

Abstract
The compound WP1066 was originally synthesized by modifying the structure of AG490, which inhibits the activation of signal transducer and activator of transcription 3 (STAT3) by directly targeting Janus kinases (JAKs). WP1066 exhibits stronger anti‐cancer activity than AG490 against malignant glioma and other cancer cells and is regarded as a promising therapeutic agent. By screening a small library of target‐known compounds, we identified WP1066 as an inhibitor of macrophage cell death induced by agonists of the NLRP3 inflammasome, an intracellular protein complex required for the processing of the proinflammatory cytokine interleukin (IL)‐1β. WP1066 strongly inhibited cell death as well as extracellular release of IL‐1β induced by inflammasome agonists in mouse peritoneal exudate cells and human leukemia monocytic THP‐1 cells that were differentiated into macrophagic cells by treatment with PMA. However, inflammasome agonists did not increase STAT3 phosphorylation, and another JAK inhibitor, ruxolitinib, did not inhibit cell death, although it strongly inhibited basal STAT3 phosphorylation. Thus, WP1066 appears to suppress macrophage cell death independently of its inhibitory effect on STAT3. In contrast, WP1066 itself induced the death of undifferentiated THP‐1 cells, suggesting that WP1066 differentially modulates cell death in a context‐dependent manner. Consistent with previous findings, WP1066 induced the death of human glioma A172 and T98G cells. However, neither ruxolitinib nor AG490, the former of which completely suppressed STAT3 phosphorylation, induced the death of these glioma cells. These results suggest that WP1066 targets cell death‐modulating molecules other than those involved in JAK‐STAT3 signaling.
Keywords: Cell death, glioma, macrophage, NLRP3 inflammasome, STAT3
Signal transducer and activator of transcription 3 (STAT3) functions mainly as a transcription factor and has been shown to be involved in tumor cell proliferation, survival and invasion.1 Activated Janus kinases (JAKs), such as JAK1 and JAK2, directly phosphorylate STAT3 at Y705, and phosphorylated STAT3 dimerizes and translocates to the nucleus. Thus, low‐molecular‐weight kinase inhibitors targeting JAKs have been regarded as promising anti‐cancer agents. WP1066 is among them and was originally synthesized as an anti‐cancer compound more potent than previous ones by modifying the structure of AG490, one of the prototypic JAK2 inhibitors.2, 3 WP1066 induces apoptosis and/or growth inhibition in a variety of cancer cells, such as those of malignant glioma, acute myelogenous leukemia, melanoma, renal cell carcinoma and oral squamous cell carcinoma, both in vitro and in vivo.3, 4, 5, 6, 7 According to ClinicalTrials.gov (https://clinicaltrials.gov), a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world, a phase I trial of WP1066 in patients with recurrent malignant glioma and brain metastasis from melanoma is ongoing.
The proinflammatory cytokine interleukin (IL)‐1β is transcriptionally induced as a precursor protein, called pro‐IL‐1β, after the activation of Toll‐like receptor (TLR) signaling in inflammatory cells such as macrophages.8 In response to a variety of pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs), the cysteine protease caspase‐1 is activated in an intracellular protein complex called the inflammasome, and activated caspase‐1 proteolytically processes pro‐IL‐1β into mature, active IL‐1β.9 Among the various inflammasomes, the NLRP3 inflammasome, which consists of NLRP3, a member of the NOD‐like receptor family, and the adaptor protein ASC (apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain), together with caspase‐1, is responsive to the broadest range of stimuli and therefore plays a central role in the regulation of IL‐1β processing.10 Finally, active IL‐1β is released from cells; however, the mechanism of extracellular release of IL‐1β remains elusive because the processing of pro‐IL‐1β occurs in the cytosol and generates active IL‐1β that lacks a secretory signal sequence.11 Thus, elucidating the mechanism of extracellular release of IL‐1β is a prerequisite for understanding the regulation of inflammation and inflammatory diseases.
In this study, we identified WP1066 as a strong inhibitor of macrophage cell death and the extracellular release of IL‐1β induced by NLRP3 inflammasome agonists. We further examined the effects of WP1066 on cell death in various contexts, with a particular focus on the relationship between its cell death‐modulating activity and its inhibitory effect against STAT3.
Materials and Methods
Reagents
Nigericin was purchased from Wako Chemical (Osaka, Japan). R837/imiquimod was purchased from InvivoGen (San Diego, CA, USA) and Tokyo Chemical Industry (Tokyo, Japan). WP1066, AG490 and ruxolitinib were purchased from Cayman Chemical (Ann Arbor, MI, USA). Lipopolysaccharide (LPS) and phorbol 12‐myristate 13‐acetate (PMA) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Ac‐YVAD‐CMK was purchased from Peptide Institute (Osaka, Japan). Chemical compounds in the SCADS Inhibitor Kit (Screening Committee of Anticancer Drugs supported by Grant‐in‐Aid for Scientific Research on Priority Area “Cancer” from the Ministry of Education, Culture, Sports, Science and Technology, Japan) were used for screening.
Cell culture
Human leukemia monocytic THP‐1 cells (American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 medium supplemented with 100 units/mL penicillin G and 0.1 mg/mL streptomycin containing 8% fetal bovine serum (FBS) under a 5% CO2 atmosphere at 37°C. THP‐1 cells were differentiated into macrophagic cells by overnight treatment with 0.1 μM PMA. Human glioma A172 cells and T98G cells (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 8% FBS and supplemented with 100 units/mL penicillin G and 0.1 mg/mL streptomycin under a 5% CO2 atmosphere at 37°C. Mouse peritoneal exudate cells (PECs) were isolated from the peritoneal cavity of 8‐ to 12‐week‐old mice 2 days after the intraperitoneal injection of 2 mL of 4% fluid thioglycollate medium (BD Diagnostic Systems, Heidelberg, Germany)12 and were further cultured in RPMI 1640 medium containing 8% FBS and supplemented with 100 units/mL penicillin G and 0.1 mg/mL streptomycin under a 5% CO2 atmosphere at 37°C for overnight. Prior to stimulation with inflammasome agonists, cells were washed twice with PBS and further cultured for 4 h in Opti‐MEM I Reduced‐Serum Medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 100 ng/mL LPS, which was the “priming” stimulus to induce the transcription of pro‐IL‐1β.
Immunoblot analysis
For immunoblot analysis of culture supernatants, culture medium was collected and centrifuged for 1 min at 860 g, and the resulting supernatants were added to the same volume of methanol and one‐fourth volume of chloroform and vigorously mixed. After incubation on ice for 15 min, the solution was centrifuged at 21 500 g for 10 min, and the upper phase of the solution was removed. The remaining solution was added to 500 μL of methanol and vigorously mixed. After incubation on ice for 15 min, the solution was centrifuged at 21 500 g for 10 min, and the supernatants were removed. Methanol was added to the pellet, and the solution was further centrifuged at 21 500 g for 10 min. The supernatants were removed, and the pellet was air‐dried and then dissolved in a buffer containing 125 mM Tris‐HCl (pH 6.8), 20% glycerol, 4% SDS and 10 mM DTT. For immunoblot analysis of cell lysates, PECs were lysed with a buffer containing 62.5 mM Tris‐HCl (pH 6.8), 10% glycerol, 2% SDS and 5 mM DTT, followed by sonication for 1 min. Other cells were lysed with a buffer containing 25 mM Tris‐HCl (pH 7.5), 150 mM NaCl, 5 mM EGTA, 1% Triton X‐100, 5 μg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, and after centrifugation at 21 500 g for 15 min the supernatants were collected as cell lysates. When detecting phospho‐Stat3, PhosSTOP Phosphatase Inhibitor Cocktail (Roche Life Science, Mannheim, Germany) was included in the lysis buffer. Cell lysates were then fractionated by SDS‐polyacrylamide gel electrophoresis and electroblotted onto polyvinylidene difluoride membranes. The membranes were probed with primary antibodies and horseradish peroxidase (HRP)‐conjugated secondary antibodies. Protein bands were visualized using the enhanced chemiluminescence system and analyzed with an ImageQuant LAS4000 (GE Healthcare, Piscataway, NJ, USA). The following primary antibodies were used in this study: anti‐IL‐1β (human specific; #12703) antibody, anti‐IL‐1β (mouse specific; #8689) antibody, anti‐phospho‐Stat3 (Tyr705) antibody, anti‐phospho‐Stat3 (Ser727) antibody, anti‐Stat3 antibody and anti‐β‐actin antibody, all from Cell Signaling (Danvers, MA, USA); anti‐cleaved IL‐1β (mouse) antibody (MBL, Nagoya, Japan); and anti‐caspase‐1 (p20) antibody (Adipogen, San Diego, CA, USA). HRP‐conjugated anti‐mouse IgG (GE Healthcare) and HRP‐conjugated anti‐rabbit IgG (Cell Signaling) were used as secondary antibodies.
Il‐1β ELISA
Culture medium was collected and centrifuged at 860 g for 1 min, and the IL‐1β level in the resulting supernatants was measured using an IL‐1β ELISA kit (Quantikine ELISA; R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Cell death assay
For propidium iodide (PI) staining, 2 μg/mL PI was added to the culture medium 10 min before cell harvest. For adherent cells, cells were dissociated with trypsin and suspended into single cells by pipetting or passing through 23G needles. The suspended cells were centrifuged at 860 g for 3 min and resuspended in PBS. The fluorescence emitted by cells was analyzed using a BD Accuri C6 flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA). To detect the level of lactate dehydrogenase (LDH) released from the cells, the Cytotoxicity LDH Assay Kit‐WST (Dojindo, Kumamoto, Japan) was used according to the manufacturer's instructions.
Results
WP1066 suppresses IL‐1β release from macrophages
To explore the mechanism of IL‐1β release from macrophages, we sought to identify target‐known low‐molecular‐weight compounds that inhibit IL‐1β release. We expected that this would be a fast approach to identify important molecules that regulate IL‐1β release. We screened the effects of 365 compounds on the release of IL‐1β from human leukemia monocytic THP‐1 cells treated with the chemical NLRP3 inflammasome agonist R837/imiquimod.13 Prior to treatment with R837, THP‐1 cells were differentiated into macrophagic cells by PMA treatment and were then primed with LPS to efficiently induce the transcription of pro‐IL‐1β. We found that WP1066 was among the strongest compounds that inhibited R837‐induced IL‐1β release from THP‐1 cells (Fig. 1a). While IL‐1β was continuously released from THP‐1 cells after R837 stimulation, 10 μM WP1066 completely suppressed IL‐1β release during the 120‐min treatment with R837 (Fig. 1b), and as little as 1 μM WP1066 suppressed IL‐1β release 60 min after stimulation (Fig. 1c). Immunoblot (IB) analysis of IL‐1β released into the culture supernatant of THP‐1 cells revealed that WP1066 also strongly suppressed the release of IL‐1β and unprocessed pro‐IL‐1β in response to the bacterial toxin nigericin, monosodium urea (MSU) crystals and low osmolarity (treatment with 2:5 diluted Opti‐MEM I Reduced‐Serum Medium), all of which have been shown to act as strong NLRP3 inflammasome agonists14, 15, 16 (Fig. 1d). These results suggest that WP1066 suppresses IL‐1β release upon NLRP3 inflammasome activation from THP‐1 cells.
Figure 1.
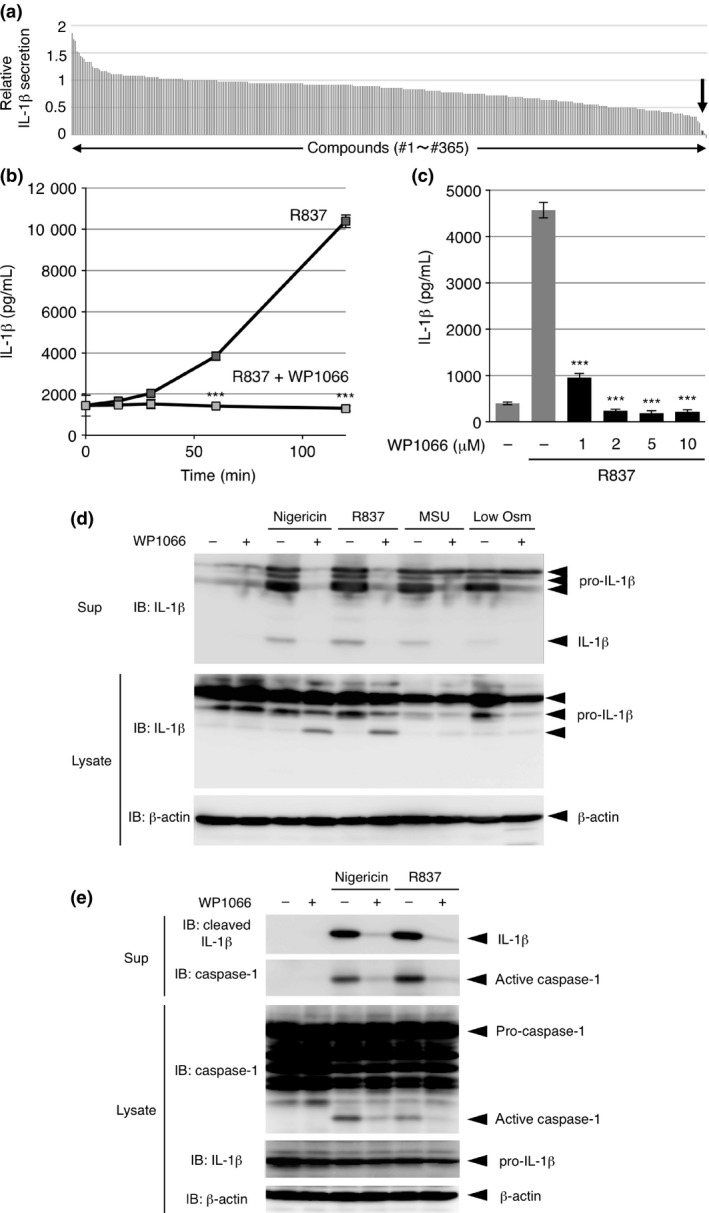
WP1066 suppresses IL‐1β release from differentiated THP‐1 cells and peritoneal exudate cells (PECs). (a) Differentiated THP‐1 cells were pre‐treated with 1% dimethylsulfoxide (DMSO) (control) or 1 μM of each compound for 30 min and treated with 10 μg/mL R837 for 2 h. IL‐1β secretion relative to control was determined by analyzing the culture supernatants with an IL‐1β enzyme linked immunosorbent assay (ELISA). Data are an average of two independent experiments and are sorted in descending order. The black bar indicated by an arrow shows the result for WP1066. (b, c) Differentiated THP‐1 cells were pretreated with 10 μM (b) or the indicated doses (c) of WP1066 for 30 min and treated with 10 μg/mL R837 for the indicated periods (b) or for 2 h (c). The culture supernatants were subjected to an IL‐1β ELISA. Data are shown as the mean ± SEM (n = 3). ***P < 0.001, Student's t‐test (b) and Dunnett's multiple comparison test (c), compared with the cells treated with R837 but not with WP1066. (d) Differentiated THP‐1 cells were pretreated with 10 μM WP1066 for 30 min and treated with 5 μM nigericin, 10 μg/mL R837, 50 μg/mL MSU, or low osmolarity (Low Osm) for 2 h. The culture supernatants (Sup) and cell lysates were subjected to IB analysis. (e) PECs were pretreated with 10 μM WP1066 for 30 min and treated with 5 μM nigericin or 10 μg/mL R837 for 2 h. The culture supernatants (Sup) and cell lysates were subjected to IB analysis.
We next examined the effect of WP1066 on PECs. WP1066 suppressed the nigericin‐ and R837‐induced release of IL‐1β together with that of active caspase‐1, as shown by IB analysis of the culture supernatant of PECs (Fig. 1e, upper two panels), suggesting that WP1066 also suppresses IL‐1β release upon NLRP3 inflammasome activation in PECs. IB analysis of PEC lysates revealed that WP1066 suppressed the nigericin‐ or R837‐induced processing and activation of caspase‐1 (Fig. 1e, lower three panels). Although we could not examine the effect of WP1066 on caspase‐1 activation in THP‐1 cells because of the unavailability of antibodies detecting active caspase‐1 in human cells, the NLRP3 inflammasome or its upstream pathway may be at least one of the targets of WP1066 in inflammasome agonist‐induced IL‐1β release.
WP1066 suppresses cell death induced by inflammasome agonists
In IB analysis of the culture supernatant of THP‐1 cells, we noticed that β‐actin was released from the cells in a time‐dependent manner similar to that of IL‐1β release after R837 stimulation (Fig. 2a). This raised the possibility that cell death was induced by inflammasome agonists, as has been shown in the case of pyroptosis, a form of cell death mediated by the activation of caspase‐1.17 In fact, the LDH release assay revealed that R837 strongly induced the death of THP‐1 cells, an effect that was suppressed by pretreatment with WP1066 (Fig. 2b). When dead cells were detected through the cellular incorporation of propidium iodide (PI), R837‐induced death and the strong inhibitory effect of WP1066 on it were confirmed in THP‐1 cells (Fig. 2c, upper graph). This R837‐induced death was strongly inhibited by the addition of high KCl to the culture medium. High KCl is known to inhibit NLRP3 inflammasome activation and cell death by blocking K+ efflux, a common trigger of NLRP3 inflammasome activation, as confirmed by the considerable reduction in released IL‐1β in the culture supernatant18, 19 (Fig. 2c, lower panel). On the other hand, the inhibition of cell death by the caspase‐1 inhibitor Ac‐YVAD‐CMK was limited, although it effectively suppressed caspase‐1 activation, as confirmed by the reduction in cleaved IL‐1β in the supernatant (Fig. 2c). We found similar results when THP‐1 cells were stimulated with nigericin (Fig. 2d). Furthermore, the R837‐induced death of PECs was suppressed by WP1066, whereas it was not suppressed but rather enhanced by Ac‐YVAD‐CMK (Fig. 2e). These results suggest that the site of action of WP1066 is not limited to NLRP3 inflammasome activation, i.e., caspase‐1 activation and that WP1066 targets the cell death‐inducing machinery that does not rely on caspase‐1 activity in macrophages.
Figure 2.
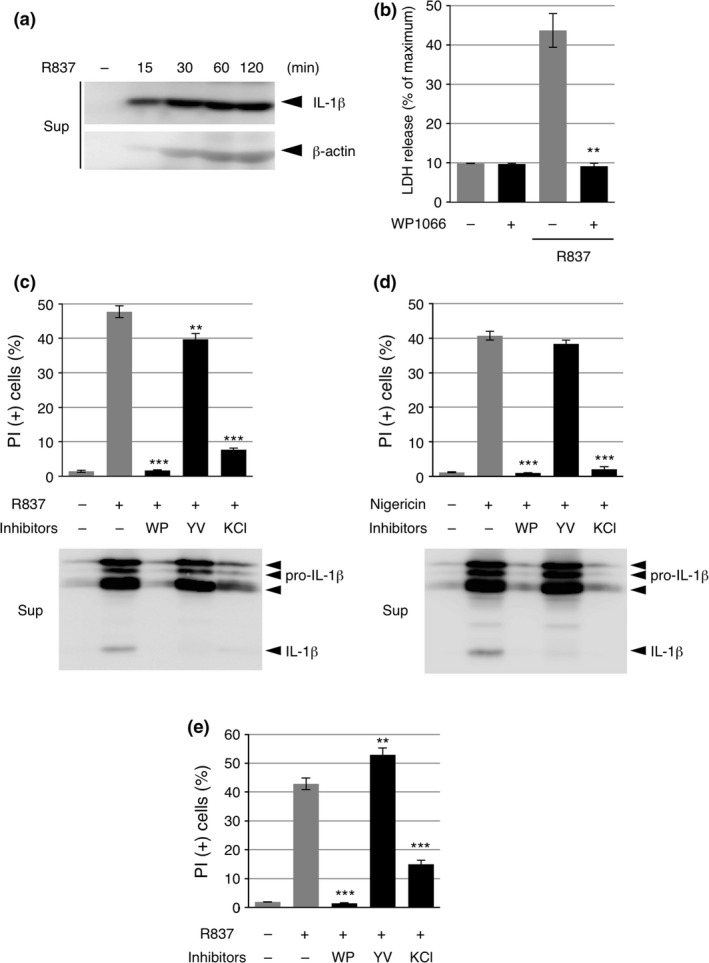
WP1066 suppresses cell death induced by inflammasome agonists. (a) Differentiated THP‐1 cells were treated with 10 μg/mL R837 for the indicated periods. The culture supernatants (Sup) were subjected to IB analysis. (b) Differentiated THP‐1 cells were pretreated with 10 μM WP1066 for 30 min and treated with 10 μg/mL R837 for 2 h. Cells were subjected to an lactate dehydrogenase (LDH) release assay. Data are shown as the mean ± SEM (n = 3). **P < 0.01, Student's t‐test, compared with the cells treated with R837 but not with WP1066. (c, d) Differentiated THP‐1 cells were pretreated with 10 μM WP1066 (WP), 20 μM Ac‐YVAD‐CMK (YV), or 60 mM KCl for 30 min and treated with 10 μg/mL R837 (c) or 5 μM nigericin (d) for 2 h. Cells were subjected to a propidium iodide (PI) assay, and the culture supernatants were subjected to IB analysis. Data are shown as the mean ± SEM (n = 3). **P < 0.01, ***P < 0.001, Dunnett's multiple comparison test, compared with the cells treated with R837 but not with any inhibitors. (e) Peritoneal exudate cells (PECs) were pretreated with 10 μM WP1066 (WP), 20 μM Ac‐YVAD‐CMK (YV), or 60 mM KCl for 30 min and treated with 10 μg/mL R837 for 2 h. Cells were subjected to a PI assay. Data are shown as the mean ± SEM (n = 3). **P < 0.01, ***P < 0.001, Dunnett's multiple comparison test, compared with the cells treated with R837 but not with any inhibitors.
Inflammasome agonists do not induce STAT3 activation
Because WP1066 has been characterized as a STAT3 inhibitor, we examined the activation state of STAT3 by monitoring its phosphorylation at Y705 in THP‐1 cells. Whereas the basal phosphorylation of Y705 (P‐Y705) was quite low in undifferentiated THP‐1 cells, P‐Y705 increased in a time‐dependent manner in response to PMA and increased further by 4‐h priming with LPS (Fig. 3a). The PMA/LPS‐induced P‐Y705 was attenuated by treatment with WP1066. The phosphorylation of STAT3 at S727, which has been reported to be required for the mitochondrial translocation of STAT3,20 did not change throughout treatment with PMA and LPS, in contrast to P‐Y705. We first expected that inflammasome agonists would further activate STAT3 to facilitate NLRP3 inflammasome activation and cell death, but quite the contrary, R837 time‐dependently decreased P‐Y705 that had been induced by pretreatment with PMA and LPS (Fig. 3b). Moreover, none of other NLRP3 inflammasome agonists examined increased STAT3 P‐Y705 (Fig. 3c).
Figure 3.
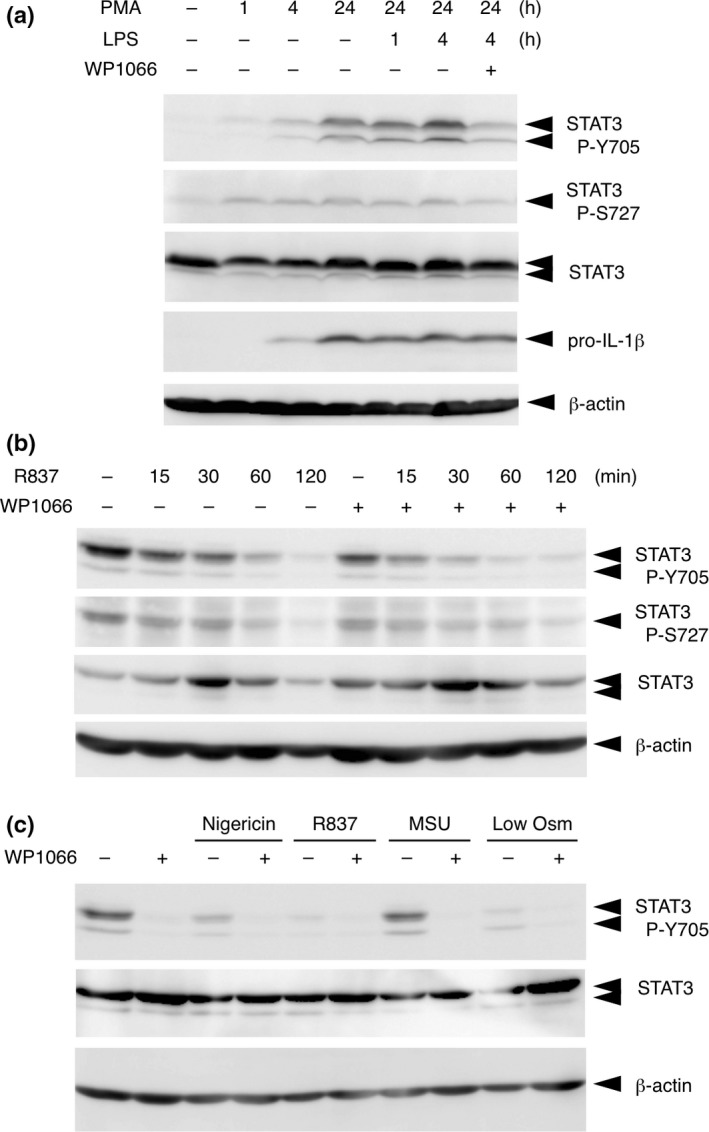
Inflammasome agonists do not induce STAT3 activation. (a) THP‐1 cells were treated with 0.1 μM phorbol 12‐myristate 13‐acetate (PMA) for the indicated periods and then treated with lipopolysaccharide (LPS) for 1 or 4 h. Cells were treated with or without WP1066 for 2 h before lysis. Cell lysates were subjected to immunoblot (IB) analysis. (b) THP‐1 cells were treated with 0.1 μM PMA for 24 h followed by treatment with 100 ng/mL LPS for 4 h and were further treated with 10 μg/mL R837 for the indicated periods. Cells were or were not treated with WP1066 30 min before treatment with R837. Cell lysates were subjected to IB analysis. (c) Differentiated THP‐1 cells were pretreated with 10 μM WP1066 for 30 min and treated with 5 μM nigericin, 10 μg/mL R837, 50 μg/mL MSU, or low osmolarity (Low Osm) for 2 h. Cell lysates were subjected to IB analysis.
WP1066 suppresses inflammasome agonist‐induced cell death independently of its inhibitory effect on STAT3
We then examined the involvement of STAT3 in IL‐1β release and cell death using different JAK inhibitors that have previously been shown to inhibit STAT3 activation. Ruxolitinib (INCB018424), a well‐characterized inhibitor of JAK1 and JAK2,21 strongly suppressed STAT3 P‐Y705 but unexpectedly did not suppress IL‐1β release from THP‐1 cells treated with R837 (Fig. 4a). The inhibitory effect of AG490, the predecessor compound of WP1066, on R837‐induced IL‐1β release was much weaker than that of WP1066 (Fig. 4b). Consistent with these results, neither ruxolitinib nor AG490 suppressed the R837‐induced death of THP‐1 cells (Fig. 4c). Thus, the inhibitory effect of WP1066 on inflammasome agonist‐induced cell death does not appear to depend on its inhibitory effect on STAT3, and WP1066 may target cell death‐inducing molecules other than those involved in JAK‐STAT3 signaling.
Figure 4.
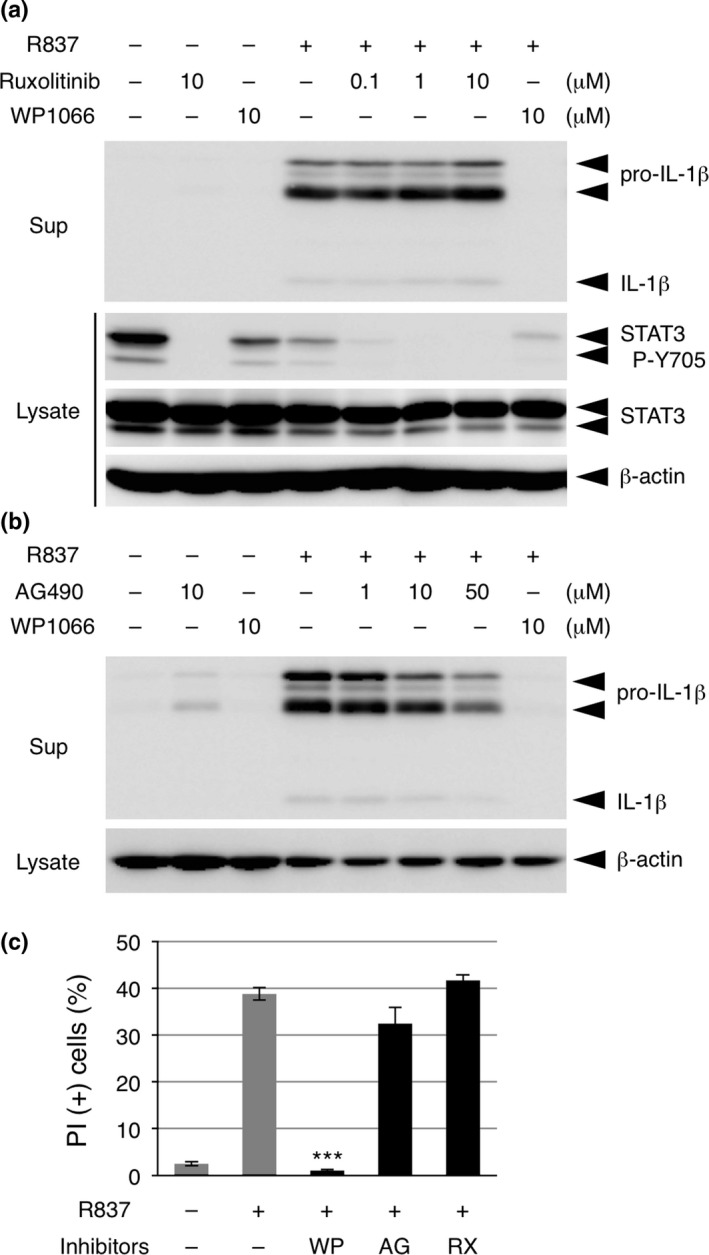
WP1066 suppresses inflammasome agonist‐induced cell death independently of its inhibitory effect on STAT3. (a) Differentiated THP‐1 cells were pretreated with the indicated doses of ruxolitinib or WP1066 for 30 min and treated with 10 μg/mL R837 for 2 h. The culture supernatants (Sup) and cell lysates were subjected to immunoblot (IB) analysis. (b) Differentiated THP‐1 cells were pretreated with the indicated doses of AG490 or WP1066 for 30 min and treated with 10 μg/mL R837 for 2 h. The culture supernatants (Sup) and cell lysates were subjected to IB analysis. (c) Differentiated THP‐1 cells were pretreated with 10 μM WP1066, AG490 (AG), or ruxolitinib (RX) for 30 min and treated with 10 μg/mL R837 for 2 h. Cells were subjected to a propidium iodide (PI) assay. Data are shown as the mean ± SEM (n = 3). ***P < 0.001, Dunnett's multiple comparison test, compared with the cells treated with R837 but not with any inhibitors.
WP1066 induces the death of undifferentiated THP‐1 cells
As described above, WP1066 has been shown to induce apoptosis in a variety of malignant cells.3, 4, 5, 6, 7 We thus examined whether WP1066 itself affected the viability of THP‐1 cells. Whereas WP1066 exhibited no obvious toxicity on PMA‐differentiated THP‐1 cells, it clearly induced the death of undifferentiated THP‐1 cells 6 h after stimulation (Fig. 5a). More prolonged treatment with WP1066 (for 12 h) still did not induce the death of differentiated THP‐1 cells but strongly induced the death of undifferentiated THP‐1 cells (Fig. 5b). These results suggest that the sensitivity of THP‐1 cells to WP1066 largely depends on their differentiation state. Similar to the inhibitory effects on inflammasome agonist‐induced cell death, neither AG490 nor ruxolitinib induced the death of undifferentiated THP‐1 cells (Fig. 5a,b), and cell viability was not correlated with the phosphorylation state of STAT3 Y705 (Fig. 5c). Thus, the cell death‐inducing activity of WP1066 may also not depend on its inhibitory effect on STAT3.
Figure 5.
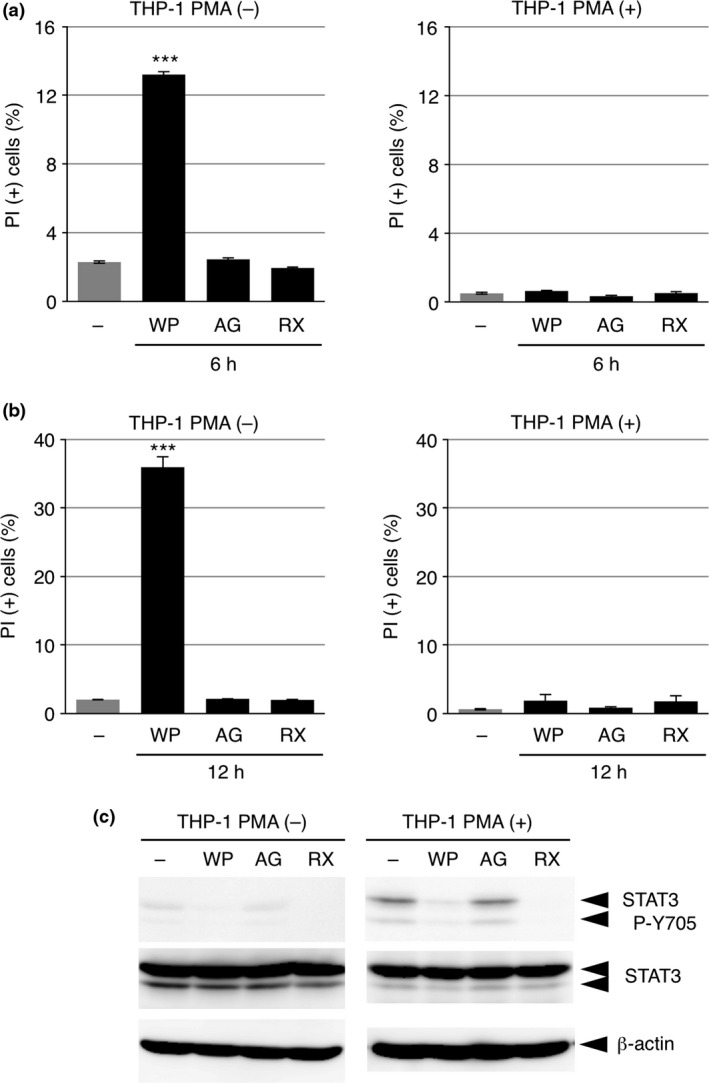
WP1066 induces the death of undifferentiated THP‐1 cells. (a, b) THP‐1 cells were treated with or without phorbol 12‐myristate 13‐acetate (PMA) overnight and then washed twice with PBS. Undifferentiated (left graphs; PMA (−)) and differentiated (right graphs; PMA (+)) THP‐1 cells were treated with 10 μM WP1066, AG490 (AG), or ruxolitinib (RX) for 6 h (a) or 12 h (b) in RPMI 1640 medium containing 8% fetal bovine serum (FBS). Cells were subjected to a propidium iodide (PI) assay. Data are shown as the mean ± SEM (n = 3). ***P < 0.001, Dunnett's multiple comparison test, compared with the untreated cells. (c) Undifferentiated (left panels; PMA (−)) and differentiated (right panels; PMA (+)) THP‐1 cells pretreated as in (a, b) were treated with 10 μM WP1066, AG490 (AG), or ruxolitinib (RX) for 1 h. Cell lysates were subjected to IB analysis.
WP1066 induces the death of glioma cells independently of its inhibitory effect on STAT3
Given that WP1066 likely induces the death of undifferentiated THP‐1 cells by targeting molecules other than those involved in JAK‐STAT3 signaling, we examined whether the well‐characterized cell death‐inducing effects of WP1066 on glioma cells indeed were dependent on its inhibitory effect on STAT3. Whereas WP1066 consistently induced death and inhibited STAT3 P‐Y705 in A172 glioma cells (Fig. 6a), it induced death but did not inhibit STAT3 P‐Y705 in another glioma cell line, T98G (Fig. 6b). In addition, neither AG490 nor ruxolitinib, the latter of which completely suppressed STAT3 P‐Y705, induced death in both cell lines. These results suggest that, contrary to previous findings, WP1066 induces the death of glioma cells independently of its inhibitory effect on STAT3.
Figure 6.
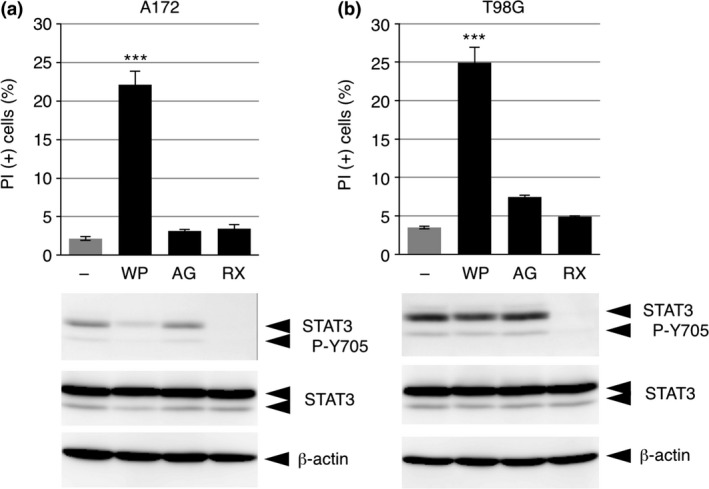
WP1066 induces the death of glioma cells independently of its inhibitory effect on STAT3. A172 cells (a) and T98G cells (b) were treated with 10 μM WP1066 (WP), AG490 (AG), or ruxolitinib (RX) for 12 h. Cells were subjected to a propidium iodide (PI) assay (upper graphs), and cell lysates were subjected to immunoblot (IB) analysis (lower panels). Data from the PI assay are shown as the mean ± SEM (n = 3). ***P < 0.001, Dunnett's multiple comparison test, compared with the untreated cells.
Discussion
In this study, we found that WP1066 strongly suppressed macrophage cell death and the extracellular release of IL‐1β induced by NLRP3 inflammasome agonists in two models of macrophages, mouse primary PECs and PMA‐differentiated human THP‐1 cells. IL‐1β is essential for an appropriate acute inflammatory response to various pathogens and injury, but its dysregulated excess release leads to sepsis and septic shock or to chronic inflammation.8, 11 Thus, the NLRP3 inflammasome is a fascinating drug target for controlling IL‐1β production. It has indeed been reported that a small‐molecule inhibitor of the NLRP3 inflammasome is effective in treating various auto‐inflammatory and autoimmune diseases.22 However, considering that the extracellular release of IL‐1β upon cell death does not totally depend on the activation state of the NLRP3 inflammasome, regulating cell death may be the ultimate way to control IL‐1β production, particularly in the case of severe inflammatory conditions, such as septic shock. In this regard, it would be interesting to see if WP1066 could also effectively suppress IL‐1β production in in vivo models of inflammation.
Although cell death appears to be one of the potential ways to release IL‐1β from cells, the mechanisms by which macrophage cell death is induced by various inflammatory stimuli, including NLRP3 inflammasome agonists, are still elusive. It has recently been reported that R837 activates the NLRP3 inflammasome by inducing robust reactive oxygen species through disturbing the quinone oxidoreductase NQO2 and mitochondrial complex I,23 but how R837 induces cell death has not been clarified. In our experiments, the caspase‐1 inhibitor Ac‐YVAD‐CMK did not suppress the R837‐ or nigericin‐induced death of THP‐1 cells or the R837‐induced death of PECs, suggesting that pyroptosis, which depends largely on caspase‐1, is not a major type of cell death in these conditions. In fact, it has recently been reported that necrosis mostly contributes to IL‐1β release at least from THP‐1 cells.24 Nevertheless, caspase‐1 has been proposed to induce necrosis independently of its catalytic activity,25 which may indeed be induced in our experiments particularly in the presence of Ac‐YVAD‐CMK (Fig. 2). Thus, we should further carefully examine the role of caspase‐1 in macrophage cell death induced by various inflammatory stimuli. Although we concluded in this study that WP1066 suppresses inflammasome agonist‐induced macrophage cell death independently of its effect on JAK‐STAT3 signaling, identification of the molecules targeted by WP1066 in stimulated macrophages will shed new light on the mechanism regulating inflammatory cell death in macrophages.
The induction of death in THP‐1 cells only in their undifferentiated state was an unexpected effect of WP1066. Interestingly, this cell death‐promoting effect of WP1066 on undifferentiated THP‐1 cells was also independent of its effect on JAK‐STAT3 signaling. Although it is unknown at the present time whether the target molecules of WP1066 differ between undifferentiated and differentiated THP‐1 cells, WP1066 generally targets molecules critical for the regulation of cell death regardless of cellular conditions. This difference in sensitivity to WP1066 between the two differentiation states of THP‐1 cells suggests that the range of cell death‐inducing stimuli differs depending on the differentiation state of the THP‐1 cells. This might account for the mechanism by which activated macrophages are prone to inflammatory death when they should release intracellular cytokines such as IL‐1β. WP1066 would also be a strong tool to use in exploring this issue.
The results that WP1066 induced death in two glioma cell lines, A172 and T98G, apparently independently of the activation state of STAT3 were unexpected. Of course, we need to examine many more cell types, including those other than glioma cells, to confirm our conclusion. However, this appears to provide an important caution with regard to the clinical use of this compound. In this case, neither the phosphorylation level of STAT3 nor the activity of JAKs may be suitable markers for the use of WP1066, at least in malignant glioma. Thus, identification of the WP1066 target molecules that are critical for cell death regulation would be a rather difficult but reliable approach to ensure the appropriate clinical use of this compound.
Disclosure
The authors have no conflict of interest.
Acknowledgments
We thank the Screening Committee of Anticancer Drugs supported by Grant‐in‐Aid for Scientific Research on Priority Area “Cancer” from The Ministry of Education, Culture, Sports, Science and Technology for the generous gift of the SCADS Inhibitor Kit. This work was supported in part by JSPS KAKENHI Grant Number JP26293016, JST PRESTO, the Mitsubishi Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, the Life Science Foundation of Japan, the Takeda Science Foundation and the Naito Foundation.
Cancer Sci 108 (2017) 520–527
Funding Information
JSPS KAKENHI (JP26293016); Naito Foundation; JST PRESTO; NOVARTIS Foundation; Takeda Science Foundation; Mitsubishi Foundation; Life Science Foundation of Japan.
References
- 1. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 2014; 14: 736–46. [DOI] [PubMed] [Google Scholar]
- 2. Meydan N, Grunberger T, Dadi H et al Inhibition of acute lymphoblastic leukaemia by a Jak‐2 inhibitor. Nature 1996; 379: 645–8. [DOI] [PubMed] [Google Scholar]
- 3. Hussain SF, Kong LY, Jordan J et al A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res 2007; 67: 9630–6. [DOI] [PubMed] [Google Scholar]
- 4. Ferrajoli A, Faderl S, Van Q et al WP1066 disrupts Janus kinase‐2 and induces caspase‐dependent apoptosis in acute myelogenous leukemia cells. Cancer Res 2007; 67: 11291–9. [DOI] [PubMed] [Google Scholar]
- 5. Kong LY, Abou‐Ghazal MK, Wei J et al A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res 2008; 14: 5759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horiguchi A, Asano T, Kuroda K et al STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br J Cancer 2010; 102: 1592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou X, Ren Y, Liu A et al STAT3 inhibitor WP1066 attenuates miRNA‐21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol Rep 2014; 31: 2173–80. [DOI] [PubMed] [Google Scholar]
- 8. Dinarello CA. Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol 2009; 27: 519–50. [DOI] [PubMed] [Google Scholar]
- 9. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16: 407–20. [DOI] [PubMed] [Google Scholar]
- 10. Wen H, Miao EA, Ting JP. Mechanisms of NOD‐like receptor‐associated inflammasome activation. Immunity 2013; 39: 432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Piccioli P, Rubartelli A. The secretion of IL‐1β and options for release. Semin Immunol 2013; 25: 425–9. [DOI] [PubMed] [Google Scholar]
- 12. Schneider M. Collecting resident or thioglycollate‐elicited peritoneal macrophages. Methods Mol Biol 2013; 1031: 37–40. [DOI] [PubMed] [Google Scholar]
- 13. Kanneganti TD, Ozoren N, Body‐Malapel M et al Bacterial RNA and small antiviral compounds activate caspase‐1 through cryopyrin/Nalp3. Nature 2006; 440: 233–6. [DOI] [PubMed] [Google Scholar]
- 14. Mariathasan S, Weiss DS, Newton K et al Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–32. [DOI] [PubMed] [Google Scholar]
- 15. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–41. [DOI] [PubMed] [Google Scholar]
- 16. Compan V, Baroja‐Mazo A, Lopez‐Castejon G et al Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012; 37: 487–500. [DOI] [PubMed] [Google Scholar]
- 17. Croker BA, O'Donnell JA, Gerlic M. Pyroptotic death storms and cytopenia. Curr Opin Immunol 2014; 26: 128–37. [DOI] [PubMed] [Google Scholar]
- 18. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007; 14: 1583–9. [DOI] [PubMed] [Google Scholar]
- 19. Munoz‐Planillo R, Kuffa P, Martinez‐Colon G, Smith BL, Rajendiran TM, Nunez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38: 1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wegrzyn J, Potla R, Chwae YJ et al Function of mitochondrial Stat3 in cellular respiration. Science 2009; 323: 793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quintas‐Cardama A, Vaddi K, Liu P et al Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010; 115: 3109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coll RC, Robertson AA, Chae JJ et al A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21: 248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gross CJ, Mishra R, Schneider KS et al K+ Efflux‐independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 2016; 45: 761–73. [DOI] [PubMed] [Google Scholar]
- 24. Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse activators of the NLRP3 inflammasome promote IL‐1β secretion by triggering necrosis. Cell Rep 2015; 11: 1535–48. [DOI] [PubMed] [Google Scholar]
- 25. Motani K, Kushiyama H, Imamura R, Kinoshita T, Nishiuchi T, Suda T. Caspase‐1 protein induces apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC)‐mediated necrosis independently of its catalytic activity. J Biol Chem 2011; 286: 33963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]