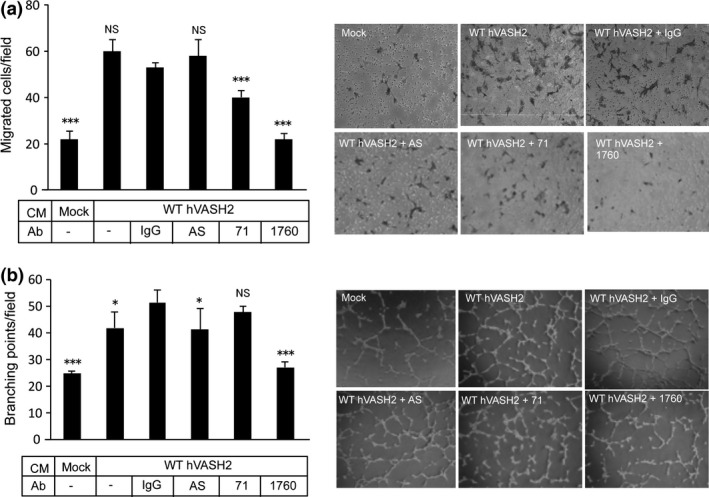
Selection of neutralizing anti‐hVASH2 monoclonal antibody (mAb). (a) Migration of HUVEC toward WT‐hVASH2 transfectants was examined by Transwell migration assay. Mouse normal IgG (5 μg/mL), antisera or mAb from the indicated clone (5 μg/mL) was added to the lower chamber. The HUVEC that migrated across the membrane were Giemsa‐stained and counted in five random fields at a magnification of ×200. Data are presented as the mean and SD (n = 6). (b) Tube formation by HUVEC on the Matrigel was determined. Mouse normal IgG (5 μg/mL), antisera or mAb from the indicated clone (5 μg/mL) was added to the cultures. Data are presented as the mean and SD (n = 6). Dunnet's test was used for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001 (vs IgG).
Antitumor effect of anti‐hVASH2 monoclonal antibody in vivo
We have previously shown that VASH2 is expressed in human ovarian cancer cells, and that it can be a therapeutic target.9, 13, 14 Here we selected clone 1760 and applied it to an in vivo xenograft model of human ovarian cancer cells. It was revealed that the i.p. administration of 25 mg/kg twice weekly for 3 weeks was optimal (Fig. 5a). The time course of clone 1760 administration is shown in Figure 5(b). Tumors were resected at day 22, and immunohistochemical analysis was then performed. Tumor angiogenesis was significantly decreased by the treatment with 25 mg/kg of clone 1760 (Fig. 5c), and tumor vessels treated with clone 1760 at this dose were more mature, as indicated by the attached αSMA‐positive mural cells (Fig. 5d).
Figure 5.
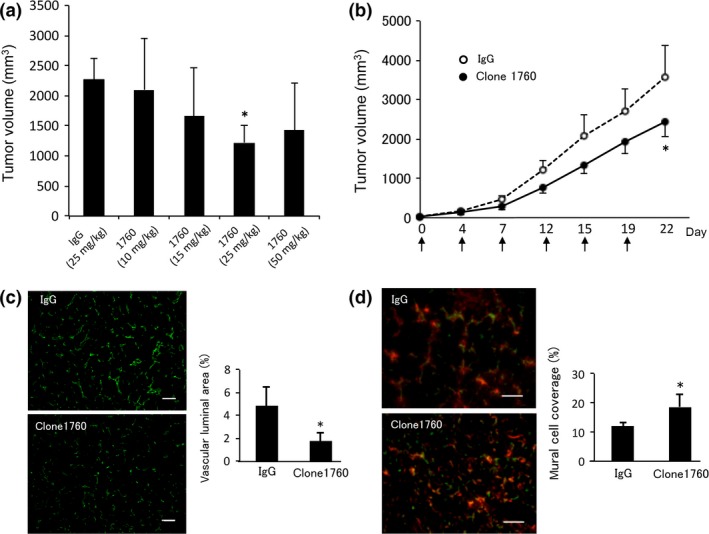
Anti‐tumor effect of clone 1760 in vivo. (a) SKOV‐3 cells were injected s.c. into the dorsal flank of BALB/c nude mice at 1 × 106 cells/mouse. Normal mouse IgG (5 mg/kg) or 10, 15, 25 or 50 mg/kg of clone 1760 was administered (i.p.) twice per week for 3 weeks; and at 22 days after treatment initiation, the final tumor volume was recorded. Experiments were repeated at least twice to confirm the reproducibility. Representative data are presented as the mean and SD (n = 4). *P < 0.05 (vs IgG). (b) SKOV‐3 cells were injected s.c. into the dorsal flank of BALB/c nude mice at 1 × 106 cells/mouse. Normal mouse IgG (5 mg/kg) or clone 1760 (25 mg/kg) was administered i.p. twice per week for 3 weeks, and tumor growth was monitored. Experiments were repeated at least twice to confirm the reproducibility. Representative data are presented as the mean and SD (n = 8). *P < 0.05 (vs IgG). (c) Tumors were resected on day 22. Tumor vessels were examined by immunohistochemically with anti‐CD31 Ab (green), and the vascular luminal area was quantified. Data are presented as the mean and SD (n = 6). *P < 0.05 (vs IgG). (d) Tumor vessels were examined by immunohistochemically with anti‐CD31 Ab (green) and anti‐αSMA Ab (red); and vessels covered by mural cells were quantified. Data are presented as the mean and SD (n = 4). *P < 0.05 (vs IgG).
Bevacizumab is used clinically for the treatment of ovarian cancers.15 Therefore, we compared the therapeutic effect of clone 1760 with that of bevacizumab. The optimal dosage of bevacizumab in this mouse model is 5 mg/kg.16, 17, 18 The results revealed that the anti‐tumor activity of clone 1760 (25 mg/kg) was equivalent to that of bevacizumab (5 mg/kg, Fig. 6a). Clone 1760 (25 mg/kg) and bevacizumab (5 mg/kg) equally inhibited tumor angiogenesis (Fig. 6b,c).
Figure 6.
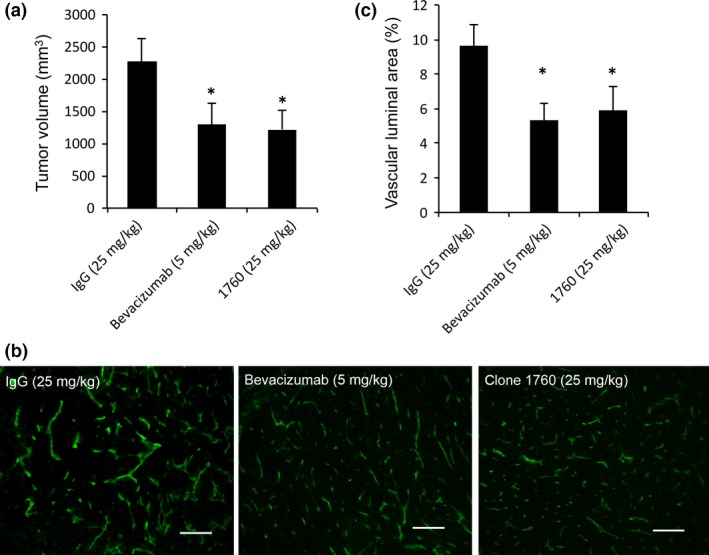
Comparison of the effect of clone 1760 with that of bevacizumab. (a) SKOV‐3 cells were injected s.c. in the dorsal flank of BALB/c nude mice at 1 × 106 cells/mouse. Mice were treated with 5 mg/kg of mouse IgG, 25 mg/kg of hVASH2 monoclonal antibody (mAb) or 5 mg/kg of bevacizumab i.p. twice per week for 3 weeks. At 22 days after treatment initiation, the mice were sacrificed; and tumor volume was subsequently determined. Experiments were repeated at least twice to confirm the reproducibility. Representative data are presented as the mean and SD (n = 8). *P < 0.05 (vs IgG). (b) Tumor blood vessels in mice treated with 5 mg/kg of mouse IgG, 25 mg/kg of hVASH2 mAb or 5 mg/kg of bevacizumab were immunostained with anti‐CD31 antibody. Bar = 50 μm. (c) Vascular luminal area was quantified. Data are presented as the mean and SD (n = 4). *P < 0.05 (vs IgG).
Finally, we examined the combined efficacy of clone 1760 and bevacizumab. However, we could not obtain any additive effect with the combination of clone 1760 and bevacizumab (Fig. 7).
Figure 7.
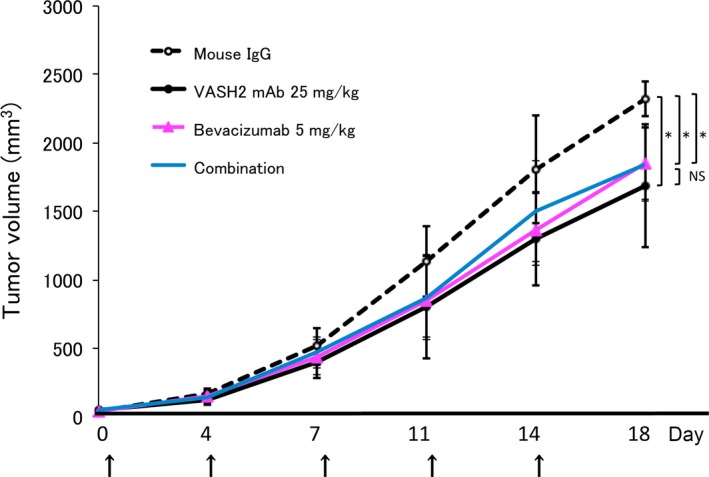
The efficacy of combination therapy with bevacizumab and clone 1760. DISS cells were injected s.c. in the dorsal flank of BALB/c nude mice at 1 × 106 cells/mouse. Mice were treated with 5 mg/kg of mouse IgG, 25 mg/kg of hVASH2 mAb, 5 mg/kg of bevacizumab or 25 mg/kg of hVASH2 mAb plus 5 mg/kg of bevacizumab i.p. twice per week, and tumor growth was monitored. Experiments were repeated at least twice to confirm the reproducibility. Representative data are presented as the mean and SD (n = 8). *P < 0.05 (vs IgG). NS, not significant.
Discussion
It has been challenging to specify the bioactive center of hVASH2 for pro‐angiogenic activity, because there is no known functional motif in the amino acid sequence of hVASH2. As one of the mAb (4E12) had neutralizing activity against hVASH1,19 we applied this information. Because of the similarity in the amino acid sequence between VASH1 and VASH2, we predicted the bioactive center of hVASH2 from the epitope for 4E12, made a mutant hVASA2, and suggested that the estimated region was responsible for the pro‐angiogenic activity of hVASH2, although our present study did not exclude the possibility that other regions of VASH2 also involved in its bioactivity. We then made a synthetic peptide that included this region of VASH2 and raised mAb against it. The number of anti‐hVASH2 peptide mAb that recognized hVASH2 protein was low, probably because the amino acid sequence of VASH2 was highly conserved between mouse and human. Nevertheless, we isolated clone 1760, a neutralizing anti‐hVASH2 mAb that blocked the VASH2‐stimulated migration of or tube formation by EC.
The expression of VASH2 is detected in various human cancers.7, 8, 9, 10, 11, 12, 13 In addition, the knockdown of VASH2 in cancer cells results in significant anti‐angiogenic and anti‐tumor effects in a mouse xenograft model.8, 9, 13 Therefore, VASH2 expressed in cancer cells has been regarded as a novel molecular target for the anti‐cancer treatment. Indeed, the local injection of hVASH2 siRNA is effective as an anti‐cancer treatment.14 Therefore, we applied clone 1760 for anti‐tumor therapy. The i.p. injection of clone 1760 into tumor‐bearing mice significantly inhibited the tumor growth and tumor angiogenesis, and the remaining tumor vessels became more mature, as indicated by the attached mural cells. The present results agree well with our previous observation that tumor vessels formed in the intestinal polyp of Apc min/+ Vash2 lacZ/LacZ mice are more mature than those formed in Apc min/+,10 although the mechanism of this vascular normalization remains to be elucidated. An important aspect regarding the success of anti‐angiogenic cancer treatment is whether or not the tumor vessels become mature (i.e. associated with mural cells) or remain immature (i.e. defective in coverage by these cells).20
We compared the effect of clone 1760 with that of bevacizumab, a humanized neutralizing anti‐VEGF‐A mAb. Our results indicated that the maximal anti‐tumor and anti‐angiogenic effects of clone 1760 were comparable to those of bevacizumab. One problem of VEGF‐A targeting treatment is damage to normal EC.21 It is well known that VEGF‐A plays an important role in the development and maintenance of EC, as haploinsufficiency of the VEGF‐A gene results in embryonic lethality,22, 23 and conditional knockout of this gene in EC causes multiple organ failure due to EC dysfunction.24 In contrast to the case for the Vegf‐A gene, Vash2 knockout mice are viable; although minor abnormality occurs in the placenta, these mice do not show a severe phenotype.25 Therefore, we predict that the targeting of VASH2 may not be deleterious.
Another difference between VASH2 and VEGF is the regulation of their expression. Whereas VEGF is induced by hypoxia, VASH2 is not induced by it but is upregulated by the aborted expression of certain microRNA, namely, mir‐200b, in cancer cells.9, 26 Thus, the expressions of these two pro‐angiogenic factors are independent. Indeed, we observed previously that VASH2 is upregulated during the transition from adenoma to adenocarcinoma in Apc min/+ mice but that the expression of VEGF in tumor cells remains unchanged during this same transition.10 This difference should be important when VASH2 is targeted for the treatment of cancers.
We tested the efficacy of combination therapy with bevacizumab and anti‐hVASH2 mAb, but we could not obtain any advantageous effects. The reason for this result is not clear at the moment. Is it possible that the VEGF‐induced tumor growth acts via VASH2, or the VASH2‐induced tumor growth acts via VEGF? Apparently, we need to know more about the molecular basis of the effect of VASH2. Nonetheless, it is suggested that bevacizumab may not be valuable for the combination with other antibodies.27
In summary, we identified the bioactive center of VASH2, established neutralizing anti‐hVASH2 mAb against it, and confirmed its anti‐tumor activity in a xenograft mouse model. Further studies are currently underway to clarify the usefulness of neutralizing anti‐hVASH2 mAb as a treatment modality for various cancer cells.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Amino acid sequences of human VASH1 and VAHS2.
{kind=link}
Acknowledgments
This work was supported by a grant from Projects for Development of Innovative Research on Cancer Therapeutics, the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Cancer Sci 108 (2017) 512–519
Funding Information
Projects for Development of Innovative Research on Cancer Therapeutics, The Ministry of Education, Culture, Sports, Science, and Technology of Japan
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 2. Dey N, De P, Brian LJ. Evading anti‐angiogenic therapy: resistance to anti‐angiogenic therapy in solid tumors. Am J Transl Res 2015; 7: 1675–98. [PMC free article] [PubMed] [Google Scholar]
- 3. Watanabe K, Hasegawa Y, Yamashita H et al Vasohibin as an endothelium‐derived negative feedback regulator of angiogenesis. J Clin Invest 2004; 114: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shibuya T, Watanabe K, Yamashita H et al Isolation and characterization of vasohibin‐2 as a homologue of VEGF‐inducible endothelium‐derived angiogenesis inhibitor vasohibin. Arterioscler Thromb Vasc Biol 2006; 26: 1051–7. [DOI] [PubMed] [Google Scholar]
- 5. Kimura H, Miyashita H, Suzuki Y et al Distinctive localization and opposed roles of vasohibin‐1 and vasohibin‐2 in the regulation of angiogenesis. Blood 2009; 113: 4810–8. [DOI] [PubMed] [Google Scholar]
- 6. Suzuki Y, Kobayashi M, Miyashita H et al Isolation of a small vasohibin‐binding protein (SVBP) and its role in vasohibin secretion. J Cell Sci 2010; 123: 3094–4101. [DOI] [PubMed] [Google Scholar]
- 7. Shen Z, Kauttu T, Seppänen H et al Vasohibin‐1 and vasohibin‐2 expression in gastric cancer cells and TAMs. Med Oncol 2012; 29: 2718–26. [DOI] [PubMed] [Google Scholar]
- 8. Xue X, Gao W, Sun B et al Vasohibin 2 is transcriptionally activated and promotes angiogenesis in hepatocellular carcinoma. Oncogene 2013; 32: 1724–34. [DOI] [PubMed] [Google Scholar]
- 9. Takahashi Y, Koyanagi T, Suzuki Y et al Vasohibin‐2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol Cancer Res 2012; 10: 1135–46. [DOI] [PubMed] [Google Scholar]
- 10. Kitahara S, Suzuki Y, Morishima M et al Vasohibin‐2 modulates tumor onset in the gastrointestinal tract by normalizing tumor angiogenesis. Mol Cancer 2014; 13: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tu M, Liu X, Han B et al Vasohibin‐2 promotes proliferation in human breast cancer cells via upregulation of fibroblast growth factor‐2 and growth/differentiation factor‐15 expression. Mol Med Rep 2014; 10: 663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim JC, Kim KT, Park JT et al Expression of vasohibin‐2 in pancreatic ductal adenocarcinoma promotes tumor progression and is associated with a poor clinical outcome. Hepatogastroenterology 2015; 62: 251–6. [PubMed] [Google Scholar]
- 13. Koyanagi T, Saga Y, Takahashi Y et al Downregulation of vasohibin‐2, a novel angiogenesis regulator, suppresses tumor growth by inhibiting angiogenesis in endometrial cancer cells. Oncol Lett 2013; 5: 1058–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koyanagi T, Suzuki Y, Saga Y et al In vivo delivery of siRNA targeting vasohibin‐2 decreases tumor angiogenesis and suppresses tumor growth in ovarian cancer. Cancer Sci 2013; 104: 1705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burger RA1, Brady MF, Bookman MA et al Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med 2011; 365: 2473–83. [DOI] [PubMed] [Google Scholar]
- 16. Mabuchi S, Terai Y, Morishige K et al Maintenance treatment with bevacizumab prolongs survival in an in vivo ovarian cancer model. Clin Cancer Res 2008; 14: 7781–9. [DOI] [PubMed] [Google Scholar]
- 17. Kim TJ, Landen CN, Lin YG et al Combined anti‐angiogenic therapy against VEGF and integrin aVb3 in an orthotopic model of ovarian cancer. Cancer Biol Ther 2009; 8: 2263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao M, Hollar S, Chambliss D et al Targeting the insulin growth factor and the vascular endothelial growth factor pathways in ovarian cancer. Mol Cancer Ther 2012; 11: 1576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyashita H, Watanabe T, Hayashi H et al Angiogenesis inhibitor vasohibin‐1 enhances stress resistance of endothelial cells via induction of SOD2 and SIRT1. PLoS One 2012; 7: e46459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10: 417–27. [DOI] [PubMed] [Google Scholar]
- 21. Khan KA, Bicknell R. Anti‐angiogenic alternatives to VEGF blockade. Clin Exp Metastasis 2016; 33: 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferrara N, Carver‐Moore K, Chen H et al Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996; 380: 439–42. [DOI] [PubMed] [Google Scholar]
- 23. Carmeliet P, Ferreira V, Breier G et al Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996; 380: 435–9. [DOI] [PubMed] [Google Scholar]
- 24. Lee S, Chen TT, Barber CL et al Autocrine VEGF signaling is required for vascular homeostasis. Cell 2007; 130: 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suenaga K, Kitahara S, Suzuki Y et al Role of the vasohibin family in the regulation of fetoplacental vascularization and syncytiotrophoblast formation. PLoS One 2014; 9: e104728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xue X, Zhang Y, Zhi Q et al MiR200‐upregulated vasohibin 2 promotes the malignant transformation of tumors by inducing epithelial‐mesenchymal transition in hepatocellular carcinoma. Cell Commun Signal 2014; 12: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arjaans M, Oude Munnink TH, Oosting SF et al Bevacizumab‐induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res 2013; 73: 3347–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Amino acid sequences of human VASH1 and VAHS2.