

Abstract
The role of HGF/SF‐MET signaling is important in cancer progression, but its relation with Helicobacter pylori‐positive gastric cancers remains to be elucidated. In total, 201 patients with primary gastric carcinoma who underwent curative or debulking resection without preoperative chemotherapy were studied. MET4 and anti‐HGF/SF mAbs were used for immunohistochemical analysis. Survival of gastric cancer patients was estimated by Kaplan–Meier method and compared with log‐rank. Cox proportional hazards models were fit to determine the independent association of MET‐staining status with outcome. The effect of live H. pylori bacteria on cell signaling and biological behaviors was evaluated using gastric cancer cell lines. MET4‐positive gastric cancers showed poorer prognosis than MET4‐negative cases (overall survival, P = 0.02; relapse‐free survival, P = 0.06). Positive staining for MET4 was also a statistically significant factor to predict poor prognosis in H. pylori‐positive cases (overall survival, P < 0.01; relapse‐free survival, P = 0.01) but not in H. pylori‐negative cases. Gastric cancers positively stained with both HGF/SF and MET4 showed a tendency of the worst prognosis. Stimulation of MET‐positive gastric cancer cells with live H. pylori bacteria directly upregulated MET phosphorylation and activated MET downstream signals such as p44/42MAPK and Akt, conferring cell proliferation and anti‐apoptotic activity. In conclusion, positive staining for MET4 was useful for predicting poor prognosis of gastric cancers with H. pylori infection. Helicobacter pylori stimulated MET‐positive gastric cancers and activated downstream signaling, thereby promoting cancer proliferation and anti‐apoptotic activity. These results support the importance of H. pylori elimination from gastric epithelial surface in clinical therapy.
Keywords: Gastric cancer, Helicobacter pylori, MET, MET4 antibody, prognosis
Oncogene MET encodes a receptor‐type tyrosine kinase, that transduces intracellular signals after reception of its only ligand, hepatocyte growth factor/scatter factor (HGF/SF).1 MET is a master signaling molecule that regulates important cancer steps, namely proliferation in the primary sites, invasion and metastasis into surrounding or remote tissues,2, 3, 4, 5 peritoneal dissemination,6, 7 angiogenesis,8 and proliferation in metastatic lesions. Clinical studies have shown that high MET expression correlates with poor prognosis of various tumors9, 10, 11, 12 (www.vai.org/met).
Reports describing the role of MET in gastric cancers have been accumulating. For instance, MET protein expression is significantly higher in gastric tumor tissue than in intestinal metaplasia, suggesting a role for MET in the process during malignant transformation of the gastric mucosa.13 Also, higher expression of MET is observed in the poorer prognosis of alpha fetoprotein‐producing gastric cancers.14 MET is actually activated in gastric carcinoma tissue, and may trigger proliferation/anti‐apoptotic signals.15 Also urokinase‐type plasminogen activator (uPA) promotes the invasive capacity of the uPA receptor‐positive gastric cancer cells and this phenotype is upregulated by the stimulation of HGF/SF‐Met signaling.16, 17 MET expression in gastric cancer cells correlates with their metastatic potential such as liver metastasis.18
Recent epidemiologic evidence indicates that Helicobacter pylori (H. pylori) infection significantly increases the risk for gastric carcinoma.19, 20 Helicobacter pylori infection induces chronic atrophic gastritis strongly enhancing the development of cancer, especially the intestinal type.21 The carcinogenicity of H. pylori is supported by the result that eradication diminishes the enhancing effects of H. pylori infection on glandular stomach carcinogenesis.22 Moreover, H. pylori infection accelerates the mutation of p53‐Rb systems and activates telomerase activity, which acts as a risk factor for gastric cancers.23 Also H. pylori bacteria augment the growth of gastric cancers via the LPS‐TLR4 pathway, whereas they attenuate the antitumor activity and IFN‐γ‐mediated cellular immunity of mononuclear cells, suggesting the inflammatory role of H. pylori infection in the proliferation and progression of gastric cancers.24 These finding may suggest some role of MET in H. pylori‐infected gastric cancers.25 However, no report has focused on the relationship between MET and H. pylori infection in clinical gastric cancers.
Recent success in the development of small molecule inhibitors against various kinases has brought a new paradigm of cancer therapy. Molecular targeting therapy against MET will be one of the most promising candidates for malignancies including gastric cancers. In this regard, an accurate evaluation of the relationship between MET expression and patient prognosis is essential for clinical application. Here we investigated the relationship between HGF/SF (MET ligand), MET expression and clinicopathological status in patients with gastric cancers.
Materials and Methods
Patients and clinicopathological characteristics
Two hundred and one patients with primary gastric carcinoma who underwent curative or debulking resection without preoperative chemotherapy at the National Defense Medical College Hospital between 1998 and 2007 were studied. Clinicopathological characteristics are shown in Table 1. For each case, all available histological sections were reviewed by two independent pathologists, and a representative block was selected for additional studies. Histological diagnosis consisted of 11 cases of papillary adenocarcinoma, 72 cases of tubular adenocarcinoma, 98 cases of poorly differentiated adenocarcinoma, six cases of signet ring cell carcinoma, 12 cases of mucinous carcinoma, and two cases of other histological types (adenosquamous carcinoma and undifferentiated carcinoma, one each). Clinical stages of the patients were evaluated according to the criteria of the Japanese Research Society for Gastric Cancer (14th edition). All specimens and clinical information were collected under Institutional Review Board‐approved protocols. Adjuvant therapy by oral anti‐cancer agents such as 5‐fluorouracil (5‐FU) and fluoropyrimidine (S‐1) were recommended in patients with stage II or stage III disease, or who had highly potential of recurrence based on the pathological findings.
Table 1.
Clinicopathological characteristics of gastric cancer patients between MET4 staining positive and negative
| Characteristic | Total | MET4 | P | |
|---|---|---|---|---|
| Positive | Negative | |||
| Number of patients | 201 | 138 | 63 | – |
| Sex | ||||
| Male | 149 | 103 | 46 | 0.808 |
| Female | 52 | 35 | 17 | |
| Age at diagnosis | 65 (36–89) | 66.0 ± 11.2 | 61.9 ± 9.5 | 0.012 |
| Maximum tumor diameter (mm) | 66.9 ± 39.6 | 68.6 ± 40.0 | 63.1 ± 38.6 | 0.360 |
| Tumor lesion | ||||
| Upper | 51 | 37 | 14 | 0.224 |
| Middle | 75 | 46 | 29 | |
| Lower | 75 | 55 | 20 | |
| Clinical stage | ||||
| I | 24 | 12 | 12 | 0.202 |
| II | 56 | 41 | 15 | |
| III | 72 | 50 | 22 | |
| IV | 49 | 35 | 14 | |
| Depth of invasion | ||||
| T1 (M, SM) | 16 | 9 | 7 | 0.318 |
| T2 (MP) | 36 | 25 | 11 | |
| T3 (SS) | 59 | 37 | 22 | |
| T4 (SE, SI) | 90 | 67 | 23 | |
| Lymph node metastasis | ||||
| N0 | 34 | 19 | 15 | 0.180 |
| N1 (1–2) | 45 | 33 | 12 | |
| N2 (3–6) | 48 | 32 | 16 | |
| N3 (≥7) | 73 | 54 | 19 | |
| NX | 1 | 0 | 1 | |
| Histology of peritoneal lavage | ||||
| CYX/0 | 172 | 114 | 58 | 0.077 |
| CY1 | 29 | 24 | 5 | |
| Peritoneal dissemination | ||||
| P0 | 169 | 113 | 56 | 0.208 |
| P1 | 32 | 25 | 7 | |
| Residual tumor | ||||
| R0 | 150 | 101 | 49 | 0.488 |
| R1 | 51 | 37 | 14 | |
| Postoperative chemotherapy | ||||
| Yes | 20 | 12 | 8 | 0.379 |
| No | 181 | 126 | 55 | |
| Histological type | ||||
| Differentiated type | 83 | 69 | 14 | 0.0002 |
| Undifferentiated type | 118 | 69 | 49 | |
| Stromal type | ||||
| med | 10 | 8 | 2 | 0.302 |
| int | 130 | 93 | 37 | |
| sci | 60 | 36 | 24 | |
| Unknown | 1 | 1 | 0 | |
| INF | ||||
| a | 9 | 6 | 3 | 0.012 |
| b | 98 | 78 | 20 | |
| c | 92 | 53 | 39 | |
| Unknown | 2 | 1 | 1 | |
| Lymphatic invasion | ||||
| ly0 | 4 | 2 | 2 | 0.019 |
| ly1 | 78 | 45 | 33 | |
| ly2 | 77 | 62 | 15 | |
| ly3 | 42 | 29 | 13 | |
| Venous invasion | ||||
| v0 | 37 | 16 | 21 | 0.0003 |
| v1 | 91 | 61 | 30 | |
| v2 | 57 | 47 | 10 | |
| v3 | 16 | 14 | 2 | |
P values less than 0.05 are underlined.
Follow‐up of the patients
Overall survival time was measured from the date of resection to the date of death due to any cause. Patients who survived until the last follow‐up were censored in our survival analyses. All patients were observed at our hospital or the outpatient clinic at 3‐ to 4‐month intervals during the first 2 years of the study and every 6 or 12 months thereafter for 3 years. After 5 years, annual follow‐up was conducted through telephone conversations with the patient, patient's family, or their practitioner. The prognosis was followed up to 10 years.
Immunohistochemical staining
To determine MET expression status by immunohistochemistry (IHC), monoclonal antibody (mAb) for MET, MET4 mAb,26 was used. Epitope sequence of MET4 revealed that DVLPEFR on amino acid residue 236–242 was the specific recognition site of MET. To retrieve MET antigen, tissue slide sections were treated at 98°C for 20 min in Target Retrieval Solution, pH9 (DAKO, Santa Clara, CA). ENVISION+ System (DAKO) was used for the secondary antibody reaction and DAB+ was used for color development. In the final step, the nuclei were lightly counterstained with Meyer hematoxylin.
Regarding the tissue staining of HGF/SF, anti‐HGF/SF mAb (clone 7‐2) was purchased from Enzo Life Sciences (Ann Arbor, MI, USA) and the samples were stained according to the company's instruction. Antigen retrieval was performed by boiling the sample slides in the 10 mM citrate buffer (pH 6) in a microwave for 7–10 min.
For the evaluation of Her2/neu IHC status HercepTest II (DAKO) was used. For the evaluation of tumor cell proliferation, we performed Ki‐67 staining using anti‐human Ki‐67 antibody (M7240, DAKO). E‐cadherin expression in the proliferative margin of tumor cells was also investigated using E‐cadherin antibody (H‐108, Santa Cruz Biotechnology, CA, USA).
Helicobacter pylori infection status
To determine the presence of Helicobacter pylori (H. pylori) infection in gastric cancers and surrounding tissues, biopsy specimens before surgery as well as all surgical specimens were subjected to IHC staining with rabbit polyclonal anti‐H. pylori antibody (DAKO). Also all medical records for clinical test for the presence of H. pylori infection were checked. After searching and testing of these items, patient's reassurance without the evidence of H. pylori infection were defined as H. pylori infection‐negative.
Statistical analyses
Comparison between patient characteristics and IHC status was performed by a χ2 test. Survival time was measured from the date of surgical operation to the date of death, or the last follow‐up. Survival outcomes were estimated according to the Kaplan–Meier product limit method and compared between groups by the log‐rank statistic. Cox proportional hazard model was used to determine the association of gastric cancer subtype with the risk of recurrence after adjustment for other significant patient and disease characteristics. All terms that were significantly associated with relapse‐free survival (P < 0.05) were considered and included in a multivariable model. Final model was based on either statistical or clinical significance. All analyses were performed using a StatView version 5.0 (SAS Institute, Inc., Cary, NC, USA).
Cell lines and H. pylori
Four MET‐expressing gastric cancer cell lines, MKN‐7, MKN‐74, NUGC‐3, and KATOIII were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS, HyClone, South Loagan, UT). CagA‐positive Helicobacter pylori (ATCC43504) was obtained from American Type Culture Collection.
Western blot, cell cycle and cell survival analysis
Western blot was performed using primary antibodies as follows: anti‐Met (C‐28, Santa Cruz Biotechnology), anti‐phospho‐Met (Tyr1234/1235, rabbit polyclonal antibodies, Cell Signaling Technology, Danvers, MA), anti‐phospho‐Akt (Ser473, 587F11, Cell Signaling Technology), anti‐phospho‐p44/42 MAPK (Thr202/Tyr204, rabbit polyclonal antibodies, Cell Signaling Technology), and anti‐β actin (AC‐15, Abcam, Cambridge, MA). For a secondary antibody HRP‐conjugated polyclonal anti‐rabbit IgG (Santa Cruz Biotechnology) was used. ECL Plus Western Blotting Detection Reagents (Amersham, GE Healthcare, Pittsburgh, PA) was used for color development.
DNA cell cycle analysis was performed using a detergent‐trypsin method and standard flow cytometry with a FACSCalibur cytometer (Becton Dickinson, San Jose, CA) and CELLQuest software. For the calculation of S‐phase fraction, ModFitLT 2.0 software (Verity) was used.
Cell survival after treatment with 5‐FU was evaluated using modified MTT assay with Cell Counting Kit‐8 (Dojindo Molecular Technologies, Inc., Tokyo, Japan).
Results
MET staining and prognosis
Immunohistochemistry analysis for MET4 staining intensity was divided into four groups (Fig. 1a). MET4‐negative cancers tended to show better prognosis than MET4‐positive cancers. However, there was no statistical difference between the four groups (Fig. 1b) and we regrouped the gastric cancer patients into two categories: MET4 negative and MET4 positive, and reanalyzed the survival plots (Fig. 2a). With MET4 mAb, a tendency of poor prognosis was established in MET positive gastric cancers (overall survival, P = 0.02; relapse‐free survival, P = 0.06).
Figure 1.
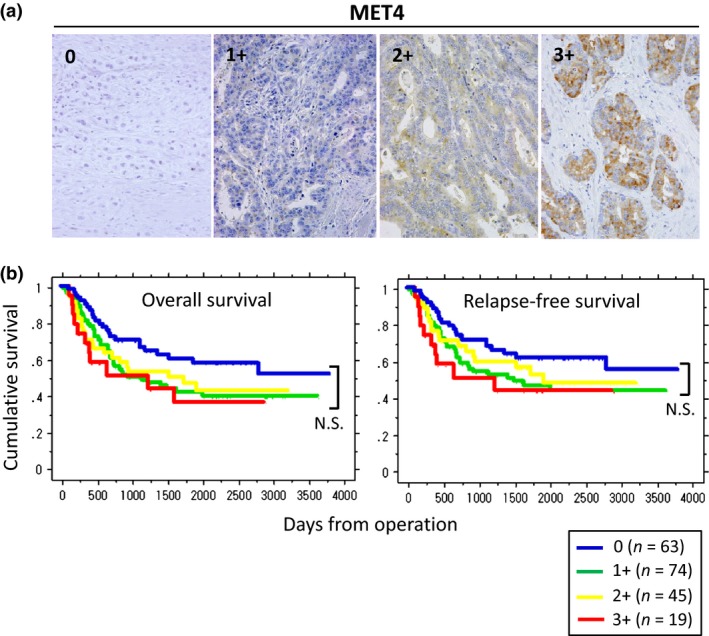
Immunohistochemical (IHC) staining of MET protein in gastric carcinomas and Kaplan–Meier survival plots by MET staining intensity. (a) IHC staining of MET with MET4 mAb. Met staining intensity is divided into four groups, 0 (negative/trace level of MET staining), 1+ (weak staining = some cells are MET positive but stained weakly), 2+ (moderate staining = many cells (more than 10%) are MET positive and stained with moderate intensity), and 3+ (strong staining = most cells are MET positive and stained with strong intensity) (original magnification, ×400). (b) Kaplan–Meier survival plots for overall survival and relapse‐free survival by MET staining intensity. Each line represents the MET status as follows: Blue (0: MET4 negative), green (1+: weakly positive), yellow (2+: moderately positive), and red (3+: strongly positive), respectively. No statistical difference in the prognosis among MET4 positive groups (1+, 2+, and 3+) is observed (N.S. = not significant).
Figure 2.
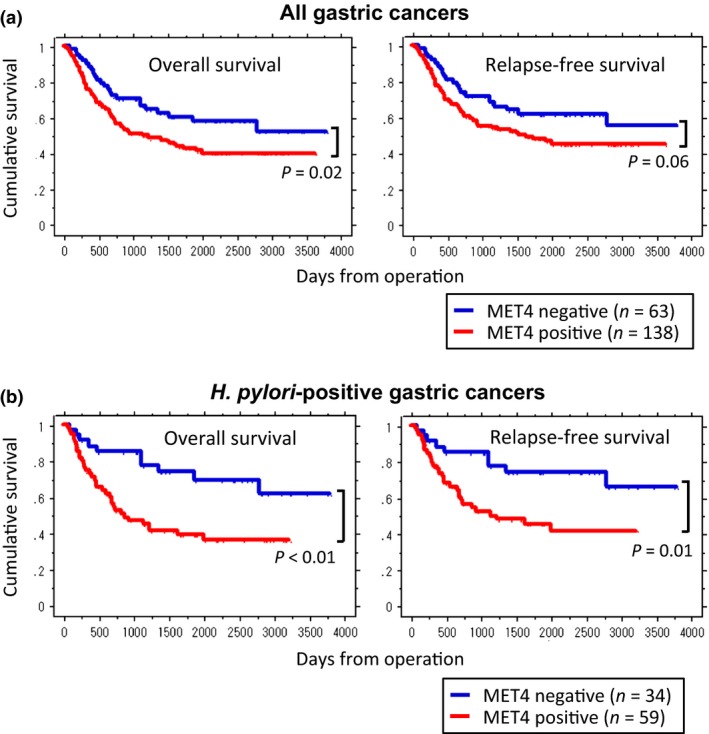
Kaplan–Meier survival plots for overall survival and relapse‐free survival by MET expression and Helicobacter pylori (H. pylori) infection status. (a) MET expression status (negative or positive) and the prognosis of all gastric cancers. To detect MET expression in gastric cancer tissues MET4 mAb was used for IHC analysis. Blue lines represent the groups of MET negative (0: MET4 negative) and red lines represent those of MET4 positive staining which includes all cases of 1+, 2+, and 3+. Immunohistochemistry (IHC) staining using MET4 mAb shows statistically significant prediction of poor overall survival (P = 0.02) and relatively poor relapse‐free survival (P = 0.06). (b) Kaplan–Meier survival by MET status in H. pylori‐positive gastric cancers. MET4 mAb shows significant prediction of poor overall survival (P < 0.01) as well as relapse‐free survival (P = 0.01).
The analysis of univariate risk factors that influence overall survival (Table S1) revealed that tumor lesion (P < 0.01), maximum tumor diameter (P < 0.01), depth of invasion (P < 0.01), lymphatic invasion (P < 0.01), vascular invasion (P < 0.01), lymph node metastasis (P < 0.01), and positive staining of MET4 (P < 0.05) were statistically significant factors. Among those factors, multivariate analysis (Table S2) indicated that tumor lesion (P < 0.05), depth of invasion (P < 0.01), lymphatic invasion (P < 0.01), and vascular invasion (P < 0.05) were recognized as independent factors for predicting poor prognosis, but positive staining of MET4 (P = 0.18) was not an independent factor contributing poor prognosis.
Clinicopathological characteristics and MET4 staining
Since MET4 mAb was reported to be a good diagnostic monoclonal antibody for human MET staining,26 we investigated the difference in the clinicopathological characteristics between MET4 positive and negative cases (Table 1). In this analysis, histopathological factors including INF (P < 0.05), lymphatic invasion (P < 0.05), and venous invasion (P < 0.01) showed positive association with MET status.
MET expression status also correlated with the percentage of Ki‐67 positive cells (Table 2). MET4 positive cases showed a higher Ki‐67 positive rate than MET4 negative cases (P < 0.01). Her2/neu overexpression was observed only in 14.4% (29 cases) and did not correlate with Ki‐67 positive rate. Most of Her2/neu‐overexpressed gastric cancers were differentiated type (24 cases out of 29 cases) and unexpectedly they showed better prognosis than Her2/neu‐negative cancers (Fig. 3).
Table 2.
Met expression, Her2/neu overexpression and Ki‐67 positive rate
| Antibody used | Staining status | Ki‐67 positive rate | P |
|---|---|---|---|
| – | – |
Mean 51.2% (Median 51.9%) |
– |
| MET4 | Met positive (n = 138) | 53.5% | 0.001 |
| Met negative (n = 63) | 46.3% | ||
| HercepTest II | Her2/neu overexpression (+) (n = 29) | 54.7% | 0.161 |
| Her2/neu overexpression (−) (n = 172) | 50.6% |
P values less than 0.05 are underlined.
Figure 3.
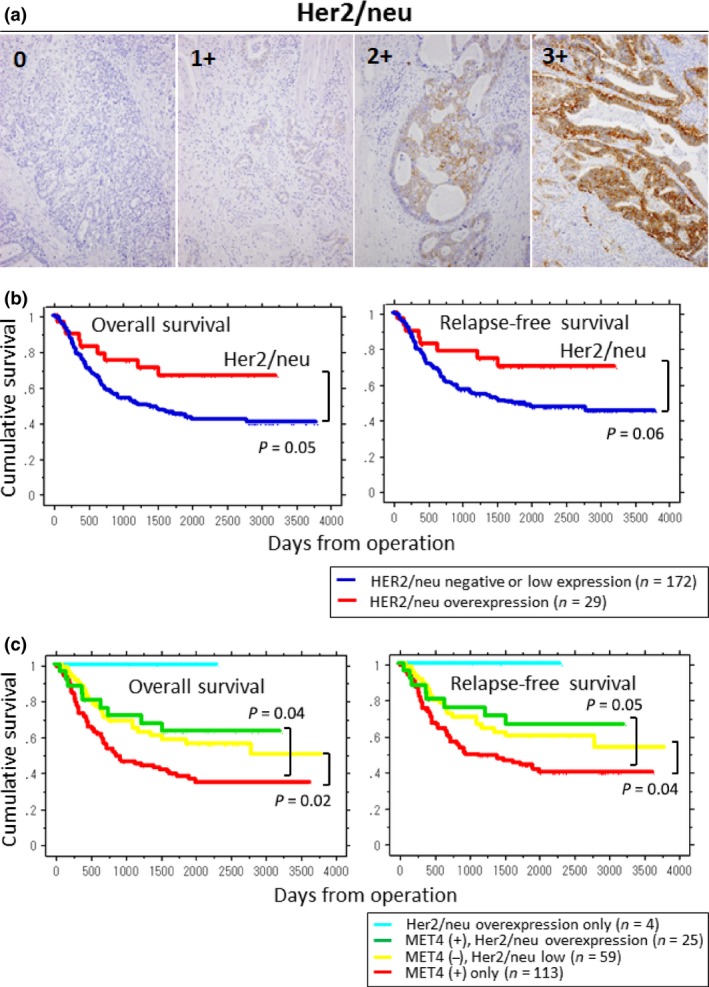
Immunohistochemistry (IHC) staining of Her2/neu and Kaplan–Meier survival plots for overall survival and relapse‐free survival by Her2/neu status. (a) For the evaluation of Her2/neu status in gastric cancers, HercepTest II (DAKO) was used for IHC analysis. Staining levels of 0 and 1+ were dealt with as negative or low expression (without Her2/neu overexpression). Staining levels of 2+ and 3+ were considered as Her2/neu overexpression. (b) Blue lines represent the groups of gastric cancers without Her2/neu overexpression and red lines represent those with Her2/neu overexpression. P‐values of overall survival and relapse‐free survival show 0.05 and 0.06, respectively. (c) Relationship among Her2/neu status, MET4 expression, and prognosis. Patients were divided into four groups according to the expression status of Her2/neu and MET4; cyan: Her2/neu overexpression only (n = 4); green: MET4 (+), Her2/neu overexpression (n = 25); yellow: MET4 (−), Her2/neu low (n = 59); and red: MET4 (+) only (n = 113). The group of MET4 (+) without Her2/neu overexpression (= MET4 (+) only) showed the poorest prognosis.
Among 201 patients tested, only 21 cases were E‐cadherin negative and undifferentiated carcinomas showed significant correlation to this property (P < 0.01). However, there was no significant difference between proliferative phenotype/vascular invasion and E‐cadherin expression. Also, there was no remarkable difference in the overall survival (P = 0.72) as well as relapse‐free survival (P = 0.99) between E‐cadherin positive and negative gastric cancers (Fig. 4).
Figure 4.
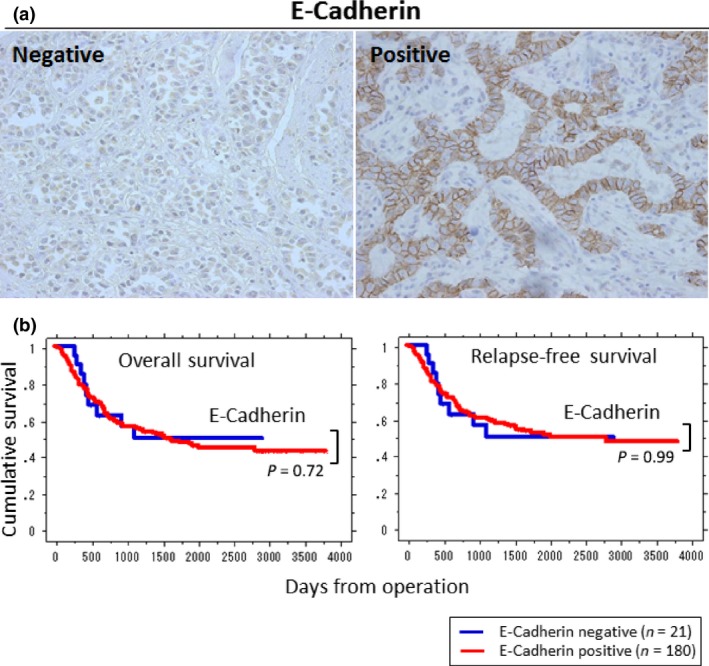
Immunohistochemistry (IHC) staining of E‐cadherin and Kaplan–Meier survival plots for overall survival and relapse‐free survival of gastric cancer patients by its status. (a) Typical IHC staining patterns of E‐cadherin (original magnification, ×400). Among 201 patients 21 were E‐cadherin negative and 180 were positive. (b) No statistical significance in the overall survival (P = 0.72) and relapse‐free survival (P = 0.99) was observed between E‐cadherin‐positive and ‐negative groups.
Helicobacter pylori infection, MET status, and prognosis
Since H. pylori infection increases the risk of gastric cancers,27 the presence of H. pylori on the gastric mucosal surface or in the gastric wall may be involved in the development of specific types of gastric cancer.28 Here, we examined the relationship between H. pylori infection and MET expression status. The presence of H. pylori infection was confirmed by the histological specimen (Fig. S1) as well as by the available clinical records. H. pylori infection did not increase the number of MET4 positive tumors (Table S3). Yet the role of H. pylori after carcinogenesis is distinct, such as the promotion of proliferation and progression of gastric cancers.24 Therefore, we investigated whether H. pylori infection affected the prognosis of gastric cancers. Interestingly, in H. pylori‐infected gastric cancers, positive MET4 staining was a statistically significant factor for predicting poor prognosis (overall survival, P < 0.01; relapse‐free survival, P = 0.01) (Fig. 2b). In contrast, in H. pylori‐negative cancers no difference was observed in the prognosis between MET‐positive and ‐negative groups (data not shown: overall survival, P = 0.88; relapse‐free survival, P = 0.95).
MET status, HGF/SF production and prognosis
HGF/SF is the only ligand for MET. Recent reports suggest that paracrine activation of MET signaling pathways by HGF/SF increases the malignancy of gastric cancer cells even without MET amplification29 and that cancer‐associated fibroblasts stimulate HGF/SF‐MET pathways and play roles as important modifiers of tumor progression.30 Therefore, HGF/SF production together with MET expression in gastric cancer tissues would be an important factor when evaluating tumor malignancy. Positive IHC staining of HGF/SF showed either intracellular pattern or diffuse pattern with extracellular staining (Fig. 5a). As was the case in the relationship between MET and H. pylori infection, no positive correlation between HGF/SF production and H. pylori infection was observed. The positive rate of H. pylori infection was 46.9% in MET4(+)/HGF/SF(+) cases, 55.8% in MET4(−)/HGF/SF(+) cases, 36.0% in MET4(+)/HGF/SF(−) cases, and 64.3% in MET4(−)/HGF/SF(−) cases, respectively. Therefore, the HGF/SF‐MET signaling in gastric cancers is considered not to affect the infectivity of H. pylori on the gastric surface mucosa. Regarding the relationship surrounding MET status, HGF/SF production, and prognosis, gastric cancers with positive staining of both MET4 and HGF/SF tended to show poorer prognosis than the groups of both negative, or MET4 or HGF/SF single positive (Fig. 5b) though statistical significance was not clearly proven.
Figure 5.
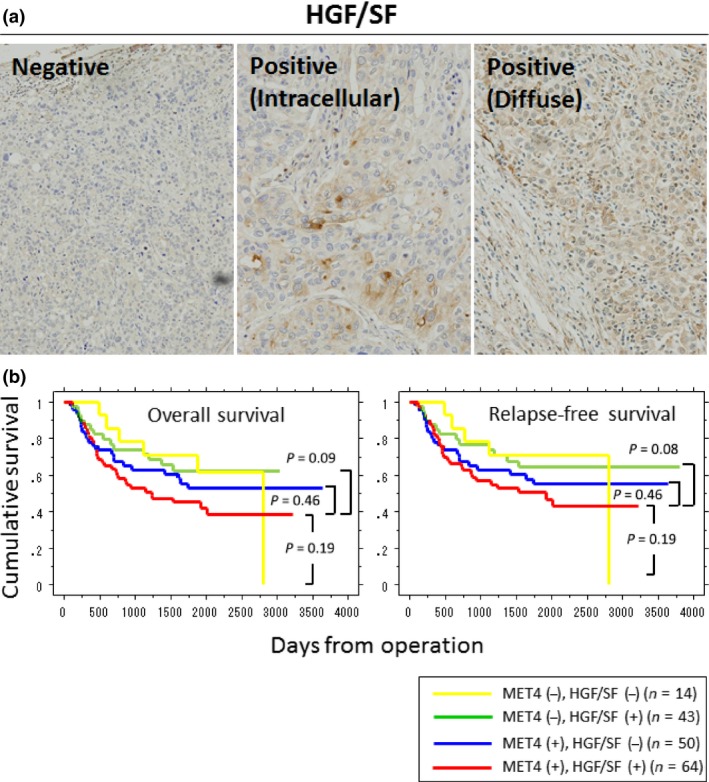
Immunohistochemistry (IHC) staining of HGF/SF and Kaplan–Meier survival plots for overall survival of gastric cancer patients by HGF/SF‐MET status. (a) Typical IHC staining patterns of HGF/SF (original magnification, ×400). HGF/SF‐positive specimens show either intracellular or diffuse staining patterns. (b) Although gastric cancers with positive staining of both MET4 and HGF/SF show a poorer prognostic tendency, no statistical significance in the overall survival as well as relapse‐free survival was observed as compared to other groups.
Effect of H. pylori stimulation on gastric cancer cell lines
Because MET expression played a significant role as a poor prognostic factor in H. pylori‐positive gastric cancers, we tested whether H. pylori bacteria might stimulate MET signaling to accelerate tumor growth. In some gastric cancer cell lines such as MKN‐45, MET activity is constitutively upregulated because of MET gene amplification (Fig. S2a). To avoid such cases we used other cell lines, MKN‐7 and MKN‐74, to see the effect of H. pylori infection both of which lack MET amplification and have wild‐type p53. Because these two gastric cancer cell lines are histologically well‐ or moderately‐differentiated, cell lines of other histological types i.e. NUGC‐3 (poorly‐differentiated adenocarcinoma) and KATO III (signet ring carcinoma) were also tested for stimulation with live H. pylori. The phosphorylation of MET after stimulation with H. pylori stimulation at multiplicity of infection (moi) = 5 (Fig. S2b) was somewhat upregulated in MKN‐7 (30 min to 240 min), MKN‐74 (15 min to 90 min), and NUGC‐3 (30–240 min) cells but not in KATO III cells. Regarding the MET downstream signaling, in MKN‐7 cells transient phosphorylation of Akt at 15–120 min and biphasic activation of p44/42MAPK were observed, suggesting the activation of this cell line by H. pylori. Also, the phosphorylation of p44/42MAPK at 15–90 min after stimulation with H. pylori was observed in MKN‐74 cells. In NUGC‐3 cells the phosphorylation of Akt at 30 to 90 min was obvious but the activation of p44/42MAPK was not as remarkable as other two cell lines. Similar to the MET phosphorylation status, obvious activation of downstream signaling in KATO III cells was not detected.
In MKN‐7 cells, dose dependency of cell signaling at 1 h after stimulation with H. pylori was tested (Fig. S3). The phosphorylation of MET downstream signals i.e. Akt and p44/42MAPK rather than MET itself was significant in response to viable H. pylori stimulation at higher moi. This may suggest that at higher moi of H. pylori cell signaling is augmented through the activation of some receptors other than MET such as toll like receptors, thereby activating intracellular cross‐talk.
To observe the effect of H. pylori stimulation on cellular activities, first DNA cell cycle status was investigated in four gastric cancer cell lines (Fig. S4a). Interestingly, the cell lines that showed marked p44/42MAPK phosphorylation, i.e. MKN‐7 and MKN‐74, displayed an increase of S‐phase fraction according to the dose of H. pylori stimulation (Fig. S4b). In addition, the survival of MKN‐7 cells in relatively high 5‐FU (100 μM) condition was enhanced in the presence of viable H. pylori (Fig. S4c), but this enhancement was not remarkable in other three cell lines (data not shown). Since strong phosphorylation of Akt in response to H. pylori was observed in MKN‐7 cells, the increased cell viability might be ascribed to an anti‐apoptotic effect of H. pylori by stimulating the Akt pathway.
Discussion
MET expression is a poor prognostic factor in some clinical cancer studies.6, 31 To see the relationship between MET expression and pathological malignancies of clinical gastric cancer cases, first we investigated clinicopathological characteristics. Although univariate analysis revealed that positive staining of MET as well as tumor size and invasive characteristics influenced the survival of gastric cancer patients, MET was not an independent risk factor proven by a multivariate analysis. Yet, positive staining of MET4 was an important factor to predict the malignancy of gastric cancers from viewpoints of their biological characteristics such as invasive nature (INF), lymphatic invasion, and venous invasion. Since there was no obvious deviation about the clinical factors between MET4‐positive and MET4‐negative groups that might affect the clinical course, MET expression in itself was considered to be a useful prognostic factor.
Most differentiated types such as pap, tub1 and tub2 expressed MET, whereas many MET negative cases were observed in poorly differentiated and signet ring cell carcinomas. Recently it has been reported that intra‐nuclear expression of C‐terminal MET fragment might be involved in the invasive/metastatic phenotype of highly malignant breast cancer cells.32 MET expression status on the cytoplasmic membrane and/or in the cytoplasm evaluated with regular MET antibodies may not necessarily correspond to the degree of tumor malignancy. Also, MET expression status is often affected by the cell cycle phase conferring the highest expression in the S‐G2/M phase and functional MET rather than overexpressed MET is important in the proliferation of tumor cells.33 Moreover, cross‐talk between MET‐mediated and EGF‐mediated signals is considered to affect tumor proliferation/invasion status.34, 35, 36 Therefore, MET expression intensity itself is not the only important factor for intracellular signaling, yet functional MET still plays a key role in the progression of gastric cancers.
Cross‐talk between MET‐mediated signaling and other intracellular pathways may affect the biological feature of cancer cells. HER2/neu, is one candidate and its importance in breast cancers as a therapeutic has received wide attention. A previous report showed that HER2/neu overexpression in gastric cancers correlated with the depth of invasion and lymph node metastasis.37 However, only 16.4% of gastric cancer cases showed HER2/neu overexpression and this marker could be applied for limited cases of MET‐positive gastric cancers. In our study, HER2/neu overexpression was observed only in 14.4% and it did not correlate with poor prognosis. Therefore, in most gastric cancers mechanisms of proliferation and invasion are considered to be independent of HER2/neu signaling.
There was no statistical correlation between MET4 staining and H. pylori infection, which suggests that these two factors are independent to each other. Interestingly, in the gastric cancer patients with obvious H. pylori infection, positive MET4 staining results indicated poorer prognosis compared with MET4 negative cases. But this prediction did not apply to the patients without H. pylori infection. In the previous studies it has been shown that H. pylori not only acts as a gastric cancer‐inducing factor but also stimulates gastric cancer cells to promote their proliferation.24, 38 Also, H. pylori bacteria are reported to provoke shedding of the surface proteins from gastric epithelial cells, and play a part in the process of epithelial mesenchymal transition.39 These data indicate that H. pylori may actively be involved in the proliferation and progression stages of gastric cancers by regulating membrane proteins such as MET. Shedding of MET may weaken the histological MET expression of some gastric cancer cells in appearance.
Since HGF/SF, the only ligand for MET, is produced in response to inflammatory processes, H. pylori infection could be considered to increase the production of HGF/SF near the infection foci.40, 41 H. pylori infection in the gastric mucosa may be one of the key carcinogenesis/proliferation factors of gastric cancers, but in our study more than half of the cases were H. pylori negative. Therefore, HGF/SF‐MET signaling is considered to be activated independently of H. pylori signals at least in those cases. Yet gastric cancers showing positive staining of both MET4 and HGF/SF revealed the tendency of the worst prognosis, suggesting the importance of HGF/SF‐MET signaling in the clinical course of gastric cancers.
Recently, Huang et al. reported that MET4 IHC staining correlated more strongly with phosphorylated MET than other anti‐MET antibodies such as SP44 and D1C2 across 18 cancer types. They concluded that MET4 IHC provides a good prediction model for MET activation and presumably a good tool to predict the response to HGF/MET inhibitors.42 Their results well support our results in this study that gastric cancers with strong MET expression evaluated by MET4 IHC staining and H. pylori stimulation have poorer prognosis than those without MET expression and the existence of H. pylori infection.
A recent paper indicated that H. pylori‐mediated cell invasion was dependent on the activation of the MET receptor.43 It was reported that the H. pylori effector protein CagA intracellularly targeted the MET receptor and promoted cellular processes leading to a forceful motogenic response.44 Also, it has been suggested that non‐phosphorylated CagA would be likely to cause sustained Met/PI3K/Akt signaling during H. pylori infection, thereby contributing to H. pylori‐associated chronic gastric proliferative and proinflammatory responses.45 Here, to see if H. pylori can activate MET and induce biological changes in gastric cancer cells, in vitro experiments were performed using viable H. pylori. We find that though the activation of MET itself was not obvious in all cases and the pattern of MET phosphorylation was different among the cell lines, H. pylori did stimulate MET signaling. Also, H. pylori stimulated downstream molecules of MET signaling such as p44/42MAPK and Akt presumably through other receptor pathways or cross‐talk, and conferred cell cycle activation and escape from apoptosis on gastric cancer cells. The results may explain why MET‐positive gastric cancers have poor prognosis only in H. pylori infection‐positive cases. Our previous papers support the idea that targeting on MET signaling may be an effective way to control tumor growth/progression.6, 46
Collectively, poor prognosis of gastric cancer patients with positive staining with MET4 is proven to be associated with H. pylori infection‐positive cases. This suggests the importance of removing these bacteria from gastric epithelial surfaces. Also, the results in this study should be taken into account when molecular targeting therapy against MET is under consideration.
Disclosure Statement
All authors certify that they have no commercial associations that might pose a conflict of interest in connection with submitted article.
Supporting information
Fig. S1. IHC staining of H. pylori in the specimens of gastric cancer patients. (a) Typical IHC staining pattern of H. pylori in a surgical specimen. Arrows indicate H. pylori bacteria on the gastric epithelial surface (original magnification, ×400). (b) Growth of H. pylori in the gastric crypts (biopsy specimen, original magnification, ×400).
Fig. S2. Effect of H. pylori stimulation on intracellular signaling of gastric cancer cell lines. (a) Western blot analysis of cultivated gastric cancer cell lines shows significant MET amplification in MKN‐45 cells but not in other gastric cancer cell lines, yet all cell lines show MET positive. (b) Time course of intracellular signaling in gastric cancer cell lines after stimulation with viable H. pylori at multiplicity of infection (moi) = 5. Cell lysates (30 μg/lane) were processed for Western blot analysis and the phosphorylation of MET, Akt and p44/42 MAPK was tested. The phosphorylation of MET after stimulation with H. pylori was observed in MNK‐7, MKN‐74, and NUGC‐3 cells but distinct activation of MET in KATO III cells was not obvious. The phosphorylation of Akt in MKN‐7 and NUGC‐3 cells as well as the phosphorylation of p44/42 MAPK in MKN‐7 and MKN‐74 cells were observed.
Fig. S3. Stimulation of MKN‐7 cells with viable H. pylori for 1 h at various bacteria‐cancer cell ratios. Cell lysates (30 μg/lane) were processed for Western blot analysis and the phosphorylation of MET, Akt and p44/42 MAPK was tested. The phosphorylation of Akt and p44/42 MAPK rather than MET was obvious at higher moi.
Fig. S4. Effect of H. pylori stimulation on cell cycle and survival of gastric cancer cell lines. (a) DNA cell cycle analysis of four gastric cancer cell lines (MKN‐7, MKN‐74, NUGC‐3, and KATO III). Cells were stimulated with viable H. pylori at moi 0 to 20 and cell cycle status was analyzed 24 h later. (b) Dose‐dependent increase of S‐phase fraction was observed in MKN‐7 and MKN‐74 cells. *P < 0.05. (c) Modified MTT assay was employed to investigate high dose 5‐FU‐induced cell death of MKN‐7 cells and its prevention by the treatment with H. pylori. Representative data are shown from the similar results of repeated experiments at least three times.
Table S1. Univariate analysis of risk factors that influence overall survival.
Table S2. Multivariate analysis of risk factors that influence overall survival.
Table S3. Helicobacter pylori infection and MET4 staining status.
Acknowledgments
The authors thank Fumiko Nakagawa (Department of Integrative Physiology and Bio‐Nano Medicine, National Defense Medical College) and Yukiko Senoo (Department of Integrative Physiology and Bio‐Nano Medicine, National Defense Medical College) for their assistance in cell culture and bioassays, and Mayumi Watanabe (Department of Anatomy, National Defense Medical College) for her assistance in immunohistochemical staining.
Cancer Sci 108 (2017) 322–330
Funding Information
Special Research Grant for National Defense Medical College, The generosity of the Jay & Betty Van Andel Foundation, Grant‐in‐Aid for Scientific Research, Japan Society for the Promotion of Science, (Grant/Award Number: ‘#23300365’ and ‘#15H04315‘)
References
- 1. Bottaro DP, Rubin JS, Faletto DL et al Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science 1991; 251: 802–4. [DOI] [PubMed] [Google Scholar]
- 2. Birchmeier C, Birchmeier W, Gherardi E et al Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4: 915–25. [DOI] [PubMed] [Google Scholar]
- 3. Thiery JP. Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 4. Royal I, Lamarche‐Vane N, Lamorte L et al Activation of cdc42, rac, PAK, and rho‐kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Mol Biol Cell 2000; 11: 1709–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shankar J, Messenberg A, Chan J et al Pseudopodial actin dynamics control epithelial‐mesenchymal transition in metastatic cancer cells. Cancer Res 2010; 70: 3780–90. [DOI] [PubMed] [Google Scholar]
- 6. Sawada K, Radjabi AR, Shinomiya N et al c‐Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res 2007; 67: 1670–9. [DOI] [PubMed] [Google Scholar]
- 7. Tomioka D, Maehara N, Kuba K et al Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res 2001; 61: 7518–24. [PubMed] [Google Scholar]
- 8. Zhang YW, Su Y, Volpert OV et al Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci USA 2003; 100: 12718–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Camp RL, Rimm EB, Rimm DL. Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer 1999; 86: 2259–65. [DOI] [PubMed] [Google Scholar]
- 10. Kammula US, Kuntz EJ, Francone TD et al Molecular co‐expression of the c‐Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett 2007; 248: 219–28. [DOI] [PubMed] [Google Scholar]
- 11. Kaposi‐Novak P, Lee JS, Gomez‐Quiroz L et al Met‐regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006; 116: 1582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kong DS, Song SY, Kim DH et al Prognostic significance of c‐Met expression in glioblastomas. Cancer 2009; 115(1): 140–8. [DOI] [PubMed] [Google Scholar]
- 13. Tang Z, Zhao M, Ji J et al Overexpression of gastrin and c‐met protein involved in human gastric carcinomas and intestinal metaplasia. Oncol Rep 2004; 11: 333–9. [PubMed] [Google Scholar]
- 14. Amemiya H, Kono K, Mori Y et al High frequency of c‐Met expression in gastric cancers producing alpha‐ fetoprotein. Oncology 2000; 59: 145–51. [DOI] [PubMed] [Google Scholar]
- 15. Inoue T, Kataoka H, Goto K et al Activation of c‐Met (hepatocyte growth factor receptor) in human gastric cancer tissue. Cancer Sci 2004; 95: 803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okusa Y, Ichikura T, Mochizuki H et al Urokinase type plasminogen activator and its receptor regulate the invasive potential of gastric cancer cell lines. Int J Oncol 2000; 17: 1001–5. [DOI] [PubMed] [Google Scholar]
- 17. Taniguchi K, Yonemura Y, Nojima N et al The relation between the growth patterns of gastric carcinoma and the expression of hepatocyte growth factor receptor (c‐met), autocrine motility factor receptor, and urokinase‐type plasminogen activator receptor. Cancer 1998; 82: 2112–22. [PubMed] [Google Scholar]
- 18. Amemiya H, Kono K, Itakura J et al c‐Met expression in gastric cancer with liver metastasis. Oncology 2002; 63: 286–96. [DOI] [PubMed] [Google Scholar]
- 19. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995; 19(Suppl. 1): S37–43. [PubMed] [Google Scholar]
- 20. Wroblewski LE, Peek RM Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 2010; 23: 713–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohata H, Kitauchi S, Yoshimura N et al Progression of chronic atrophic gastritis associated with Helicobacter pylori infection increases risk of gastric cancer. Int J Cancer 2004; 109(1): 138–43. [DOI] [PubMed] [Google Scholar]
- 22. Shimizu N, Ikehara Y, Inada K et al Eradication diminishes enhancing effects of Helicobacter pylori infection on glandular stomach carcinogenesis in Mongolian gerbils . Cancer Res 2000; 60: 1512–4. [PubMed] [Google Scholar]
- 23. Lan J, Xiong YY, Lin YX et al Helicobacter pylori infection generated gastric cancer through p53‐Rb tumor‐suppressor system mutation and telomerase reactivation. World J Gastroenterol 2003; 9(1): 54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chochi K, Ichikura T, Kinoshita M et al Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide‐toll‐like receptor 4 pathway whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin Cancer Res 2008; 14: 2909–17. [DOI] [PubMed] [Google Scholar]
- 25. Segal ED, Cha J, Lo J et al Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori . Proc Natl Acad Sci USA 1999; 96: 14559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Knudsen BS, Zhao P, Resau J et al A novel multipurpose monoclonal antibody for evaluating human c‐Met expression in preclinical and clinical settings. Appl Immunohistochem Mol Morphol 2009; 17(1): 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Konturek PC, Konturek SJ, Brzozowski T. Helicobacter pylori infection in gastric cancerogenesis. J Physiol Pharmacol 2009; 60: 3–21. [PubMed] [Google Scholar]
- 28. Kudo Y, Morohashi S, Takasugi K et al Histopathological phenotypes of early gastric cancer and its background mucosa. Biomed Res 2011; 32: 127–34. [DOI] [PubMed] [Google Scholar]
- 29. Zhao L, Yasumoto K, Kawashima A et al Paracrine activation of MET promotes peritoneal carcinomatosis in scirrhous gastric cancer. Cancer Sci 2013; 104: 1640–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu X, Chen X, Zhou Q et al Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer Lett 2013; 335(1): 128–35. [DOI] [PubMed] [Google Scholar]
- 31. Tsarfaty I, Alvord WG, Resau JH et al Alteration of Met protooncogene product expression and prognosis in breast carcinomas. Anal Quant Cytol Histol 1999; 21: 397–408. [PubMed] [Google Scholar]
- 32. Matteucci E, Bendinelli P, Desiderio MA. Nuclear localization of active HGF receptor Met in aggressive MDA‐MB231 breast carcinoma cells. Carcinogenesis 2009; 30: 937–45. [DOI] [PubMed] [Google Scholar]
- 33. Liu SI, Lui WY, Mok KT et al Effect of hepatocyte growth factor on cell cycle and c‐met expression in human gastric cancer cells. Anticancer Res 1997; 17: 3575–80. [PubMed] [Google Scholar]
- 34. Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Targeting MET as a strategy to overcome crosstalk‐related resistance to EGFR inhibitors. Lancet Oncol 2009; 10: 709–17. [DOI] [PubMed] [Google Scholar]
- 35. Puri N, Salgia R. Synergism of EGFR and c‐Met pathways, cross‐talk and inhibition, in non‐small cell lung cancer. J Carcinog 2008; 7: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reznik TE, Sang Y, Ma Y et al Transcription‐dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol Cancer Res 2008; 6(1): 139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mizutani T, Onda M, Tokunaga A et al Relationship of C‐erbB‐2 protein expression and gene amplification to invasion and metastasis in human gastric cancer. Cancer 1993; 72: 2083–8. [DOI] [PubMed] [Google Scholar]
- 38. Janas B, Orkisz S, Bartel H et al Proliferative activity of gastric epithelial cells in Helicobacter pylori infected children. Folia Histochem Cytobiol 2000; 38: 91–6. [PubMed] [Google Scholar]
- 39. Schirrmeister W, Gnad T, Wex T et al Ectodomain shedding of E‐cadherin and c‐Met is induced by Helicobacter pylori infection. Exp Cell Res 2009; 315: 3500–8. [DOI] [PubMed] [Google Scholar]
- 40. Franke R, Muller M, Wundrack N et al Host‐pathogen systems biology: logical modelling of hepatocyte growth factor and Helicobacter pylori induced c‐Met signal transduction. BMC Syst Biol 2008; 2: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suzuki M, Suzuki H, Masaoka T et al Helicobacter pylori eradication treatment modulates epithelial cell proliferation and tissue content of hepatocyte growth factor in the gastric mucosa. Aliment Pharmacol Ther 2004; 20(Suppl. 1): 158–64. [DOI] [PubMed] [Google Scholar]
- 42. Huang F, Ma Z, Pollan S et al Quantitative imaging for development of companion diagnostics to drugs targeting HGF/MET. J Pathol Clin Res 2016; 2: 210–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oliveira MJ, Costa AC, Costa AM et al Helicobacter pylori induces gastric epithelial cell invasion in a c‐Met and type IV secretion system‐dependent manner. J Biol Chem 2006; 281: 34888–96. [DOI] [PubMed] [Google Scholar]
- 44. Churin Y, Al‐Ghoul L, Kepp O et al Helicobacter pylori CagA protein targets the c‐Met receptor and enhances the motogenic response. J Cell Biol 2003; 161: 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki M, Mimuro H, Kiga K et al Helicobacter pylori CagA phosphorylation‐independent function in epithelial proliferation and inflammation. Cell Host Microbe 2009; 5(1): 23–34. [DOI] [PubMed] [Google Scholar]
- 46. Shinomiya N, Gao CF, Xie Q et al RNA interference reveals that ligand‐independent met activity is required for tumor cell signaling and survival. Cancer Res 2004; 64: 7962–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. IHC staining of H. pylori in the specimens of gastric cancer patients. (a) Typical IHC staining pattern of H. pylori in a surgical specimen. Arrows indicate H. pylori bacteria on the gastric epithelial surface (original magnification, ×400). (b) Growth of H. pylori in the gastric crypts (biopsy specimen, original magnification, ×400).
Fig. S2. Effect of H. pylori stimulation on intracellular signaling of gastric cancer cell lines. (a) Western blot analysis of cultivated gastric cancer cell lines shows significant MET amplification in MKN‐45 cells but not in other gastric cancer cell lines, yet all cell lines show MET positive. (b) Time course of intracellular signaling in gastric cancer cell lines after stimulation with viable H. pylori at multiplicity of infection (moi) = 5. Cell lysates (30 μg/lane) were processed for Western blot analysis and the phosphorylation of MET, Akt and p44/42 MAPK was tested. The phosphorylation of MET after stimulation with H. pylori was observed in MNK‐7, MKN‐74, and NUGC‐3 cells but distinct activation of MET in KATO III cells was not obvious. The phosphorylation of Akt in MKN‐7 and NUGC‐3 cells as well as the phosphorylation of p44/42 MAPK in MKN‐7 and MKN‐74 cells were observed.
Fig. S3. Stimulation of MKN‐7 cells with viable H. pylori for 1 h at various bacteria‐cancer cell ratios. Cell lysates (30 μg/lane) were processed for Western blot analysis and the phosphorylation of MET, Akt and p44/42 MAPK was tested. The phosphorylation of Akt and p44/42 MAPK rather than MET was obvious at higher moi.
Fig. S4. Effect of H. pylori stimulation on cell cycle and survival of gastric cancer cell lines. (a) DNA cell cycle analysis of four gastric cancer cell lines (MKN‐7, MKN‐74, NUGC‐3, and KATO III). Cells were stimulated with viable H. pylori at moi 0 to 20 and cell cycle status was analyzed 24 h later. (b) Dose‐dependent increase of S‐phase fraction was observed in MKN‐7 and MKN‐74 cells. *P < 0.05. (c) Modified MTT assay was employed to investigate high dose 5‐FU‐induced cell death of MKN‐7 cells and its prevention by the treatment with H. pylori. Representative data are shown from the similar results of repeated experiments at least three times.
Table S1. Univariate analysis of risk factors that influence overall survival.
Table S2. Multivariate analysis of risk factors that influence overall survival.
Table S3. Helicobacter pylori infection and MET4 staining status.