

Abstract
Chronic intestinal inflammation accompanies familial adenomatous polyposis (FAP) and is a major risk factor for colorectal cancer in patients with this disease, but the cause of such inflammation is unknown. Since retinoic acid (RA) plays a critical role in maintaining immune homeostasis in the intestine, we hypothesized that altered RA metabolism contributes to inflammation and tumorigenesis in FAP. To assess this hypothesis, we analyzed RA metabolism in the intestines of patients with FAP as well as APCMin/+ mice, a model that recapitulates FAP in most respects. We also investigated the impact of intestinal RA repletion and depletion on tumorigenesis and inflammation in APCMin/+ mice. Tumors from both FAP patients and APCMin/+ mice displayed striking alterations in RA metabolism that resulted in reduced intestinal RA. APCMin/+ mice placed on a vitamin A deficient diet exhibited further reductions in intestinal RA with concomitant increases in inflammation and tumor burden. Conversely, restoration of RA by pharmacological blockade of the RA-catabolizing enzyme CYP26A1 attenuated inflammation and diminished tumor burden. To investigate the effect of RA deficiency on the gut immune system, we studied lamina propria dendritic cells (LPDCs) since these cells play a central role in promoting tolerance. APCMin/+ LPDCs preferentially induced Th17 cells, but reverted to inducing Tregs following restoration of intestinal RA in vivo or direct treatment of LPDCs with RA in vitro. These findings demonstrate the importance of intestinal RA deficiency in tumorigenesis and suggest that pharmacological repletion of RA could reduce tumorigenesis in FAP patients.
Keywords: Familial Adenomatous Polyposis, APCMin/+ mice, colorectal cancer, dendritic cells, retinoic acid
Introduction
Several lines of epidemiological evidence show that chronic intestinal inflammation drives the formation and growth of intestinal adenomas and carcinomas (1). Intestinal inflammation accompanies familial adenomatous polyposis (FAP), an inherited disorder in which numerous adenomas develop in the intestine and eventually undergo malignant transformation. In FAP, as in the majority of sporadic colorectal cancers, tumorigenesis is initiated by mutations in the tumor suppressor gene, APC (2–4), which is part of the destruction complex that phosphorylates β-catenin, targeting it for ubiquitination and subsequent proteolytic degradation (5). In cells lacking functional APC, the stabilization and nuclear accumulation of β-catenin leads to dysregulated signaling that initiates the multi-hit progression to carcinoma (2–4). Although most patients with this disorder eventually require colectomy, chronic therapy with nonsteroidal anti-inflammatory drugs can slow adenoma growth and forestall malignant transformation (6). In the APCMin/+ mouse model, which recapitulates FAP in most respects, chronic intestinal inflammation mediated by a loss of IL10-secreting Treg cells and an increase in IL17A-producing Th17 cells promotes tumor growth that is dependent upon IL17A (7–9). The genetic events involved in the progression of disease in this model, like FAP, are well delineated. Nonetheless, the molecular and cellular mechanisms that induce inflammation have not been elucidated.
The vitamin A metabolite retinoic acid (RA) plays critical roles in embryonic development, fertility, and vision (10). In addition, it is a key regulator of the immune system (11). For example, RA is required for the generation of inducible regulatory T cells (Tregs), which help to maintain immune homeostasis in sites such as the intestine (12–15). Bioactive RA is derived from vitamin A (retinol) through a multistep process that involves several enzymes including alcohol dehydrogenases (ADH), short-chain dehydrogenase reductases (RDH) and retinaldehyde dehydrogenases (RALDH) (10,16). RA is then catabolized to inactive metabolites by cytochrome p450 (CYP) family members, mainly CYP26A1.
In light of the importance of RA in regulating immunity and its potential role in modulating intestinal inflammation, we investigated the impact of this metabolite on tumorigenesis and inflammation in human FAP and APCMin/+ mice. The experiments described in this report reveal that RA metabolism is abnormal in both APCMin/+ mice and FAP, resulting in reduced RA concentrations. We find that restoration of normal intestinal RA concentrations ameliorates tumorigenesis in APCMin/+ mice. Our data show that lamina propria dendritic cells (LPDC) isolated from the small intestine, which normally contribute to the maintenance of intestinal tolerance by inducing Tregs, are sensitive to changes in RA concentration. Whereas LPDCs in healthy control mice induce the formation of Tregs, APCMin/+ LPDCs induce proinflammatory Th17 cells, but revert to a tolerogenic phenotype when RA is reconstituted, in vivo.
Materials and Methods
Mouse breeding and care
All animal experiments described herein were approved by the Stanford University Institutional Animal Care and Use Committee. Breeding pairs of APCMin/+ male and WT C57BL/6 female mice were purchased from The Jackson Laboratory and bred on-site. OT-II TCR transgenic Rag−/− mice were purchased from Taconic. All mice were housed in an American Association for the Accreditation of Laboratory Animal Care accredited animal facility, and maintained in pathogen-free conditions on standard rodent chow ad libitum unless otherwise stated.
Isolation of DCs
Mice were euthanized and small intestines were excised while removing fat and mesentery. Peyer’s patches were excised and intestines were filleted. Luminal contents and epithelial cells were removed by vigorous stirring in HBSS in petri dishes. The intestinal tissue was then cut into ~5 mm sections that were transferred into a glass scintillation vial (Fisher) containing 10% FCS, 2.5 mM EDTA, 2 mM HEPES in HBSS containing a magnetic stir bar. Vials were placed into a 37°C incubator on a 9-position magnetic stir plate at 400 rpm for 20 minutes. Tissue pieces were then filtered through a sieve to remove residual epithelial cells and tissue fragments were collected back into the vial. This wash was repeated once. Subsequently, tissue fragments were minced very finely in the scintillation vial and then digested in 200 collagen digestion units/mL of Type VIII Collagenase (Sigma-C2139) and 40 U/mL DNase I (Sigma) in RPMI containing 10% FCS with antibiotics and antifungals again in the 37° C incubator with stirring. Tissue fragments were digested twice and after each time liberated cells were filtered through 70 μm strainers to obtain single cell suspensions that were stored on ice. The remaining pieces of intestine were placed in RPMI containing 10% FCS in the scintillation vial and shaken vigorously to free remaining cells. Spleen and mesenteric lymph nodes were digested with collagenase Type IV (Worthington). For purification of DCs, cells were magnetically enriched using CD11c+ selection kits (Stem Cell Technologies), stained with mAbs CD45.2-PE Cy7 (Clone 104, Biolegend), CD49b-FITC (Clone DX5, BD Biosciences), CD3e-FITC (Clone 17A2, Biolegend), CD19-FITC (Clone eBio1D3, eBiosciences), CD11b-Pacific Blue (Clone M1/70, Biolegend), CD11c-APC Cy7 (Clone N418, Biolegend), major histocompatibility complex (MHC) II-PE (Clone M5/114.15.2, eBiosciences), and EpCAM and propidium iodide (PI), and then sorted using a FACS Aria (BD Biosciences). In some cases, DCs were additionally sorted using anti-CD103 mAb (Clone 2E7, Biolegend).
Flow cytometric analysis
Isolated LP cells from the small intestine and mesenteric lymph node (MLN) cells were resuspended in 1% BSA in PBS (FACS buffer). After Fc blockade with anti-FcγRIII/II (BD Biosciences), cells were stained with Live/Dead Blue (Invitrogen) or PI, and mAbs against Thy1.2 and CD4 (Biolegend). Intracellular Foxp3 and cytokines were stained using mAbs against Foxp3, IL10, IL17A (eBioscience) per manufacturer’s instructions. IL17 intracellular cytokine staining in freshly isolated LP was performed after stimulating LP cells for 5 hours in PMA, ionomycin and brefeldin A. Flow cytometric analysis was performed on a LSRII flow cytometer (BD Biosciences).
Immunoblotting
Tissue lysates were prepared using RIPA lysis buffer. The following primary antibodies were used for immunoblotting: anti-ALDH1A1 (Protein tech, 15910-1-AP), anti-ALDH1A2 (Abcam, ab75674), and anti-β-actin (Cell Signaling, 8457S). Primary and secondary HRP-labeled antibodies were used at 1:500 and 1:2000 dilutions respectively. Detection was performed with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific).
T-cell differentiation assay
2×104 sorted CD11chi MHCII+ DCs were cocultured with 1×105 MACS-enriched CD4+CD62L+Foxp3− naïve T cells from the spleen and lymph nodes of OT-II TCR-transgenic mice, along with OVA323-339 peptide (New England Peptide) and 1ng/ml recombinant human transforming growth factor (TGF)-β1 (Peprotech). Unless otherwise stated, a 200 nM – 1μM peptide range was used for the standard Treg induction assay, while a 200 μM peptide concentration was used for Th17 induction. After 96 hr, cells were harvested and analyzed for intracellular Foxp3 or intracellular cytokines. For the latter, cells were restimulated at the 90 hr coculture time point with plate-bound anti-CD3 (145-2C11) and anti-CD28 (37.51) (Biolegend), with or without brefeldin A (BFA) for 6 hr. In experiments where BFA was not added, culture supernatants were assayed for the indicated cytokines using kits from eBioscience according to the manufacturer’s instructions. Where indicated, 10 nM all-trans RA (Sigma) or 1μM LE540 (Wako Chemicals) was added to culture wells. In some experiments, DCs were incubated with 10 nM RA for 18 hr, washed extensively and cocultured with freshly isolated OT-II T cells.
Quantitation of gene expression using real-time PCR
Total RNA from purified IECs was extracted using RNeasy kits (Qiagen), and DNase-treated total RNA was reverse-transcribed using High-Capacity Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. Expression of RALDH1A1, RALDH1A2, CYP26A1 and CTBP1 was determined by quantitative PCR with Power SYBR Green PCR Master Mix (Applied Biosystems) per manufacturer’s instructions using a 7900HT real-time PCR instrument (Applied Biosystems). Ubiquitin levels were measured in a separate reaction and used to normalize the data.
Quantitation of tissue retinoids
Retinyl esters (RE), all-trans retinol (ROL), and all-trans RA were extracted from the duodenum, jejunum, and ileum as described (17,18). RA was quantified by LC/MS with atmospheric pressure chemical ionization. RE and ROL were quantified by HPLC (19). Tissues were harvested and retinoids handled under yellow light using only glass laboratory equipment. Results were normalized to per gram tissue weight.
Histology
Histological studies utilized immunofluorescence staining. 11 normal and 8 FAP patient samples were analyzed. For human stains, primary antibodies, all from PTG, were β-catenin (51067-2-AP), CTBP1 (Human Protein Atlas (HPA) HPA018987), RALDH1A1 (15910-1-AP), RALDH1A2 (HPA010022), CYP26A1 (HPA C6498). Chicken secondary antibodies include anti-rabbit Alexa647 (Invitrogen). The same primary antibodies were used for mouse stains, except for RALDH1A2 (Abcam ab96060). Secondary antibodies include DyLight™ 649 goat anti-Armenian hamster (Biolegend 405505) and goat anti-rabbit Alexa549 (Invitrogen). Images were collected using a Leica DM2500 confocal laser scanning microscope, and analyzed using LAS AF software.
Drug treatment
40 ppm liarozole (Tocris Pharmaceuticals) or 8 ppm talarozole (custom-synthesized by Acme Biosciences) was incorporated into a base diet (4 IU/g of vitamin A) by Research Diets. A vitamin A deficient (0 IU/g) diet (VAD) was also utilized. Hematocrits were measured every 2 weeks, while weights were measured every week. At the point of euthanasia, adenomas were enumerated in a blinded fashion using a stereomicroscope at 10X magnification, from the gastro-duodenal junction to the ceco-colic junction.
Statistics
The Mann-Whitney U test was performed in Prism (GraphPad) to analyze all experimental data unless otherwise stated. P < 0.05 = *; P < 0.01 = **; P < 0.001 = ***.
Results
Altered RA metabolism contributes to an RA deficit in intestinal adenomas
To assess RA metabolism in patients with FAP and in APCMin/+ mice, we analyzed the expression of key enzymes regulating RA synthesis and catabolism in intestinal tissue (Fig. 1A and B). In these tissue sections, tumors are distinguished by constitutive β-catenin expression (Supplementary Fig. S1A). Overall, samples from a total of 11 normal colon and 8 FAP adenoma patients were analyzed. The results revealed reduced expression of RALDH1A1 and RALDH1A2 in adenomatous epithelia relative to healthy tissue in both human FAP and APCMin/+ adenomas (Fig. 1A and Fig. 1B). In agreement with the observation that CYP26A1 mRNA is upregulated in APCMin/+ adenomas (20), CYP26A1 protein was upregulated in FAP adenomas compared to normal colon (Fig. 1A). Elevated expression of CTBP1 has been reported to inactivate RDH (21) and as shown in Fig. 1A, CTBP1 was upregulated in FAP and APCMin/+ adenomas. Quantification of fluorescence intensity confirmed a clear upregulation of CYP26A1 and downregulation of RALDH1A1 in diseased crypts of FAP patient samples compared to healthy tissue (Supplementary Fig. S1B). These results were validated in mice by measurement of mRNA isolated from sorted WT and APCMin/+ intestinal epithelial cells (Fig. 1B).
Figure 1. Intestinal adenomas in FAP patients and APCMin/+ mice exhibit similar defects in RA metabolism, contributing to a local RA deficit.

WT □; APCMin/+ ■.
(A) Immunofluorescence stains of RALDH1A1, RALDH1A2, CTBP1 and CYP26A1 (red) in human tissue sections with adenomas demarcated from surrounding healthy tissue by a dashed line. Shown are samples from a representative normal colon and a representative FAP adenoma out of a total of 11 normal and 8 FAP patient samples analyzed. Images for the FAP column show serial sections of the same adenoma with adjacent normal grossly uninvolved tissue. (B) The left panel shows immunofluorescence stains of RALDH1A1, RALDH1A2 and CTBP1 in a representative APCMin/+ mouse small intestine at late-stage disease. Dashed lines outline adenoma (tumor) tissue surrounded by uninvolved tissue. The magnification bar shown is 100 μm. All the images in (A) and (B) have the same magnification and were captured using the same exposure time and fluorescence settings for each protein of interest. The right panel shows the expression of RALDH1A1, RALDH1A2, and CTBP1 in FACS purified 18-week old WT and APCMin/+ intestinal epithelial cells (IECs) normalized to ubiquitin b, as assayed by qPCR on total RNA. Data are representative of 3 independent qPCR experiments, with IECs pooled from 3 sorts per time point using at least 5 WT and at least 3 APCMin/+ mice per sort. (C) Immunoblots for RALDH1A1 and RALDH1A2 using lysates of tumor and healthy tissue surrounding the tumors from 4 18-week-old APCMin/+ mice. (D) Mean concentrations of RA (with SEM) per gram tissue. RA was quantified by LC/MS in the duodenum, jejunum and ileum of 18-week old WT and APC Min/+ mice, with 5 mice per strain. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Retinoic acid metabolizing enzymes from tumors dissected away from surrounding tissue were analyzed by western blot to determine protein expression. In agreement with immunofluorescence observations, tumors exhibited local alterations in expression of RA metabolizing enzymes relative to surrounding tissue (Fig. 1C and Supplementary Fig. S1C). In order to determine how the size, and consequently the age, of tumors correlated with alterations in expression of RA metabolizing enzymes, tumors were dissected, pooled based on size, RNA isolated from them, and qRT-PCR performed. The smallest and most recently formed tumors exhibited the greatest elevation of CYP26A1 expression, whereas the larger tumors contained CYP26A1 RNA comparable to normal tissue (Supplementary Fig. S1D). Conversely, RALDH1A1 RNA was reduced in tumors of all sizes, whereas RALDH1A2 RNA was consistently elevated in tumors of all sizes (Supplementary Fig. S1D). Thus, CYP26A1 was elevated in small/early adenomas and RALDH1A1 was consistently reduced in tumors of all sizes.
The net effect of diminished expression of RA-synthesizing enzymes and increased expression of RA-catabolizing enzymes suggested a local RA deficit in intestinal polyposis. To test this possibility we used a highly sensitive assay based on quantitative mass spectrometry to measure RA. The results revealed reductions in RA concentrations throughout the intestine of APCMin/+ mice by comparison to WT mice (Fig. 1D). The reduction of RA was observed at early-stage disease and persisted through later stages of disease (data not shown). Retinol (ROL), and vitamin A in the storage form retinyl esters (RE) were also measured. Although retinol was reduced in the APCMin/+ ileum, tissues from WT and APCMin/+ mice had comparable amounts of retinyl esters, indicating that the RA deficit is not due to a lack of vitamin A absorption or storage (Supplementary Fig. S1E). Nevertheless, this deficit did not only occur in tumors as hepatic stores of RA were also reduced (Supplementary Fig. S1F). Taken together, these data suggest a RA deficit in FAP and APCMin/+ intestine, likely explained by a combination of reduced synthesis and excess breakdown of the molecule local to the tumor environment.
Effects of vitamin A-deficient (VAD) diet and vitamin A restoration on tumorigenesis
We utilized APCMin/+ mice to assess the impact of altering the concentration of intestinal RA on tumorigenesis. Eight-week-old APCMin/+ mice were placed on a VAD diet for 6 weeks to test the effect of a further reduction in RA. Intestinal RA measurements confirmed that VAD exacerbated RA loss, with a reduction in the ileum, the main site of tumor formation in this model (Fig. 2A). Compared to mice on the base diet, mice on a VAD diet developed more tumors in all three regions of the small intestine, contributing to an increase in overall tumor number (Fig. 2B). They also lost more weight than base diet-treated control mice and trended towards lower hematocrits as well (Fig. 2C).
Figure 2. Vitamin-A deficient diet (VAD) reduces intestinal RA and exacerbates disease, while CYP26A1 inhibition restores intestinal RA levels and ameliorates disease.

Base ■; VAD
 ; liarozole □. Groups of 8 week-old APC Min/+ mice were placed on base diet, VAD diet, base diet containing liarozole 40 ppm for 6 consecutive weeks. (A) As in Fig. 1D, RA was quantified by LC/MS, with 5 mice per dietary treatment. Mean number of tumors (B), mean change in percentage body weight and hematocrit (C) at 14 weeks are shown for APCMin/+ mice given these treatments. (D) Additional groups of 8 week-old APCMin/+ mice were fed chow containing talarozole 8ppm for 6 weeks, and intestinal tumors enumerated. For (A–D), data for VAD-fed mice are aggregated from 2 independent experiments, with 4 mice per experiment; data for liarozole-treated mice are aggregated from 4 independent experiments, with at least 4 mice per experiment. For (D), data for talarozole-treated mice are aggregated from 2 independent experiments, with 5 mice per experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
; liarozole □. Groups of 8 week-old APC Min/+ mice were placed on base diet, VAD diet, base diet containing liarozole 40 ppm for 6 consecutive weeks. (A) As in Fig. 1D, RA was quantified by LC/MS, with 5 mice per dietary treatment. Mean number of tumors (B), mean change in percentage body weight and hematocrit (C) at 14 weeks are shown for APCMin/+ mice given these treatments. (D) Additional groups of 8 week-old APCMin/+ mice were fed chow containing talarozole 8ppm for 6 weeks, and intestinal tumors enumerated. For (A–D), data for VAD-fed mice are aggregated from 2 independent experiments, with 4 mice per experiment; data for liarozole-treated mice are aggregated from 4 independent experiments, with at least 4 mice per experiment. For (D), data for talarozole-treated mice are aggregated from 2 independent experiments, with 5 mice per experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate the possibility that restoring intestinal RA might have a beneficial impact on tumorigenesis, we administered RA intraperitoneally (i.p.) twice weekly for 6 weeks to APCMin/+ mice, beginning at 8 weeks of age and using the same protocol that has been reported to attenuate ileitis in a mouse model of Crohn’s disease (22). However, this regimen did not increase RA concentrations in APCMin/+ mice (Supplementary Fig. S2A). Such direct RA administration correspondingly did not improve disease outcome in terms of tumor burden (Supplementary Fig. S2B) or other disease indicators such as body weight and hematocrit (data not shown).
Given the tight physiological control of RA levels, a potential explanation for the failure of RA i.p. to increase RA was that administered RA is rapidly catabolized by the upregulated CYP26A1 in APCMin/+ tissue (20). On this basis, we evaluated an alternative strategy to reconstitute intestinal RA by targeting the upregulated CYP26A1 with liarozole, an inhibitor of this enzyme (23). When 8-week-old APCMin/+ mice were fed chow containing liarozole at 40 ppm for 6 weeks, RA was restored in the ileum of liarozole-treated mice (Fig. 2A). Moreover, compared to base diet-treated controls, liarozole-fed APCMin/+ mice exhibited a reduction in overall tumor burden (Fig. 2B), as well as a substantial increase in body weight and a lesser decrease in hematocrit (Fig. 2C). To confirm the detrimental effects of upregulated CYP26A1 and validate our results with liarozole, we treated APCMin/+ mice with talarozole, a highly selective inhibitor of CYP26 that is structurally distinct from liarozole.(24) Consistent with the results from liarozole treatment, talarozole improved disease outcome (Fig. 2D).
APCMin/+ LPDCs preferentially induce Th17 cells
Given the importance of DCs in maintaining immune tolerance in the intestine, we hypothesized that low RA in the intestine of APCMin/+ mice might affect local DC function. At steady state, DCs in the gut consist of three phenotypically distinct populations: CD103+CD11b−, CD103+CD11b+ and CD103−CD11b+ DCs. No significant differences in the frequencies of the three main DC subsets were observed in the small intestine LP (SI-LP) of APCMin/+ mice compared to WT mice (Supplementary Fig. S2C). Further studies of SI-LPDCs from APCMin/+ mice revealed that, although their expression of costimulatory molecules was similar to that of WT SI-LPDCs (Supplementary Fig. S2D), they secreted much more pro-inflammatory cytokines such as TNFα, IL6 and IL12p40 under basal conditions and in response to a panel of Toll-like receptor agonists (Supplementary Fig. S2E). As CD103+ LPDCs are the main cells responsible for generating Tregs in the intestinal environment (12), we sorted LPDCs into CD103+ and CD103− subsets (sorting strategy shown in Supplementary Fig. S3) and assessed the role of each subset in a conventional TGFβ-dependent Treg induction assay (12,14,15). This assay utilizes a relatively low concentration of peptide as this is important for Treg differentiation (25–27). Although we attempted to sort the highest CD11c-expressing cells from the lamina propria, it is possible that the CD103− cell population had some contaminating macrophages. APCMin/+ CD103+ LPDCs only induced 25% of the Foxp3+ T cells compared to their WT counterparts (Fig. 3A). In contrast, CD103− LPDCs from WT and APCMin/+ mice induced Foxp3+ T cells equally (Fig. 3A). Both CD103− and CD103+ APCMin/+ LPDCs induced more Th17 cells compared to the WT LPDC subsets (Fig. 3A). Consistent with these results, supernatants obtained from whole APCMin/+ LPDC-T-cell cocultures contained only one-sixth of the IL10 compared to WT LPDC cocultures (Fig. 3B). Moreover, there was a concomitant and similarly dramatic increase in IL17A (Fig. 3B), an inflammatory cytokine that plays an important role in adenoma development (9). Since Th17 differentiation requires IL6 in addition to TGFβ (28) and no exogenous IL6 was added to our cultures, we measured IL6 in coculture supernatants and, as expected, found more in APCMin/+ LPDC cocultures compared to WT cocultures (Fig. 3B).
Figure 3. Exposure of APCMin/+ LPDCs to RA, in vitro, reverses their inflammatory phenotype.

(A) To assess Treg induction capacity, LPDCs, defined as PI− EpCAM− CD45+ Lin- CD11chi MHCII+ (Supplementary Fig. S3), were further sorted into CD103+ and CD103− subsets and cocultured with CD4+CD62L+Foxp3− naïve T cells from OT-II mice, along with OVA323- 339 peptide and TGFβ, and the frequency of Foxp3+ and IL17A+ T cells was determined after 4 days. Shown are the mean frequencies of Foxp3+ T cells and IL17A+ T cells induced in these cultures, representative of 3 independent experiments. (B) Whole LPDC–T-cell cocultures were stimulated with plate-bound anti-CD3 and anti-CD28 for the last 6 hr of coculture and IL10, IL17A and IL6 were assayed in the supernatants. The experiment shown is representative of 4 independent experiments. (C–E) All-trans RA or the RAR antagonist LE540 was added to whole CD11c+ LPDC-T-cell coculture wells at the initiation of coculture, and induced Foxp3+ T cells are shown in representative dot plots and bar graphs (C–D). (E) Cells were stimulated with plate-bound anti-CD3 and anti-CD28 for the last 6 hr of coculture. Bar graphs show mean production of IL17A in culture supernatants harvested at the end of coculture. For (C–E), results shown are representative of 4 independent experiments. (F–G) CD103+ and CD103− LPDCs were incubated with all-trans RA (+RA) or in media alone (control) for 18 hr, washed extensively and cocultured with naive T cells as before. Representative dot plots and bar graphs show the mean frequency of the induced IL17A+CD4+ T cells. For (F–G), results shown are representative of 3 independent experiments. For (A–G), DCs are pooled from 5 WT and 3 APCMin/+ mice per experiment, all at 18 weeks of age. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
RA reverses the inflammatory phenotype of APCMin/+ LPDCs in vitro
APCMin/+ LPDCs expressed less RALDH1A2 compared to WT LPDCs (Supplementary Fig. S4), possibly reducing their RA synthesis capacity and exacerbating their proinflammatory phenotype. Therefore, we hypothesized that the pro-inflammatory phenotype of APCMin/+ LPDCs could be modulated by RA exposure. To assess this we added RA or the RA receptor antagonist LE540 to LPDC-T-cell cocultures. The addition of RA to APCMin/+ LPDC–T-cell cocultures restored the induction of Foxp3+ CD4 T cells to WT coculture numbers (Fig. 3C and D) and also strongly inhibited the production of IL17A (Fig. 3E). In contrast, LE540 abrogated Foxp3 induction in both WT and APCMin/+ LPDC cocultures (Fig. 3C and D) and greatly enhanced IL17A production in APCMin/+ cocultures (Fig. 3E). To determine whether the normalizing effects of RA were due to direct action on DCs, CD103+ and CD103− LPDCs from WT and APCMin/+ mice were incubated with 10 nM RA for 18 hours, extensively washed, and then cocultured with OT-II T cells to assess their effects on T-cell differentiation. RA treatment completely reversed the proinflammatory phenotype of both APCMin/+ LPDC subsets, reducing their induction of Th17 cells to WT levels (Fig. 3F and G). Although induction of Foxp3+CD4+ T cells by RA-treated APCMin/+ LPDCs trended toward WT levels, this increase did not reach statistical significance (data not shown). Taken together, these results indicate that RA can act directly on APCMin/+ LPDCs to suppress their Th17-polarizing capacity.
Th17 inflammation is exacerbated by VAD diet and alleviated by RA restoration
Since altered RA concentrations in the intestine have been linked to mucosal inflammation, we sought to determine whether modulation of RA in vivo via VAD diet or blockade of RA catabolism via liarozole treatment affected Th17 frequency. Mice maintained on a VAD diet had an almost three-fold increase in Th17 cells relative to untreated control mice, whereas liarozole-treated APCMin/+ mice had a nearly 75% reduction in the number of Th17 cells (Fig. 4). These findings show that repletion of RA reverses the proinflammatory phenotype of LPDCs, attenuates Th17-driven inflammation and ameliorates disease, whereas depletion of RA exacerbates inflammation and disease.
Figure 4. Restoration of RA in vivo reverses the proinflammatory phenotype of LPDCs of APCMin/+ mice.
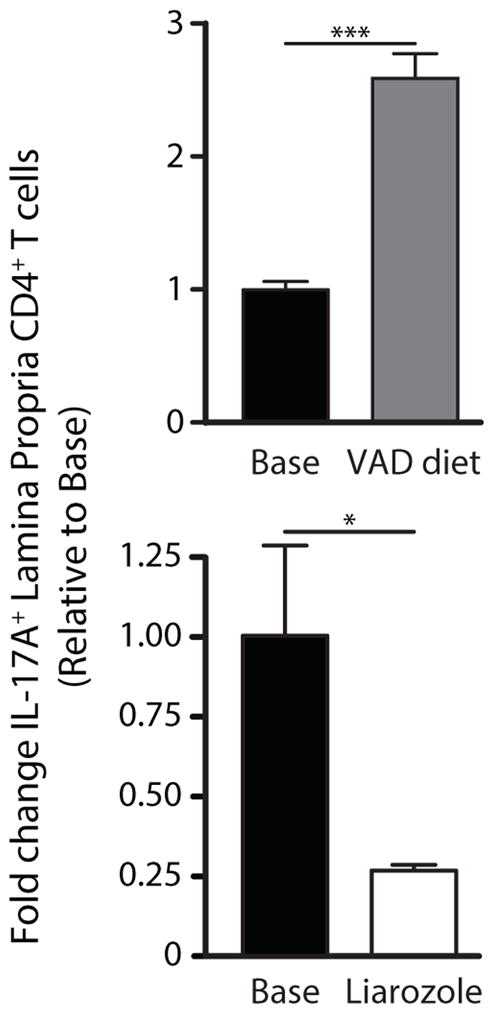
Fold change in the mean frequency of IL17A+ CD4+ T cells in LP from APCMin/+ mice treated with liarozole, VAD or base diet, as described in Fig. 2. The experiment shown is representative of 2 independent experiments, with 4 mice per diet. DCs obtained were pooled from all mice on the same diet in each experiment. *, P < 0.05; ***, P < 0.001.
Ablation of DCs reduces tumor burden in APCMin/+ mice
To directly assess the role of DCs in tumor progression, we generated bone marrow (BM) chimeras using donor cells in which diphtheria toxin A (DTA) is activated in CD11c-expressing cells (CD11c:DTA mice) (29). Chimeras generated with CD11c-DTA BM exhibited greatly reduced numbers of DCs in the SI-LP compared to chimeras with WT and APCMin/+ BM (Fig. 5A). As shown in Fig. 5B, DC depletion in APCMin/+ mice resulted in a decrease in total tumor frequency relative to mice reconstituted with APCMin/+ BM. Reconstitution with WT BM also led to a reduction in tumor number, although the reduction did not reach statistical significance. These results directly demonstrate that DCs in APCMin/+ mice exacerbate tumor progression, and are consistent with our finding that a factor in the gut microenvironment, which we believe to be RA, regulates these cells.
Figure 5. Ablation of CD11c+ DCs in APC Min/+ mice reduces adenoma formation.

WT □; CD11c-DTA
 ; APCMin/+ ■. 6 week-old APCMin/+ mice were lethally irradiated and reconstituted with WT, APCMin/+ or CD11c-DTA BM. (A) Representative dot plots and bar graphs depict DC populations in the SI-LP of CD11c-DTA BM-reconstituted APC Min/+ mice compared to those reconstituted with WT or APCMin/+ BM, using 3 mice per group. (B) Scatter plots of tumors in the small intestine enumerated at 24 weeks, as aggregated from 2 independent experiments, with at least 4 mice per group per experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
; APCMin/+ ■. 6 week-old APCMin/+ mice were lethally irradiated and reconstituted with WT, APCMin/+ or CD11c-DTA BM. (A) Representative dot plots and bar graphs depict DC populations in the SI-LP of CD11c-DTA BM-reconstituted APC Min/+ mice compared to those reconstituted with WT or APCMin/+ BM, using 3 mice per group. (B) Scatter plots of tumors in the small intestine enumerated at 24 weeks, as aggregated from 2 independent experiments, with at least 4 mice per group per experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
Our study quantitatively demonstrates RA deficiency in the intestinal tissue of a murine model of spontaneous familial intestinal polyposis. In addition, we report that restoration of intestinal RA attenuates inflammation with a concomitant reduction in polyp formation. Although RA inhibits the growth of a variety of tumor cell lines, including osteosarcoma (30), breast (31,32), thyroid (33), and even head and neck (34) cancer, such in vitro findings lack the clinical relevance of the APCMin/+ model. In an azoxymethane-induced rat model of colon cancer, 9-cis-RA reduces the multiplicity of aberrant crypt foci in the colon (35). This finding is consistent with our findings, although the model of colon cancer utilized was chemically induced, and may not recapitulate the physiological condition as well as the APCMin/+ model. Whereas these previous studies focused on the anti-proliferative and differentiating effects of RA, we highlight its potent capacity to modulate the tumor immune profile. RA repletion alone can attenuate inflammation as well as impede disease progression in the APCMin/+ model.
The APCMin/+ model differs from FAP in one major respect, the principal location of tumors (ileum vs. colon). Nonetheless, the model is considered highly relevant to FAP due to its similarities in virtually all other aspects of the disease. Moreover, anti-inflammatory drugs and other agents that reduce inflammation and tumor growth in APCMin/+ mice also reduce tumor growth in the colon of FAP patients (36). Our data show that affected colons from FAP patients express inflammatory markers and abnormalities in RA metabolism consistent with those seen in the APCMin/+ small intestine. Although we saw substantial decreases in RA concentration in all sections of the APCMin/+ small intestine, it remains to be understood why the majority of polyps develop in the ileum, with few (<3) polyps ever observed in the colon.
We found that APCMin/+ mice have an RA deficit in their intestines. The reduction was due to both diminished synthesis and excessive breakdown of the molecule. Expression of the RALDH enzymes that promote RA production was reduced in APCMin/+ intestinal epithelial cells (IECs), along with a marked accumulation of CTBP1, which normally suppresses RDH (21). Constitutive expression of β-catenin downstream of the APC mutation upregulates the major RA catabolic enzyme, CYP26A1 (20). Indeed, we observed upregulated CYP26A1 expression in APCMin/+ and FAP adenomas, consistent with previous findings in sporadic colorectal carcinomas and the intestine of APC-mutant zebrafish embryos (20). Taken together, these results point to several mechanisms that cooperate to decrease RA concentration in the tumor milieu.
The role of intestinal inflammation in the development of adenomas in the APCMin/+ model of spontaneous neoplasia is well-documented. However, the few immunological studies performed on APCMin/+ mice and related APC mutation models have focused almost exclusively on altered T-cell responses, with little attention directed at the underlying cause of these changes (7,9,37–39). Our findings suggest that proinflammatory LPDCs in these mice may play an important role in inducing and maintaining Th17 cells. This contrasts with the LPDCs of healthy mice, which do not promote inflammation but instead maintain immune tolerance through the generation of Tregs (12,14,15,40). Stimulation with various TLR ligands causes LPDCs to produce more IL6 and TNF when compared to LPDCs from wild-type mice. It is well known that efficient induction of Th17 cells requires TGFβ and IL6 (41). The addition of TGFβ to cultures simultaneously reduced IL6 release from LPDCs as well as stimulated the generation of IL17-producing Th17 cells. Given the widely reported pleiotropism of TGFβ proteins, this seemingly contradictory effect is not surprising and potentially represents a control mechanism for restricting the induction of Th17 cells. When proinflammatory LPDCs from APCMin/+ mice are cultured in the presence of RA, in vitro, or exposed to increased concentrations of RA in vivo, they revert to tolerogenic cells. Although these findings do not prove that LPDCs are the only immune cells affected by RA deficiency, they highlight their importance in promoting the intestinal inflammation that characterizes this disorder.
A subset of intestinal macrophages also expresses CD11c and MHC II (42). As such, these cells may have been included in our DC-enriched LP preparations, although their contribution to intestinal inflammation in APCMin/+ mice is unknown. Regardless, the induction of Th17 cells by LP antigen-presenting cells might explain the dependence of tumor growth on IL17 in the APCMin/+ intestine (9) and potentially represents a critical control point in shaping the inflammatory milieu that drives tumor growth.
Our studies also examined the impact of RA metabolism on intestinal DC subsets. The data indicate that both CD103+ and CD103− LPDC subsets are responsive to local fluctuations in RA concentration and adopt a proinflammatory phenotype in the presence of reduced RA. The CD103+ LPDCs isolated from APCMin/+ mice were more phenotypically altered than the CD103− LPDCs, following in vitro and in vivo modulation of RA concentrations. Although our studies indicate that RA can act directly on APCMin/+ LPDCs to reverse their inflammatory phenotype, RA may impact intestinal inflammation through additional mechanisms. One potential mechanism involves IL22, which is a potent inducer of pathological inflammation (43) that promotes intestinal tumorigenesis in APCMin/+ mice (44). The secreted receptor that inhibits IL22, IL22BP, is constitutively produced by mouse intestinal CD11b+CD103+ DCs (45). RA can elicit the expression of IL22BP in human immature monocyte-derived DCs (45), suggesting that RA may regulate intestinal tolerance in vivo via expression of IL22BP by the mouse CD11b+CD103+ DC subset. In another study, mice given a pan-RAR antagonist or subjected to radiation-induced mucosal injury to obtain an acute VAD state selectively lost the CD11b+CD103+ DC subset (46). These findings suggest that CD11b+CD103+ DCs are particularly dependent on RA for their function. Whether the effect of RA on APCMin/+ LPDCs depends on IL22 remains to be determined.
Large i.p. doses of RA did not reduce tumor burden or impact GI inflammation, which initially puzzled us, but was explained by the lack of increased RA concentration in the intestine which, in turn, is likely due to rapid catabolism of the injected RA by upregulated CYP26A1. This result is consistent with a prior report of orally administered RA not benefiting and, in fact worsening, disease in APCMin/+ mice (47). In contrast to systemically administered RA, our study showed that the CYP26A1 inhibitor, liarozole, increased intestinal RA, reduced Th17 inflammation, and ameliorated disease. Because liarozole can affect other CYP enzymes in addition to CYP26A1, we studied talarozole, a third generation retinoic acid metabolism-blocking agent. Like liarozole, talarozole inhibits CYP26A1 from hydroxylating carbon 4 of RA (23), thus preventing the catabolism of RA. However, in contrast to liarozole, talarozole has virtually no effect on other CYP-dependent activities (48) and is specific to certain isoforms of CYP26A1. Moreover, it is structurally unrelated to liarozole and generates different metabolites (23). As shown in our study, talarozole, like liarozole, ameliorated disease, confirming that tumor development induced by intestinal inflammation can be reversed by inhibiting RA breakdown.
Conversely, a VAD diet resulted in more pronounced DC-mediated inflammation and exacerbation of all disease parameters. A standard protocol for achieving vitamin A deficiency in mice is to place pregnant dams on a 0 IU/g vitamin A diet beginning day 7–10 of gestation (49). However, we observed that RA concentrations were further reduced from baseline in APCMin/+ mice fed a VAD diet for 6 weeks starting at 8 weeks of age, suggesting that the existing RA deficiency in the APCMin/+ intestine may make these animals more susceptible to the effects of VAD. In contrast, WT mice subjected to the same VAD diet and protocol did not exhibit any signs of inanition and appeared to tolerate this diet well (data not shown).
In studies of a mouse model of Crohn’s disease in which TNFα is constitutively produced, Collins and colleagues showed that RA concentrations are reduced in this model and RA supplementation attenuates ileitis (22). Like APCMin/+ mice, regulatory LPDCs were reduced in frequency in this model and recovered with RA treatment, but proinflammatory LPDCs were not found. Also, whereas oral RA proved effective in the Crohn’s model, it was ineffective in APCMin/+ mice, suggesting that the mechanism responsible for the RA deficit in these two models is distinct. Despite these differences, the combined results suggest that RA deficiency may underlie several inflammatory disorders of the intestine.
Although we did not measure RA in FAP tissues, an RA deficit likely occurs in this disease, based on the severe vitamin A metabolism defects seen in FAP intestine. Given their many shared features, it is reasonable to hypothesize that the beneficial impact of RA reconstitution on APCMin/+ disease might also be seen in FAP. Attempts to increase RA in the intestine of patients with inflammatory bowel disorders must be undertaken with caution, since RA in combination with IL15 has been reported to induce proinflammatory DCs in a mouse model of celiac disease and pharmacological retinoid treatment may be a risk factor for inflammatory bowel disease (50,51). Nonetheless, our results provide a strong rationale for studying CYP26A1 inhibitors in appropriate patients. Liarozole and talarozole have been evaluated in clinical trials for diseases unrelated to FAP or colorectal cancer and are apparently well-tolerated (52,53). These or other agents that safely increase intestinal RA have the potential to reverse inflammation and reduce tumor burden as observed here in APCMin/+ mice.
Supplementary Material
Acknowledgments
We thank D. Jones for secretarial assistance and M. Alonso and C. Benike for critical review of the manuscript.
Grant Support
These studies were supported by National Institutes of Health grants CA141468 and HL075462 (E.G.E.). T.R.P. is supported by NIH NRSA F30CA196145.
Footnotes
Disclosures: The authors have declared that no conflict of interest exists.
References
- 1.Ullman TA, Itzkowitz SH. Intestinal Inflammation and Cancer. Gastroenterology. 2011;140(6):1807–16.e1. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Nilbert MC, Vogelstein B, Bryan TM, Levy DB, Smith KJ, et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science. 1991;251(4999):1366–70. doi: 10.1126/science.1848370. [DOI] [PubMed] [Google Scholar]
- 3.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 4.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359(6392):235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 5.Abu-Abed SS, Beckett BR, Chiba H, Chithalen JV, Jones G, Metzger D, et al. Mouse P450RAI (CYP26) Expression and Retinoic Acid-inducible Retinoic Acid Metabolism in F9 Cells Are Regulated by Retinoic Acid Receptor γ and Retinoid X Receptor α. Journal of Biological Chemistry. 1998;273(4):2409–15. doi: 10.1074/jbc.273.4.2409. [DOI] [PubMed] [Google Scholar]
- 6.Lynch PM. Pharmacotherapy for inherited colorectal cancer. Expert Opin Pharmacother. 2010;11(7):1101–8. doi: 10.1517/14656561003698123. [DOI] [PubMed] [Google Scholar]
- 7.Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, et al. CD4+CD25+ Regulatory Lymphocytes Induce Regression of Intestinal Tumors in ApcMin/+ Mice. Cancer Research. 2005;65(10):3998–4004. doi: 10.1158/0008-5472.CAN-04-3104. [DOI] [PubMed] [Google Scholar]
- 8.Gounaris E, Blatner NR, Dennis K, Magnusson F, Gurish MF, Strom TB, et al. T-Regulatory Cells Shift from a Protective Anti-Inflammatory to a Cancer-Promoting Proinflammatory Phenotype in Polyposis. Cancer Res. 2009;69(13):5490–97. doi: 10.1158/0008-5472.CAN-09-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(12):5540–4. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9(7):541–53. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 11.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204(8):1765–74. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317(5835):256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 15.Sun C-M, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. The Journal of Experimental Medicine. 2007;204(8):1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. Journal of Neurobiology. 2006;66(7):606–30. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 17.Kane MA, Folias AE, Wang C, Napoli JL. Quantitative Profiling of Endogenous Retinoic Acid in Vivo and in Vitro by Tandem Mass Spectrometry. Analytical Chemistry. 2008;80(5):1702–08. doi: 10.1021/ac702030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kane MA, Folias AE, Wang C, Napoli JL. Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: a potential mechanism of ethanol toxicity. FASEB J. 2010;24(3):823–32. doi: 10.1096/fj.09-141572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kane MA, Folias AE, Napoli JL. HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Analytical Biochemistry. 2008;378(1):71–79. doi: 10.1016/j.ab.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shelton DN, Sandoval IT, Eisinger A, Chidester S, Ratnayake A, Ireland CM, et al. Up-regulation of CYP26A1 in Adenomatous Polyposis Coli-Deficient Vertebrates via a WNT-Dependent Mechanism: Implications for Intestinal Cell Differentiation and Colon Tumor Development. Cancer Res. 2006;66(15):7571–77. doi: 10.1158/0008-5472.CAN-06-1067. [DOI] [PubMed] [Google Scholar]
- 21.Nadauld LD, Phelps R, Moore BC, Eisinger A, Sandoval IT, Chidester S, et al. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J Biol Chem. 2006;281(49):37828–35. doi: 10.1074/jbc.M602119200. [DOI] [PubMed] [Google Scholar]
- 22.Parés X, Farrés J, Kedishvili N, Duester G. Medium- and short-chain dehydrogenase/reductase gene and protein families. Cellular and Molecular Life Sciences. 2008;65(24):3936–49. doi: 10.1007/s00018-008-8591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verfaille CJ, Vanhoutte FP, Blanchet-Bardon C, Van Steensel MA, Steijlen PM. Oral liarozole vs. acitretin in the treatment of ichthyosis: a phase II/III multicentre, double-blind, randomized, active-controlled study. British Journal of Dermatology. 2007;156(5):965–73. doi: 10.1111/j.1365-2133.2006.07745.x. [DOI] [PubMed] [Google Scholar]
- 24.Baert B, De Spiegeleer B. Local skin pharmacokinetics of talarozole, a new retinoic acid metabolism-blocking agent. Skin Pharmacol Physiol. 2011;24(3):151–9. doi: 10.1159/000323012. [DOI] [PubMed] [Google Scholar]
- 25.Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. The Journal of experimental medicine. 2010;207(8):1701–11. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long SA, Rieck M, Tatum M, Bollyky PL, Wu RP, Muller I, et al. Low-dose antigen promotes induction of FOXP3 in human CD4+ T cells. Journal of immunology. 2011;187(7):3511–20. doi: 10.4049/jimmunol.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. Journal of immunology. 2009;183(8):4895–903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Vaglenova J, Martínez SE, Porté S, Duester G, Farrés J, Parés X. Expression, localization and potential physiological significance of alcohol dehydrogenase in the gastrointestinal tract. European Journal of Biochemistry. 2003;270(12):2652–62. doi: 10.1046/j.1432-1033.2003.03642.x. [DOI] [PubMed] [Google Scholar]
- 30.Yang QJ, Zhou LY, Mu YQ, Zhou QX, Luo JY, Cheng L, et al. All-trans retinoic acid inhibits tumor growth of human osteosarcoma by activating Smad signaling-induced osteogenic differentiation. International journal of oncology. 2012;41(1):153–60. doi: 10.3892/ijo.2012.1426. [DOI] [PubMed] [Google Scholar]
- 31.Zhu WY, Jones CS, Kiss A, Matsukuma K, Amin S, De Luca LM. Retinoic acid inhibition of cell cycle progression in MCF-7 human breast cancer cells. Experimental cell research. 1997;234(2):293–9. doi: 10.1006/excr.1997.3589. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Yang W, Uytingco MS, Christakos S, Wieder R. 1,25-Dihydroxyvitamin D3 and all-trans-retinoic acid sensitize breast cancer cells to chemotherapy-induced cell death. Cancer Res. 2000;60(7):2040–8. [PubMed] [Google Scholar]
- 33.Hoffmann S, Rockenstein A, Ramaswamy A, Celik I, Wunderlich A, Lingelbach S, et al. Retinoic acid inhibits angiogenesis and tumor growth of thyroid cancer cells. Molecular and cellular endocrinology. 2007;264(1–2):74–81. doi: 10.1016/j.mce.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Lim YC, Kang HJ, Kim YS, Choi EC. All-trans-retinoic acid inhibits growth of head and neck cancer stem cells by suppression of Wnt/beta-catenin pathway. Eur J Cancer. 2012;48(17):3310–8. doi: 10.1016/j.ejca.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Zheng Y, Kramer PM, Olson G, Lubet RA, Steele VE, Kelloff GJ, et al. Prevention by retinoids of azoxymethane-induced tumors and aberrant crypt foci and their modulation of cell proliferation in the colon of rats. Carcinogenesis. 1997;18(11):2119–25. doi: 10.1093/carcin/18.11.2119. [DOI] [PubMed] [Google Scholar]
- 36.Corpet DE, Pierre F. Point: From animal models to prevention of colon cancer. Systematic review of chemoprevention in min mice and choice of the model system. Cancer Epidemiol Biomarkers Prev. 2003;12(5):391–400. [PMC free article] [PubMed] [Google Scholar]
- 37.Coletta PL, Müller AM, Jones EA, Mühl B, Holwell S, Clarke D, et al. Lymphodepletion in the ApcMin/+ mouse model of intestinal tumorigenesis. Blood. 2004;103(3):1050–58. doi: 10.1182/blood-2003-03-0707. [DOI] [PubMed] [Google Scholar]
- 38.Rao VP, Poutahidis T, Ge Z, Nambiar PR, Horwitz BH, Fox JG, et al. Proinflammatory CD4+ CD45RB(hi) lymphocytes promote mammary and intestinal carcinogenesis in Apc(Min/+) mice. Cancer Res. 2006;66(1):57–61. doi: 10.1158/0008-5472.CAN-05-3445. [DOI] [PubMed] [Google Scholar]
- 39.Coletta PL, Muller AM, Jones EA, Muhl B, Holwell S, Clarke D, et al. Lymphodepletion in the ApcMin/+ mouse model of intestinal tumorigenesis. Blood. 2004;103(3):1050–8. doi: 10.1182/blood-2003-03-0707. [DOI] [PubMed] [Google Scholar]
- 40.Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med. 2005;202(8):1051–61. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tada Y, Asahina A, Fujita H, Mitsui H, Torii H, Watanabe T, et al. Differential effects of LPS and TGF-beta on the production of IL-6 and IL-12 by Langerhans cells, splenic dendritic cells, and macrophages. Cytokine. 2004;25(4):155–61. doi: 10.1016/j.cyto.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 42.Mowat AM, Bain CC. Mucosal macrophages in intestinal homeostasis and inflammation. J Innate Immun. 2011;3(6):550–64. doi: 10.1159/000329099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445(7128):648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 44.Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491(7423):259–63. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin JC, Beriou G, Heslan M, Chauvin C, Utriainen L, Aumeunier A, et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol. 2013 doi: 10.1038/mi.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, et al. Retinoic acid controls the homeostasis of pre-cDC–derived splenic and intestinal dendritic cells. The Journal of Experimental Medicine. 2013 doi: 10.1084/jem.20122508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mollersen L, Paulsen JE, Olstorn HB, Knutsen HK, Alexander J. Dietary retinoic acid supplementation stimulates intestinal tumour formation and growth in multiple intestinal neoplasia (Min)/+ mice. Carcinogenesis. 2004;25(1):149–53. doi: 10.1093/carcin/bgg176. [DOI] [PubMed] [Google Scholar]
- 48.Stoppie P, Borgers M, Borghgraef P, Dillen L, Goossens J, Sanz G, et al. R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodents. J Pharmacol Exp Ther. 2000;293(1):304–12. [PubMed] [Google Scholar]
- 49.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–38. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 50.Reddy D, Siegel CA, Sands BE, Kane S. Possible association between isotretinoin and inflammatory bowel disease. Am J Gastroenterol. 2006;101(7):1569–73. doi: 10.1111/j.1572-0241.2006.00632.x. [DOI] [PubMed] [Google Scholar]
- 51.DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471(7337):220–24. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seidmon EJ, Trump DL, Kreis W, Hall SW, Kurman MR, Ouyang SP, et al. Phase I/II dose-escalation study of liarozole in patients with stage D, hormone-refractory carcinoma of the prostate. Ann Surg Oncol. 1995;2(6):550–6. doi: 10.1007/BF02307090. [DOI] [PubMed] [Google Scholar]
- 53.Verfaille CJ, Thissen CA, Bovenschen HJ, Mertens J, Steijlen PM, van de Kerkhof PC. Oral R115866 in the treatment of moderate to severe plaque-type psoriasis. J Eur Acad Dermatol Venereol. 2007;21(8):1038–46. doi: 10.1111/j.1468-3083.2007.02158.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.