

Abstract
Vasohibin‐2 (VASH2) is a homolog of VASH1, an endothelium‐derived angiogenesis inhibitor. Vasohibin‐2 is mainly expressed in cancer cells, and has been implicated in the progression of cancer by inducing angiogenesis and tumor growth. Although VASH2 has been recently reported to be involved in epithelial–mesenchymal transition (EMT), its precise roles are obscure. The aim of the present study was to clarify the role of VASH2 in the EMT of cancer cells in relation to transforming growth factor‐β (TGF‐β) signaling, which is a major stimulator of EMT. Decreased expression of VASH2 in ovarian cancer cells significantly repressed the expression of TGF‐β type I receptor, namely activin receptor‐like kinase 5. Transforming growth factor‐β1‐induced phosphorylation of Smad2 and Smad3 was markedly decreased in VASH2 knockdown cells while the expression of Smad2 and Smad3 was unchanged. Accordingly, the responses to TGF‐β1 shown by promoter assay and plasminogen activator inhibitor type 1 expression were significantly attenuated in VASH2 knockdown cells. Furthermore, knockdown of VASH2 in cancer cells abrogated the TGF‐β1‐induced reduced expression of epithelial markers including E‐cadherin, and the elevated expression of mesenchymal markers including fibronectin, ZEB2, and Snail2, suggesting that endogenous VASH2 is required for TGF‐β1‐induced EMT. In accordance with these results, the effects of TGF‐β1 on cell morphology, migration, invasion, and MMP2 expression were also abrogated when VASH2 was knocked down. These results indicate that VASH2 played a significant role in the EMT by modulating the TGF‐β signaling. We propose that VASH2 would be a novel molecular target for the prevention of EMT in cancers.
Keywords: ALK5, EMT, ovarian cancer, TGF‐β, vasohibin‐2
Epithelial–mesenchymal transition (EMT) is a complex cellular program that regulates changes in cell morphology and function during embryogenesis and tissue development, and it is also involved in cancer progression and metastasis.1, 2, 3 The EMT is characterized by loss of cell–cell contact with decreased expression of epithelial markers such as E‐cadherin, increased expression of mesenchymal markers such as fibronectin and vimentin, elongated cell morphology, and increased cell motility and invasiveness.4 Various factors have been implicated in this process, and they act in a sequential or cooperative manner. Among them, transforming growth factor‐β (TGF‐β) is a principal factor that induces the EMT by propagating intracellular signaling pathways, namely, those involving Smads, and by activating certain transcriptional factors such as Snails and ZEBs, which repress the transcription of the E‐cadherin gene.5, 6, 7, 8
Transforming growth factor‐β is present in the tumor microenvironment, and is considered to be the most potent inducer of the EMT.9 Transforming growth factor‐β is a homodimeric multifunctional cytokine involved in the regulation of proliferation, differentiation, migration, and survival of many different cell types.10 Three isoforms of TGF‐β are present in mammals, TGF‐β1, TGF‐β2, and TGF‐β3, which show partly overlapping as well as distinct functions.11, 12 Transforming growth factor‐β signals are transduced by TGF‐β type II receptor (TβRII) and TGF‐β type I receptors (TβRI), namely activin receptor‐like kinase 5 (ALK5). Following binding of TGF‐β to TβRII, ALK5 is recruited and phosphorylated, which leads to activation of downstream signals that are transduced by intracellular effectors, termed Smads.13, 14 Activation of ALK5 induces the phosphorylation of Smad2 and Smad3, followed by their translocation to the nucleus in a complex with the common Smad, Smad4, to regulate the transcription of various target genes.15
Previously we searched for and isolated novel angiogenesis regulators that we designated as vasohibin‐1 (VASH1) and vasohibin‐2 (VASH2).16 Vasohibin‐1 is an endothelium‐derived angiogenesis inhibitor, whereas VASH2 is a homolog of VASH1, which acts as an angiogenesis stimulator.17, 18 Importantly, VASH2 is expressed by various cancer cells, including ovarian cancer cells, and it promotes tumor angiogenesis and tumor growth.18 Overexpression of VASH2 enhances tumor angiogenesis and tumor progression, whereas knockdown of VASH2 suppresses them.18 Furthermore, when Apc min/+ mice are crossed with Vash2 lacZ/lacZ mice, the number of intestinal tumors are significantly decreased. That decrease is associated with normalization of tumor vessels.19 These results suggest a paracrine effect of VASH2 on tumor angiogenesis.
Recently, it was reported VASH2 promotes malignant transformation through promoting the EMT in hepatocellular carcinoma.20 However, the biological role of VASH2‐dependent regulation of EMT has not yet been identified. Here we hypothesized VASH2 to be involved in the regulation of TGF‐β signaling and examined this possibility. Our present study disclosed that the knockdown of VASH2 downregulated the expression of ALK5. Accordingly, the TGF‐β signaling was significantly attenuated, with a reduction in the TGF‐β‐induced EMT, a mechanism that has not been previously identified.
Materials and Methods
Antibodies used for immunodetection of proteins
We used the following antibodies for Western blot analysis: E‐cadherin (3195; Cell Signaling Technology [CST], Danvers, MA, USA), vimentin (5741; CST), fibronectin (610077; BD Biosciences, Franklin Lakes, NJ, USA), ALK5 (sc‐398; Santa Cruz Biotechnology, Dallas, TX, USA), Smad2/3 (3102; CST), phosphorylated Smad2 (Ser465/467) (3108; CST), Smad3 (9513; CST), phosphorylated Smad3 (Ser423/425) (9513; CST), β‐actin (A5441; Sigma‐Aldrich, St. Louis, MO, USA), α‐tubulin (CBL270; Merck Millipore, Darmstadt, Germany), and hemagglutinin (HA)‐tag (M132‐3; Medical & Biological Laboratories, Nagoya, Japan). For immunocytochemistry analysis, antibodies recognizing E‐cadherin (610181; BD Transduction Laboratories, San Jose, CA, USA), SM22α (ab14106; Abcam, Cambridge, UK), anti‐mouse IgG (H+L) Alexa Fluor 488‐conjugated (A‐11001; Life Technologies) or anti‐Rabbit IgG (H+L) Alexa Fluor 594‐conjugated (A‐21207; Life Technologies, Carlsbad, CA, USA) were used.
Cell culture
DISS and SKOV3 cells and VASH2 knocked‐down (sh‐VASH2) clonal cell lines from DISS cells were described previously.21 Cells were maintained in DMEM (Wako, Saitama, Japan) with 10% FBS (Sigma‐Aldrich) and penicillin–streptomycin (Wako). For experimental treatments, the serum was reduced to 0.5% and 10 ng/mL TGF‐β1 (Wako) or 5 μM SB431542 (Wako) was added.
Immunostaining
For confocal microscopic analysis, the siVASH2 and siControl DISS transfected cells, or shVASH2 and sh‐control DISS cells, were seeded on coverslips in a 12‐well plate at a density of 12 × 104 cells and cultured overnight at 37°C in DMEM with 10% FBS. The next cells were starved for 4 h in DMEM containing 0.5% FBS, followed by treatment with or without 10 ng/mL TGF‐β1 for 72 h. Cells were then fixed with ice‐cold mixture of methanol‐acetone (1:1) for 30 s, blocked with 1% BSA, and incubated with anti‐E‐cadherin (1:200 dilution) and anti‐SM22α (1:1000 dilution) antibodies overnight at 4°C. The next day, cells were incubated with secondary antibodies conjugated either with Alexa Fluor 488 (1:500 dilution) or Alexa Fluor 594 (1:500 dilution) for 1 h at room temperature to visualize stained molecules. After extensive washing, cells were mounted in ProLong Diamond with DAPI (Life Technologies). The images were captured under a confocal microscope (FV10i; Olympus, Tokyo, Japan).
Western blot analysis
Cells were lysed in RIPA buffer containing 0.1% SDS (Nacalai Tesque, Kyoto, Japan) and PhosSTOP phosphatase inhibitor cocktail (Roche, Basel, Switzerland). The protein concentration was determined by the Lowry protein assay (Bio‐Rad Laboratories, Hercules, CA, USA). Equal amounts of proteins were separated by SDS‐PAGE and transferred to PVDF membranes (Bio‐Rad Laboratories). The membranes were then probed with the desired antibodies. Chemiluminescence detection was carried out by using Immobilon Western Chemiluminescent HRP substrate (Merck Millipore) and LAS‐4000 (Fuji Photo Film, Tokyo, Japan).
RNA interference
Small interfering RNA specific for human VASH2 (sense, CACUCUGAAUGAAGUGGGCUAUCAA; Thermo Fisher Scientific) and a negative control oligonucleotide (stealth siRNA; Thermo Fisher Scientific, Waltham, MA, USA) were used. Cells were reverse‐transfected with siRNAs by using Lipofectamine RNAi MAX transfection reagent (Thermo Fisher Scientific) at a final concentration of 10 nM.
Adenovirus infection
For the overexpression of ALK5, we used adenovirus vectors encoding HA‐tagged constitutively active form of human ALK‐5 (AdALK5‐TD).22 The shVASH2 cells were plated in 35‐mm dishes at 2 × 105 cells and cultured overnight at 37°C in DMEM with 10% FBS. The following day, medium was replaced by DMEM containing 2% FBS and cells were infected with AdALK5‐TD or AdLacZ at a final MOI of 100. Cells were incubated for an additional 48 h, then RT‐PCR and Western blot analysis were carried out.
Quantitative real‐time RT‐PCR
For preparation of total RNA, 2 × 105 cells were cultured in 6‐well tissue culture plates. Total RNA was isolated by using an RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. First‐strand cDNA was prepared from 1 μg total RNA using ReverTra Ace (ToYoBo, Osaka, Japan). The quantitative real‐time RT‐PCR was undertaken by using a CFX96 real‐time PCR detection system (Bio‐Rad Laboratories) according to the manufacturer's instructions. Each mRNA level was measured as a fluorescent signal corrected according to the signal for β‐actin.
Transfection and luciferase assays
The TGF‐β‐responsive reporter gene vector ((CAGA)9‐Luc) and pRL‐TK vector were kindly provided by Professor Dan Rifkin (New York University, School of Medicine, New York, NY, USA) and Professor Scott Friedman (Mount Sinai, School of Medicine, New York, NY, USA), respectively. DISS and SKOV3 cells were transiently transfected with the (CAGA)9‐Luc and the internal control pRL‐TK vector by using FuGENE HD (Promega, Madison, WI, USA). The cells were stimulated with 10 ng/mL TGF‐β1, and luciferase activities were quantified 24 h later by a dual luciferase assay (Promega). Values were normalized with the Renilla luciferase activity expressed from pRL‐TK. Luciferase values shown in the figures are the average of three independent experiments performed in duplicate.
Migration assay and invasion assay
The migration potential of DISS cells was determined by using a 24‐well Transwell with an 8.0‐μm Pore Polycarbonate Membrane Insert (Corning, New York, NY, USA). Following preincubation for 16 h in DMEM and 0.5% FCS, the cells were treated for 24 h with 5 ng/mL TGF‐β1. Subsequently, the cells were added at 1 × 104 cells to the upper chamber (insert); the lower chamber was filled with DMEM 1% FCS. The DISS cells were allowed to migrate for 6 h, and those that had migrated across the filter were fixed in methanol and stained with DAPI. The number of total cells that had migrated was counted. The invasion potential was determined by using a 24‐well Matrigel invasion chamber (Corning). Following incubation in DMEM 0.5% FCS, the cells were treated with 5 ng/mL TGF‐β1 for 48 h. Then 1 × 104 cells were added to the upper chamber, after which the lower chamber was filled with DMEM 10% FCS. The cells were incubated for 24 h, and invasive cells were fixed, stained, and counted.
Statistical analysis
The results were expressed as the mean and standard deviation.15 Data were analyzed using the unpaired Student's t‐test. In all statistical analyses, a P‐value < 0.05 was considered statistically significant.
Results
Knockdown of VASH2 reduces ALK5 expression
Given that VASH2 is involved in the EMT,20 we sought its mechanism by using human serous ovarian carcinoma cell lines DISS and SKOV3. As the principal regulator of the EMT is TGF‐β, we examined whether VASH2 could affect the TGF‐β signaling pathway. When VASH2 in DISS or SKOV3 cells was knocked down by its siRNA, the expression of ALK5 was significantly decreased, whereas TβRII expression was maintained (Fig. 1a–d). We further used the DISS cell line stably transfected with shVASH2 (shVASH2 cells) and its control mock cells, and confirmed the significant reduction in ALK5, but not TβRII, expression (Fig. 1e,f).
Figure 1.
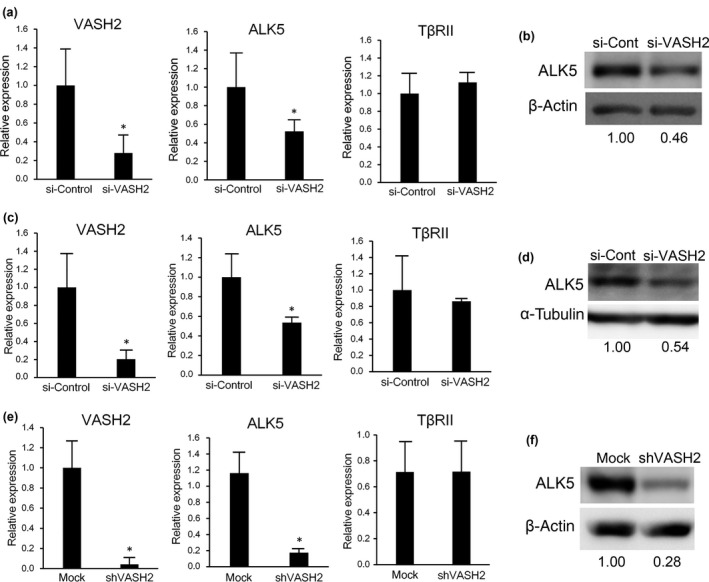
Vasohibin‐2 (VASH2) knockdown reduces the expression of activin receptor‐like kinase 5 (ALK5). (a) Quantitative real‐time RT‐PCR analysis of VASH2, ALK5, and transforming growth factor‐β type II receptor (TβRII) expression in DISS cells transfected with either control or VASH2 siRNA was carried out 24 h after transfection. Values were normalized to the β‐actin mRNA level. (b) Western blotting of ALK5 in DISS cells transfected with either control (si‐Cont) or VASH2 siRNA was carried out 24 h after transfection. β‐Actin in cell lysate was used as a loading control. (c) Quantitative real‐time RT‐PCR analysis of VASH2, ALK5, and TβRII expression in SKOV3 cells transfected with either control or VASH2 siRNA was undertaken 4 days after transfection. Values were normalized to the β‐actin mRNA level. (d) Western blotting of ALK5 in SKOV3 cells transfected with either control (si‐Cont) or VASH2 siRNA was undertaken 4 days after transfection. α‐Tubulin in cell lysates was used as a loading control. (e) Quantitative real‐time RT‐PCR analysis of VASH2, ALK5, and TβRII expression in DISS cells transfected with mock or shVASH2 was carried out. (f) Western blotting of ALK5 in DISS cells transfected with control mock or shVASH2 was carried out. β‐Actin in cell lysates was used as a loading control. The intensity of each band was determined by densitometry. Values indicate the fold change of ALK5 level normalized to β‐actin. (a,c,e) Mean and SDs are shown (*P < 0.05).
Vasohibin‐2 is required for activation of TGF‐β1 signaling pathway
As ALK5 is requisite for TGF‐β signaling, we investigated whether VASH2 knockdown would attenuate the TGF‐β signaling. We used the (CAGA)9‐Luc reporter for the assessment of TGF‐β signaling. When VASH2 was knocked down by its siRNA in SKOV3 cells, TGF‐β1 stimulated luciferase activity was significantly decreased (Fig. 2a). Plasminogen activator inhibitor type 1 (PAI‐1) is a representative target induced by TGF‐β. When VASH2 was knocked down by siRNA in SKOV3 cells, the TGF‐β1‐stimulated induction of PAI‐1 was significantly decreased (Fig. 2b). These changes were confirmed in the cell line stably transfected with shVASH2 (Fig. 2c,d). The construct drives the expression of the luciferase gene through a promoter containing CAGA boxes to which activated Smad2/3 binds.23 We therefore examined the status of Smad2 and Smad3. Smad2 and Smad3 protein levels were almost identical between mock and shVASH2 cells. But when those cells were stimulated with TGF‐β1, the phosphorylation of Smad2 and Smad3 was markedly repressed in shVASH2 cells (Fig. 2e). These findings support the idea that downregulation of ALK5 by VASH2 knockdown attenuated TGF‐β signaling.
Figure 2.
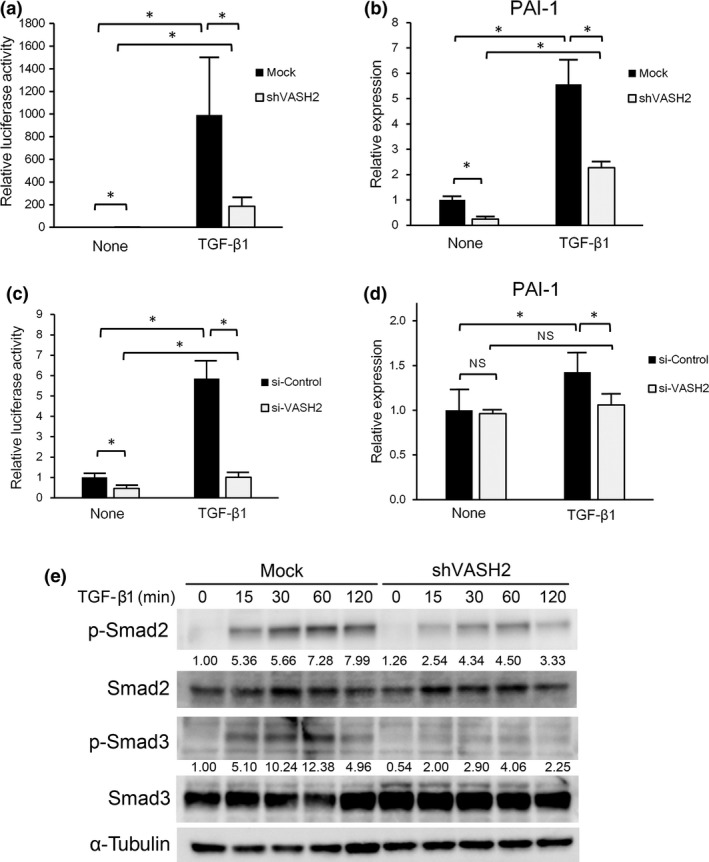
Vasohibin‐2 (VASH2) is required for transforming growth factor‐β (TGF‐β) signaling. (a) SKOV3 cells were cotransfected with the (CAGA)9‐Luc construct and either control siRNA or VASH2 siRNA. Three days after this procedure, cells were treated or not with TGF‐β1 for 24 h, and the (CAGA)9‐Luc reporter activity was then quantified. (b) SKOV3 cells transfected with control siRNA or VASH2 siRNA. Three days after this procedure, cells were treated or not with TGF‐β1 for 24 h. Thereafter, quantitative real‐time RT‐PCR analysis of plasminogen activator inhibitor type 1 (PAI‐1) expression was carried out. Values were normalized to the β‐actin mRNA level. (c) Control mock or shVASH2 transfected cells established from DISS were transfected with the (CAGA)9‐Luc construct. Twenty four hours after this procedure, cells were treated or not with TGF‐β1 for 24 h, and the (CAGA)9‐Luc reporter activity was then quantified. (d) DISS cells transfected with mock or shVASH2 were treated or not with TGF‐β1 for 24 h. Quantitative real‐time RT‐PCR analysis of PAI‐1 expression was then carried out. Values were normalized to the β‐actin mRNA level. (e) DISS cells transfected with mock or shVASH2 were treated with TGF‐β1 for the indicated times then Western blotting for pSmad2, total Smad2, pSmad3, and total Smad3 was undertaken. α‐Tubulin in the cell lysates was used as a loading control. The intensity of each band was determined by densitometry. Values indicate the fold change of pSmad2 and pSmad3 levels normalized to total Smad2 and Smad3, respectively. Mean and SDs are shown (*P < 0.05). N.S., not significant.
Vasohibin‐2 is required for EMT induced by TGF‐β1
We next tested whether VASH2 was required for the EMT induced by TGF‐β1. The mRNA expression of E‐cadherin was increased, whereas that of fibronectin and SM22α was decreased in shVASH2 cells. Epithelial–mesenchymal transition is accompanied by the induction of ZEB and Snail families. Among them, ZEB2 and Snail2 levels were decreased in shVASH2 cells. Moreover, TGF‐β1 decreased the mRNA expression of E‐cadherin and increased that of fibronectin, vimentin, SM22α, ZEB2, and Snail2 in mock cells, but those responses were abrogated in shVASH2 cells (Fig. 3a). Western blot analysis showed that E‐cadherin expression was increased, whereas that of fibronectin was decreased, in shVASH2 cells. Moreover, when cells were stimulated with TGF‐β1, E‐cadherin was decreased, and fibronectin was increased, in mock cells, but those responses were abrogated in shVASH2 cells (Fig. 3b). These changes were confirmed in the cells transfected with control or VASH2 siRNA (Fig. 3c). Significant changes in the expression of E‐cadherin, fibronectin, SM22α, ZEB2, and Snail2 in unstimulated shVASH2 cells raised the question as to whether or not those changes were dependent on endogenous TGF‐β. To test this, we used SB431542, an inhibitor of ALK5. SB431542 significantly increased the expression of E‐cadherin and decreased that of fibronectin, SM22α, ZEB2, and Snail2 (Fig. 3d). Alternatively, we undertook a rescue experiment by transfecting shVASH2 cells with AdALK5‐TD to overexpress constitutively active ALK5 (Fig. 3e). This transfection decreased the expression of E‐cadherin and increased those of fibronectin and SM22α in shVASH2 cells (Fig. 3f). Collectively, these data indicate that VASH2 is required for the expression of ALK5, which is essential for the signaling of TGF‐β for EMT.
Figure 3.
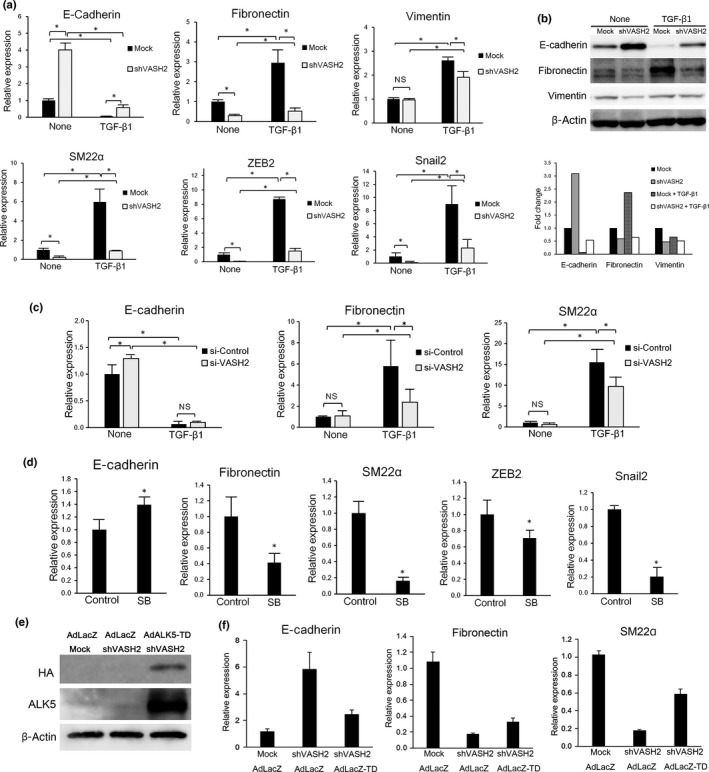
Downregulation of vasohibin‐2 (VASH2) blocks transforming growth factor‐β1 (TGF‐β1)‐induced gene regulation in relation to epithelial–mesenchymal transition. (a) DISS cells transfected with mock or shVASH2 were treated or not with TGF‐β1 for 24 h. Thereafter, quantitative real‐time RT‐PCR analysis of E‐cadherin, fibronectin, vimentin, SM22α, ZEB2, and Snail2 expression was carried out. Values were normalized to the β‐actin mRNA level. (b) DISS cells transfected with mock or shVASH2 were treated or not with TGF‐β1 for 24 h. Thereafter, Western blotting of E‐cadherin, fibronectin, and vimentin was carried out. β‐Actin in the cell lysate was used as a loading control. The densitometric analysis of Western signals is shown in the column graph. The bars indicate the fold change of E‐cadherin, fibronectin, and vimentin expression, normalized to β‐actin and relative to untreated mock. (c) DISS cells transfected with control siRNA or VASH2 siRNA were treated or not with TGF‐β1 for 24 h. Thereafter, quantitative real‐time RT‐PCR analysis of E‐cadherin, fibronectin, and SM22α expression levels was carried out. Values were normalized to the β‐actin mRNA level. (d) DISS cells were treated or untreated (control) with 5 μM SB431542 (SB) for 48 h. Thereafter, quantitative real‐time RT‐PCR analysis of E‐cadherin, fibronectin, SM22α, ZEB2, and Snail2 expression levels was carried out. Values were normalized to the β‐actin mRNA level. (e) Cells were infected with AdLacZ or AdALK5‐TD and incubated for 48 h, then Western blotting of hemagglutinin (HA)‐tag and activin receptor‐like kinase 5 (ALK5) was carried out. (f) Cells were infected with AdLacZ or AdALK5‐TD and incubated for 48 h, then quantitative real‐time RT‐PCR analysis of E‐cadherin, fibronectin, and SM22α expression was carried out. Values were normalized to the β‐actin mRNA level. (a,c,d,f) Mean and SDs are shown (*P < 0.05).
Immunofluorescence staining showed that E‐cadherin expression was increased, whereas that of SM22α was decreased, in shVASH2 cells. Moreover, when cells were stimulated with TGF‐β1, E‐cadherin was decreased and SM22α was increased in the mock cells, but those responses were abrogated in shVASH2 cells. The reduced responses to TGF‐β1 were further confirmed in cells transfected with control or VASH2 siRNA (Fig. 4).
Figure 4.
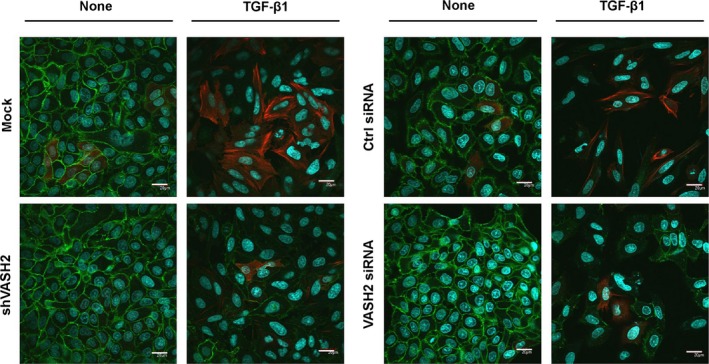
Downregulation of vasohibin‐2 (VASH2) blocks transforming growth factor‐β1 (TGF‐β1)‐induced protein modulation in relation to epithelial–mesenchymal transition. Immunofluorescence staining of E‐cadherin (green) and SM22α (red) was compared between mock and shVASH2 transfectant cells established from DISS, or between siControl (Ctrl) and siVASH2. Nuclei were stained with DAPI (blue). Scale bar = 20 μm.
Vasohibin‐2 knockdown attenuates TGF‐β1‐induced cell invasion
Epithelial–mesenchymal transition induces morphological changes of epithelial cells from cobblestone‐like structures to elongated spindle shapes. Mock cells became elongated when they were treated with TGF‐β1 (Fig. 5a). We did not observe significant differences in morphology of shVASH2 cells from mock cells in the absence or presence of TGF‐β1 (Fig. 5a).
Figure 5.
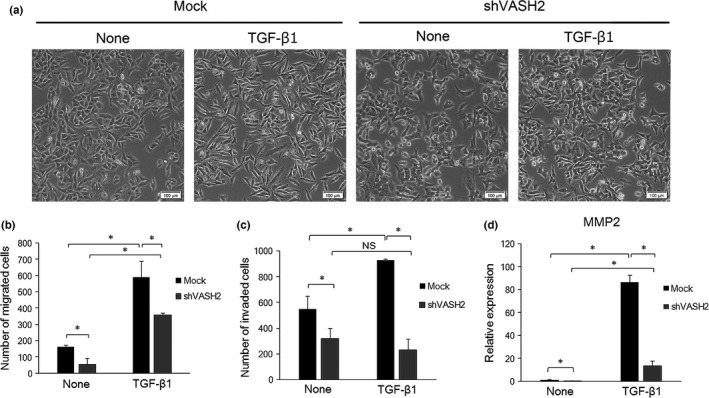
Downregulation of vasohibin‐2 (VASH2) blocks transforming growth factor‐β1 (TGF‐β1)‐induced morphological changes, cell migration, and invasion. (a,b) DISS cells transfected with mock or shVASH2 were treated or not with TGF‐β1 for 24 h. (a) Cells were observed under an optical microscope. Scale bar = 100 μm. (b) Transwell migration assay was carried out. Total number of cells that had migrated were stained with DAPI and counted in duplicate. (c) DISS cells transfected with mock or shVASH2 were treated or not with TGF‐β1 for 48 h then the Matrigel invasion assay was undertaken. Total number of cells that invaded the gel were stained with DAPI and counted in duplicate. (d) DISS cells transfected with mock or shVASH2 were treated or not with TGF‐β1 for 24 h, then quantitative real‐time RT‐PCR analysis of MMP2 expression was carried out. Values were normalized to the β‐actin mRNA level. Mean and SDs are shown (*P < 0.05). (b,c) Data shown are representative of three independent experiments.
As EMT also increases the migration and invasion of epithelial cells, we then examined the roles of endogenous VASH2 in cell migration and invasion. Transforming growth factor‐β1‐induced cell migration was observed in both mock and shVASH2 cells and VASH2 knockdown decreased the number of migrated cells regardless of TGF‐β stimulation (Fig. 5b). In contrast, VASH2 knockdown repressed the TGF‐β1‐induced increase in DISS invasion ability (Fig. 5c). Increased invasion ability during EMT is accompanied with increased expression of MMP family members. It is known that MMP2 degrades the ECM,24 which was induced by TGF‐β1. In accordance with the decreased invasive ability in shVASH2 cells, TGF‐β1‐induced MMP2 expression was attenuated by knockdown of VASH2 (Fig. 5d).
Discussion
Vasohibin‐2 is expressed in cancer cells and stimulates tumor angiogenesis in a paracrine manner.18 Here we examined the effect of VASH2 on cancer cells themselves with regards to the EMT. As TGF‐β is a critical factor that promotes the EMT for cancer invasiveness and metastasis, we investigated the role of VASH2 in relation to TGF‐β signaling. The knockdown of VASH2 in cancer cells repressed the expression of ALK5; this repression abrogated the downstream signaling of TGF‐β for the EMT. Activin receptor‐like kinase 5 is critical for the TGF‐β signaling pathway, as it activates Smad2/3, and activated Smad2/3 recognizes promoter sequences with a core CAGA motif. Although it has a critical function in TGF‐β signaling, little is known about the gene regulation for the expression of ALK5. Our present study reveals for the first time the requirement of VASH2 for the expression of ALK5 and its downstream signaling of TGF‐β for EMT in cancers. Further study is required to elucidate the mechanism that drives the regulation of expression of ALK5 by VASH2.
Due to its central role in TGF‐β signaling, ALK5 is emerging as a promising target for the blockade of the TGF‐β signaling pathway. Indeed, the inhibition of ALK5 is regarded as a promising therapeutic approach. Galunisertib (LY2157299 monohydrate) is an oral small‐molecule inhibitor of ALK5 that specifically downregulates the phosphorylation of Smad2 and abrogates the activation of the downstream pathway. Moreover, galunisertib shows antitumor activity in tumor‐bearing animal models such as those for breast, colon, and lung cancers, as well as hepatocellular carcinoma.25 We have been proposing VASH2 to be a molecular target for cancer treatment. Knockdown of VASH2 by the local injection of siRNA against VASH2 in vivo inhibits tumor growth.26 Based on the present study, we now propose the possibility that knockdown of VASH2 inhibits not only tumor angiogenesis but also the EMT of cancer cells. This possibility should be clarified in vivo in the future.
The phylogenic tree of the vasohibin family of proteins reveals that vasohibin genes are highly conserved among species.27 Lower organisms possess one common ancestral vasohibin gene, whereas vertebrates have Vash1 and Vash2, suggesting that this common ancestral vasohibin gene became divided into Vash1 and Vash2 during evolution. Blood vessels are organized from vertebrates during evolution, which indicates that the original function of the ancestral vasohibin was unrelated to the regulation of vascular formation, and that vasohibins later became applied to the vasculature in vertebrates. For that reason, it is possible that vasohibins in vertebrates have preserved the original functions of the ancestral vasohibin, and that the effect of VASH2 on the EMT might be one such function.
In our previous reports, we investigated the correlation between VASH2 and microRNAs (miR), and observed an inverse correlation between VASH2 and miR‐200b in human ovarian cancer tissue.21 This inverse correlation was confirmed in human hepatocellular carcinoma.20 It is well known that miR‐200b is involved in the regulation of the EMT by suppressing the expression of ZEB and that this microRNA balances the TGF‐β signaling for the EMT.28, 29 Our present data further suggest an intimate interaction among VASH2, TGF‐β, and miR‐200b for the regulation of EMT.
It has been reported that ALK5 expression is induced by inflammatory cytokines including tumor necrosis factor‐α during EMT of A549 lung carcinoma cells.30 While the mechanisms behind the regulation of ALK5 expression by various signals have not been elucidated, it is of interest to investigate in the future whether the regulation of ALK5 expression by VASH2 involves signals mediated by inflammatory cytokines.
In summary, our present study revealed that VASH2 is a novel player in EMT by modulating TGF‐β signaling. We propose VASH2 to be a possible molecular target for the prevention of EMT in cancers.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgment
This work was supported by a Grant‐in‐Aid for Scientific Research (B) (16H04689) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Cancer Sci 108 (2017) 419–426
Funding Information
Ministry of Education, Culture, Sports, Science, and Technology of Japan (16H04689).
References
- 1. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest 2009; 119: 1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT‐inducing transcription factors. Nat Cell Biol 2014; 16: 488–94. [DOI] [PubMed] [Google Scholar]
- 3. Wendt MK, Smith JA, Schiemann WP. Transforming growth factor‐beta‐induced epithelial‐mesenchymal transition facilitates epidermal growth factor‐dependent breast cancer progression. Oncogene 2010; 29: 6485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15: 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Comijn J, Berx G, Vermassen P et al The two‐handed E box binding zinc finger protein SIP1 downregulates E‐cadherin and induces invasion. Mol Cell 2001; 7: 1267–78. [DOI] [PubMed] [Google Scholar]
- 6. Bolos V, Peinado H, Perez‐Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E‐cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 2003; 116(Pt 3): 499–511. [DOI] [PubMed] [Google Scholar]
- 7. Sanchez‐Tillo E, Lazaro A, Torrent R et al ZEB1 represses E‐cadherin and induces an EMT by recruiting the SWI/SNF chromatin‐remodeling protein BRG1. Oncogene 2010; 29: 3490–500. [DOI] [PubMed] [Google Scholar]
- 8. Tam WL, Weinberg RA. The epigenetics of epithelial‐mesenchymal plasticity in cancer. Nat Med 2013; 19: 1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu J, Lamouille S, Derynck R. TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res 2009; 19: 156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts AB, Sporn MB. Physiological actions and clinical applications of transforming growth factor‐beta (TGF‐beta). Growth Factors 1993; 8(1): 1–9. [DOI] [PubMed] [Google Scholar]
- 11. Waite KA, Eng C. From developmental disorder to heritable cancer: it's all in the BMP/TGF‐β family. Nat Rev Genet 2003; 4: 763–73. [DOI] [PubMed] [Google Scholar]
- 12. Morikawa M, Derynck R, Miyazono K. TGF‐β and the TGF‐β family: context‐dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol 2016; 8: a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature 2003; 425: 577–84. [DOI] [PubMed] [Google Scholar]
- 14. ten Dijke P, Hill CS. New insights into TGF‐beta‐Smad signalling. Trends Biochem Sci 2004; 29: 265–73. [DOI] [PubMed] [Google Scholar]
- 15. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF‐beta type I receptors. EMBO J 2002; 21: 1743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watanabe K, Hasegawa Y, Yamashita H et al Vasohibin as an endothelium‐derived negative feedback regulator of angiogenesis. J Clin Invest 2004; 114: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shibuya T, Watanabe K, Yamashita H et al Isolation and characterization of vasohibin‐2 as a homologue of VEGF‐inducible endothelium‐derived angiogenesis inhibitor vasohibin. Arterioscler Thromb Vasc Biol 2006; 26: 1051–7. [DOI] [PubMed] [Google Scholar]
- 18. Kimura H, Miyashita H, Suzuki Y et al Distinctive localization and opposed roles of vasohibin‐1 and vasohibin‐2 in the regulation of angiogenesis. Blood 2009; 113: 4810–8. [DOI] [PubMed] [Google Scholar]
- 19. Kitahara S, Suzuki Y, Morishima M et al Vasohibin‐2 modulates tumor onset in the gastrointestinal tract by normalizing tumor angiogenesis. Mol Cancer 2014; 13: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xue X, Zhang Y, Zhi Q et al MiR200‐upregulated Vasohibin 2 promotes the malignant transformation of tumors by inducing epithelial‐mesenchymal transition in hepatocellular carcinoma. Cell Commun Signal 2014; 12: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takahashi Y, Koyanagi T, Suzuki Y et al Vasohibin‐2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol Cancer Res 2012; 10: 1135–46. [DOI] [PubMed] [Google Scholar]
- 22. Wieser R, Wrana J, Massague J. GS domain mutations that constitutively activate T beta RI, the downstream signaling component in the TGF‐beta receptor complex. EMBO J 1995; 14: 2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta‐inducible elements in the promoter of human plasminogen activator inhibitor‐type 1 gene. EMBO J 1998; 17: 3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenny HA, Lengyel E. MMP‐2 functions as an early response protein in ovarian cancer metastasis. Cell Cycle 2009; 8: 683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herbertz S, Sawyer JS, Stauber AJ et al Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor‐beta signaling pathway. Drug Des Devel Ther 2015; 9: 4479–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koyanagi T, Suzuki Y, Saga Y et al In vivo delivery of siRNA targeting vasohibin‐2 decreases tumor angiogenesis and suppresses tumor growth in ovarian cancer. Cancer Sci 2013; 104: 1705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sato Y. The vasohibin family: a novel family for angiogenesis regulation. J Biochem 2013; 153(1): 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gregory PA, Bert AG, Paterson EL et al The miR‐200 family and miR‐205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601. [DOI] [PubMed] [Google Scholar]
- 29. Gregory PA, Bracken CP, Smith E et al An autocrine TGF‐beta/ZEB/miR‐200 signaling network regulates establishment and maintenance of epithelial‐mesenchymal transition. Mol Biol Cell 2011; 22: 1686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu X. Inflammatory cytokines augments TGF‐beta1‐induced epithelial‐mesenchymal transition in A549 cells by up‐regulating TbetaR‐I. Cell Motil Cytoskelet 2008; 65: 935–44. [DOI] [PubMed] [Google Scholar]