

Abstract
The iron chelator deferoxamine (DFO), approved for the treatment of iron overload, has been examined as a therapeutic in a variety of conditions which iron may exacerbate. To evaluate the potential of DFO-bearing PEG-like Nanoprobes (DFO-PNs) as therapeutics, we determined their pharmacokinetics (PK) in normal mice, and imaged their accumulation in a tumor model and in models of transient brain ischemia and inflammation. DFO-PNs consist of a DFO, a Cy5.5 and PEG (5 kDa or 30 kDa) attached to Lys-Cys scaffold. Tumor uptake of a [89Zr]:DFO-PN(10) (30 kDa PEG, diameter 10 nm) was imaged by PET, surface fluorescence, and fluorescence microscopy. DFO-PN(10) was internalized by tumor cells (fluorescence microscopy) and by cultured cells (by FACS). [89Zr]:DFO-PN(4.3) (5 kDa PEG, diameter 4.3 nm) concentrated at incision generated inflammations but not at sites of transient brain ischemia. DFO-PNs are fluorescent, PK tunable forms of DFO that might be investigated as anti-tumor or anti-inflammatory agents.
Graphical abstract
Deferoxamine (DFO) PEG-like Nanoprobes consist of a DFO and Cy5.5 bearing peptide core surrounded by a 10 kDa PEG, with the DFO binding 89Zr for PET or Fe for therapeutic applications.
Introduction
The iron chelator deferoxamine (DFO), approved for the treatment of iron overload, has been examined for possible therapeutic benefit in a wide range of conditions which iron availablility may exacerbate. Because iron is an essential element for cell growth and division, DFO (and other iron chelators) have been examined as anticancer agents1–7. DFO has been used to reduce inflammation in cardiac ischemia8,9, nerve compression10, traumatic brain injury11, and rheumatoid arthritis12,13. DFO has shown activity in pre-clinical models of stroke 14,15 and been evaluated for this purpose in clinical trials 16. Finally DFO has been considered to enhance the elimination and detoxify metals ranging from aluminum17 to the radioactive actinides18,19. DFO is highly non-toxic with daily IV doses of 3–4 grams employed to treat chronic iron overload (manufacturer’s package insert). The many potential therapeutic applications and lack of toxicity of DFO caused us to investigate its use as a component of PEG-like Nanoprobes (PNs), and to image the uptake of the resulting DFO bearing PNs in disease models.
The modular PN design (Figure 1a), and the associated stepwise synthesis (Scheme 1), allow the independent variation of three functional groups (R1, R2 and R3,) and hence the independent variation of fluorescence, pharmacokinetics (PK), and metal chelating activity. PNs20 employ a constant, central Lys-Cys peptide (black) to which three variable functional groups (R1, R2, R3) are attached. At the N-terminus, the R1 functional group (blue) is a chelator, either the DOTA as previously 20, or deferoxamine (DFO) as is used here. The R2 functional group at the cysteine thiol (red) and has been fluorescein, Cy3, or IR-783 20, or the Cy5.5 group as is used here. The R3 functional group (purple) can be a PEG ranging from 2 kDa to 40 kDa 20, and provides PK tunability. With PEG’s of 5 kDa or greater, PEG blocks fluorochrome mediated interactions 21 and determines PN size 20 and, hence, the resulting nanomaterials are said to be “PEG-like” and are termed PEG-like Nanoprobes, (PNs).
Figure 1. Modular design of PEG-like Nanoprobes (PNs) and DFO bearing PNs or DFO-PNs.

a) Variable functional group components (R1, R2, R3) are attached to a fixed Lys-Cys scaffold. Examples of R groups are given in the text. b) DFO-PNs use a 5 kDa PEG or a 30 kDa PEG to surround a central DFO and Cy5.5 bearing peptide.
Scheme 1.

Synthesis of DFO-PEG-like Nanoprobes. Reagents and conditions: a: Fmoc peptide synthesis; b: succinic anhydride, DIPEA/DMF/25°C, 1 h; c: deferoxamine mesylate salt, DIPEA/DMSO/PyBOP, 25°C, overnight; d: TFA/water/triisopropylsilane/ethanedithiol = 9:0.25:0.25:0.5; e: Cy5.5-maleimide, DMSO, DIPEA; f: mPEG-5KDa-NHS(7a) or mPEG-30KDa-NHS(7b), DMSO, DIPEA; g: 89Zr-oxalate, pH 8, room temperature, 2 h.
Here we show that deferoxamine (DFO) can replace a DOTA at the R1 position, yielding DFO-PNs. In addition, we describe PNs of differing sizes sharing an R1 of DFO and an R2 of Cy5.5. When R3 is a 5 kDa PEG, a small PN with a diameter of 4.3 nm (Figure 1b) is obtained; it is denoted “DFO-PN(4.3)”. When R3 is a 30 kDa PEG, a PN denoted “DFO-PN(10),” with a diameter of 10 nm is obtained. The PN design employs three independently variable R groups and a constant Lys-Cys scaffold, allowing nanoprobe variation and optimization.
Imaging the accumulation of DFO-PNs in different disease models as a guide to conditions of possible therapeutic benefit reflects in part the fact that DFO-PNs differ from other drug-polymer conjugates in their lack of a release requirement. With most drug-polymer therapeutics, the release of a low molecular weight drug (analogous to DFO) from a larger, targeting carrier (e.g. a DFO-PN) is required for a pharmacological effect 22,23. The lack of DFO release for a metal chelation, and hence for a chelation-based pharmacological activity, is evident from DFO-antibody conjugate chemistry, where DFO binds 89Zr before or after conjugation to the biomolecule 24,25. DFO’s many therapeutic applications and lack of toxicity, as well as the lack of a DFO release requirement for a pharmacological activity, suggest that imaging the accumulation of DFO-PNs in differerent patholgies might guide the development of new iron chelation therapeutics.
Our goals were three-fold: (i) to demonstrate the modular PEG-like Nanoprobe (PN) chemical design, developed previously with DOTA20 can accommodate DFO, (ii) to demonstrate DFO-PNs provide pharmacokinetic (PK) tunable forms of DFO and, (iii) to image the accumulation of DFO-PNs in three disease models: tumors, transient ischemic brain lesions, and sites of inflammation.
Results and Discussion
1. Synthesis of DFO-PN(4.3) and DFO-PN(10)
DFO was attached to the N-terminus of a solid phase Cys-Lys peptide (Scheme 1) through a succinamide linker, followed by release from the solid phase. A solution phase reaction (e) between maleimide bearing Cy5.5 and the cysteine thiol yielded the (DFO)Lys-Cys(Cy5.5) peptide (compound 6). A second solution phase reaction (f) between an NHS-ester of a 5 kDa PEG (7a) and lysine side chain yielded compound 8a or DFO-PN(4.3), while a 30 kDa PEG (7b) yielded 8b or DFO-PN(10)). The C-terminal amide is not reactive.
The synthetic strategy employs solid phase reactions (reactions a–c of Scheme 1), followed by a site selective, solution phase between a maleimide-bearing Cy5.5 and cysteine thiol (e). A second site selective reaction between a NHS-bearing PEG and the lysine amine yields the DFO-PN, which written in peptide notation would be (DFO)Lys(PEG)-Cys(Cy5.5). Final purification by gel filtration separates any low molecular weight peptide (compound 6) from larger DFO-PNs (compounds 8a or 8b). DFO-PNs bind 89Zr4+ with a 2 h room temperature incubation (see supplement), indicating metal binding is not blocked by PEG attachment.
The globular protein equivalent volume of DFO-PNs was determined by fast protein liquid chromatography (FPLC) gel filtration, using globular protein standards with conversion to nm diameters as described 26. The 5 kDa PEG yielded DFO-PN(4.3), and the 30 kDa PEG yielded DFO-PN(10), where 4.3 and 10 are diameters in nanometers determined by gel filtration using globular protein standards as calibrators. Albumin has a diameter of 5.4 nm so DFO-PN(4.3) is smaller and DFO-PN(10) larger than albumin. Dynamic light scattering and FPLC nanoprobe dimensions are similar, see Figure S3 of Guo et al., reference 20.
2. Biodistribution of the 89Zr labeled PNs, [89Zr]:DFO-PN(10) and [89Zr]:DFO-PN(4.3)
The percent injected dose (ID) per organ at 24 h and 48 h for [89Zr]:DFO-PNs is shown in Figure 2a. The more slowly eliminated [89Zr]:DFO-PN(10) had substantial radioactivity in the blood at 24 h (17.9±3.5%) and 48 h (8.0±1.1%), see blue arrow. In contrast, the smaller [89Zr]:DFO-PN(4.3) had less than 0.9% ID in blood at these time points. With both [89Zr]:DFO-PNs, the liver (green arrow) was the major organ of retention (Figure 2a), though hepatic radioactivity was modest; with [89Zr]:DFO-PN(10) it was 8.6±1.0 %ID at 48 h post injection (see green arrow). Kidney uptake (red arrow) was less than 0.90 %ID, which is low for a radiolabeled peptide as discussed below. Tumor accumulation (red arrow, Figure 2b) was evident with [89Zr]:DFO-PN(10) but not with smaller DFO-PN.
Figure 2. Biodistribution [89Zr]:DFO-PNs.
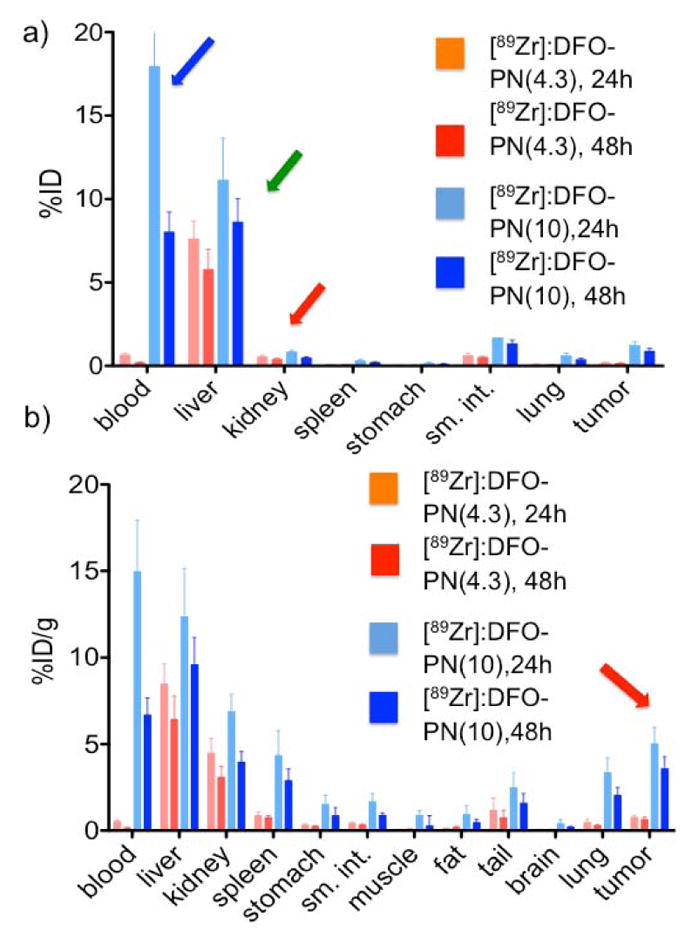
a) Biodistribution of [89Zr]:DFO-PNs as percent of injected dose (ID) per organ at 24 h and 48 h are shown in (a), and as tissue concentrations (%ID/g) in (b). Values are means ± 1 SD, n =6. Arrows highlight organ data discussed in the text.
3. Pharmacokinetics of DFO-PN(10) and DFO-PN(4.3) by serum fluorescence and whole body radioactivity
After IV injections of DFO-PNs, serum fluorescence versus time data were fit to a biexponential equation (Figure 3a), which indicated a fast and a slow kinetic process were operative. A two compartment PK model (Figure 3b) was employed, and the three microkinetic constants (kvas perm, Kvas return, kelim,) of the model were obtained 27. (Details are provided in Experimental Procedures.) Rate constants (h−1) or the corresponding half-lives (units of h) are provided for the three microconstants in Table 1. Individual data points from multiple animals taken at the indicated times are shown in Figure 3a.
Figure 3. Pharmacokinetics of DFO-PNs by serum fluorescence and whole body radioactivity.

a) Time dependence of serum fluorescence was fit to a biexponential equation, see Experimental Procedures. Data are single time points from 5 or 6 animals, approximately 3 time points per animal. b) The two compartment pharmacokinetic model used to analyze (a) provides the three microkinetic constants given in Table 1. Whole body radioactivity and elimination is shown for [89Zr]:DFO-PN(4.3) in (c) and [89Zr]:DFO-PN(10) in (d). Values are means ± 1SD, n= 3.
Table 1.
Pharmacokinetic Constants of DFO-PN(4.3) and DFO-PN(10) By Serum Fluorescence* and Whole Body Radioactivity**
| Constant | DFO-PN(4.3) rate, h−1 | DFO-PN(4.3) half-life, h | DFO-PN(10) rate, h−1 | DFO-PN(10) half-life, h |
|---|---|---|---|---|
| kvas perm* | 6.33 | 0.11 | 0.38 | 1.83 |
| kvas return* | 4.54 | 0.15 | 0.63 | 1.11 |
| kelim* | 1.56 | 0.44 | 0.067 | 10.31 |
| keiim** | 0.81 | 0.85 | 0.052 | 13.5 |
By blood fluorescence from Figure 3a.
By whole body radioactivity from Figure 2a and 2b.
To compare the Kelim from serum fluorescence with the elimination of [89Zr]:DFO-PN(10) and [89Zr]:DFO-PN(4.3), these PNs were injected and whole body radioactivity determined (Figures 3c and 3d). These data were fit to single exponential decay functions with the values of Kelim given in Table 1.
As shown in Table 1, the elimination half-lives from the two-compartment treatment serum fluorescence data (Figure 3a) and from whole body radioactive measurements (Figures 3c, 3d) were similar. Both the smaller DFO-PN(4.3) and larger DFO-PN(10) equilibrated with the extracellular compartment (see values of kvas perm and kvas return) with the kvas perm of the larger DFO-PN(10) of 0.38 h−1 (half-life of 1.83 h). DFO-PN(4.3) had elimination half-lives of less than 1 hour by both techniques, while the larger DFO-PN(10) had elimination half-lives of 10.3 h and 13.5 h by fluorescence and whole body radioactivity, respectively. The similar elimination kinetics obtained from serum fluorescence and from whole body radioactivity supports (i) the use of the two-compartment model (Figure 3b) and, (ii) the assumption that PNs are eliminated as in tact nanoprobes. Additional support for the circulation and excretion of PN’s as intact nanoprobes was obtained from their circulation after injection at their pre-injection sizes and from their stability in mouse serum (see Figures 2 and 720.
Figure 7. [89Zr]:DFO-PN(4.3) PET/CT Images and TTC staining of Ischemic rat brains.

SUV parametric images of [89Zr]:DFO-PN are shown in a transaxial (a) and a coronal (b) view of an ischemic brain with the nose denoted “N.” Cross-hairs indicate a small region of probe accumulation. (c) Coronal TTC stained sections run from anterior (top, left and down) to posterior (bottom right). A small region of tracer uptake (a & b) was within the ischemic region (c). A second rat had no tracer uptake (d & e) within the TTC defined ischemic region (f).
3. Imaging DFO-PN(10) tumor uptake by surface fluorescence
The time-dependent surface fluorescence of mice bearing two mCherry/HT-29 tumors per mouse is shown in Figure 4a, with mCherry fluorescence indicating tumor locations. The 2 h image reflected the DFO-PN(10)’s long vascular phase, while the 24 h image was a mixed vascular/tumor uptake phase. By 48 hours, the agent had cleared sufficiently for a tumor-associated fluorescence to be apparent. To further compare tumor mCherry fluorescence and DFO-PN(10) fluorescence, multichannel surface fluorescence/X-ray images of the tumor were taken post-sacrifice with skin removed (Figure 4b). Removal of skin also serves to mimic the possible clinical use of this DFO-PN(10) for the intraoperative determination of tumor margins by fluorescence.
Figure 4. Surface fluorescence imaging of the tumor uptake with DFO-PN(10).

a) Phases of DFO-PN(10) pharmacokinetics. Animals bearing two mCherry HT-29 tumors each (purple) were injected with DFO-PN(10) (green) and surface fluorescence images made at the indicated times. b) White light, surface fluorescence and X-ray images of tumor with skin removed at 48 h. DFO-PN(10) accumulated in the tumor, as shown by the overlay of tumor fluorescence (purple) and DFO-PN(10) fluorescence (yellow) which resolved to give white color in the fused image.
4. Imaging [89Zr]:DFO-PN(10) in a tumor model by PET/CT
The fate of [89Zr]:DFO-PN(10) is shown by coronal and transverse PET/CT images, slice selected to maximize tumor visualization (Figure 5a–c) or sagittal images selected to maximize bladder visualization (Figure 5d–e). Red arrows show tumors in 5c and bladder in 5d, 5e and 5f. Bladder radioactivity was seen at all times after injection, with decreasing amounts seen with increasing times after injection.
Figure 5. PET/CT image slices of [89Zr]:DFO-PN(10) accumulation in tumor and bladder.

Slices were selected to maximize tumor (A–C) or bladder visualization (D–F). (A–C) Axial (above dashed line) and coronal (below dashed line) image slices. Solid line on axial slice indicates the tumor location and confirms tumor uptake of the corresponding coronal slice and vice versa. Red arrows indicate tumor. (D–F) Sagittal slices demonstrate radioactivity concentration in the bladder, indicative of urinary excretion of the radiotracer. PET images displayed using color scale with units of standardized uptake value (SUV). Red arrow indicates bladder.
5. Uptake of DFO-PN(10) by mCherry/HT-29 tumor and cells
An advantage of our DFO-PNs is that unlike other iron chelators 28–30, their disposition in tumors or their internalization by cultured cells can be determined by fluorescence. The excitation and emission maxima of Cy5.5, 675 and 695 nm, respectively, are distinct from naturally occurring fluorochromes and readily detectable by FACS lasers or by fluorescent microscopy.
Dual channel fluorescence microscopy of tumor sections (Figure 6a) revealed the co-localization of DFO-PN(10) and mCherry fluorescence, indicating the uptake of DFO-PN(10) by mCherry/HT-29 cells. This was confirmed with the slow uptake of DFO-PN(10) by cultured mCherry/HT-29 cells using FACS (Figures 6b to 6f). The uptake of DFO-PN(10) by FACS correlated with the uptake seen by surface fluorescence (Figure 4a) and PET (Figures 5a–5c).
Figure 6. Uptake of DFO-PN(10) by the HT-29(mCherry) tumor and cells.

a) Dual channel fluorescence microscopy of HT-29(mCherry) of two tumor sections showing mCherry fluorescence (green, tumor cells) and DFO-PN(10) fluorescence (red). Nuclei are blue. Scale bar is 20 microns. b–e) Dual wavelength FACS scatter grams for DFO-PN(10) uptake by cultured HT-29(mCherry) at the indicated times. mCherry and DFO-PN’s Cy5.5 fluorescence are on the x- and y-axes, respectively. f) Time dependence of DFO-PN Cy5.5 cell fluorescence from figures b–e.
6. Imaging [89Zr]:DFO-PN(4.3) in transient rat brain ischemia
Figure 7 shows PET/CT images from two of five animals imaged, with images acquired 48 h after transient ischemia induced by a middle artery occlusion, with the injection of [89Zr]:DFO-PN(4.3) performed 24 h after ischemia. In one of five rats, [89Zr]:DFO-PN(4.3) accumulated in the ischemic region as marked with cross hairs of the transaxial image of Figure 7a and coronal image of Figure 7b. The corresponding coronal view of TTC (triphenyl tetrazolium) staining showing the ischemia generated is provided in Figure 7c. A more typical response (4 of 5 animals) is shown in Figures 7d–e where an animal exhibited no [89Zr]:DFO-PN(4.3) accumulation within the ischemic volume evident from TTC (Figure 7f).
7. Imaging [89Zr]:DFO-PN(4.3) accumulation at incision generated sites of inflammation with the rat brain ischemia model
While imaging the brain ischemia of Figure 7, an accumulation of [89Zr]:DFO-PN(4.3) at the incision sites at the neck and ultrasound Doppler probe levels, used to verify ischemic damage, was noted as shown in Figure 8. Doppler probe sites had mean SUVs of 0.26–0.36 and max SUVs of 0.46–0.76, while filament placement sites had SUVs with mean 0.35–0.45 and max of 0.65–0.92 (n=5). The magnitude of the incision SUV values was appreciable, being similar to those of DFO-PN(10) in the tumor. Given the rapid blood clearance of DFO-PN(4.3) (Figure 3, Table 1), DFO-PN(4.3) rapidly accumulates and is retained at incision generated sites of inflammation.
Figure 8. [89Zr]:DFO-PN(4.3) PET images of incision site injury.

Coronal (left) and sagital (right) images were slice selected to highlight activity at sites of inflammation in the ischemia model shown in Figure 7. Injury was due to Doppler probe placement, or incision for insertion of occlusion catheter. Animals shown here are the same as Fig. 7a–c (top) and 7d–f (bottom).
Discussion
The combination of the PNs made here with those made earlier 20 has now employed the central Lys-Cys scaffold (Figure 1a) with two R1 chelators (DFO or DOTA), four R2 fluorochromes (IR-783, fluorescein, Cy3, Cy5.5) and six R3 PEGs (2, 5,10, 20, 30, 40 kDa). This PN modular design, a Lys-Cys scaffold and three variable functional groups, generates nanoprobes with both variable metal binding properties and tunable PK whose disposition in biological systems can be ascertained by fluorescence, SPECT or PET.
Our model where the PEG surrounds the (DFO)Lys-Cys(Cy5.5) peptide, Figure 1b, is supported by the unusual properties PNs achieve in vivo. Many fluorochromes, including the drugs fluorescein and ICG, bind to albumin and undergo clearance by the liver’s hepatobiliary transport system 31–33. In contrast, PNs circulate at their PEG determined sizes, evident when PN fluorescence of serum after injection is analyzed by FPLC fluorescence 20. Though bearing a version of Cy5.5 that lacks sulfate groups, and which is highly hydrophobic, DFO-PNs do not behave like albumin-binding fluorochromes in vivo. (We employed a Cy5.5 from Lumiprobe that lacks the sulfate groups of the Cy5.5 from GE Healthsciences.) In addition, though using a Lys-Cys peptide scaffold, radiolabeled DFO-PNs do not show the high renal accumulation typical of radiolabeled peptides. Radiolabeled peptides are substrates for renal peptide transporters causing high renal accumulation 34–36. In contrast, both [89Zr]:DFO-PNs had less than 0.2 %ID in the kidney (Figure 3a).
The DFO-PN model of PEG surrounding a (DFO)Lys-Cys(Cy5.5) peptide endows DFO-PNs with an adsorption, distribution, metabolism and excretion (ADME) not seen with other nanomaterials. DFO-PNs have a high vascular permeability (see values of kvas perm and Kvas return in Table 1), a renal elimination pathway (Figures 5d–5f), a high whole body elimination (Figures 3c and 3d), and low renal and hepatic retention (Figure 2a). Though DFO-PNs bear a peptide and fluorochrome, they behave like neither in biological systems and hence we term them “PEG-like” nanoprobes.
By employing DFO as the chelator in PN design, we obtain a high molecular weight, iron chelator whose disposition in biological systems can be determined by fluorescence. DFO-PN(10) fluorescence indicated uptake by the mCherrry/HT-29 cells within a tumor, an observation supported by the uptake seen with the corresponding cells in culture and determined by FACS. We have avoided using the term “enhanced permeability and retention effect” (EPR effect) for the accumulation of DFO-PN(10) in our tumor model because of its high normal capillary permeability, evident from its half-life of 1.83 h (Table 1). The accumulation of DFO-PN(10) in the mCherry/HT-29 tumor appears to result from a high vascular permeability, followed by tumor cell fluid phase pinocytosis, since PEG covers the core peptide and there are no known receptors for PEG. Here the slow elimination of DFO-PN(10) provides time for a slow pinocytotic uptake in vivo, while a similar uptake was seen with cultured cells by FACS. Recently it has been shown Ras-transformed tumor cells use macropinocytosis to transport extracellular protein into the cell, which undergoes proteolytic degradation, yielding amino acids 37. The HT-29 line overexpresses a wild-type version of H-ras 38 38. The ability of tumor cells to internalize DFO-PN(10), together with the anti-proliferative activity of iron chelators noted above, suggests it might have a cytostatic activity of considerable interest.
The accumulation of DFO-PN(10) measured by PET and fluorescence in the HT-29 xenograft and corresponded cultured cells supports further studies with this PN in additional tumor models, The expectation that DFO-PNs would accumulate in regions of ischemic insult was based on the fact that ischemia leads to blood brain barrier breakdown and the accumulation of polymeric dextrans in ischemic regions 39–41. However, DFO-PN(10) did not accumulate and our results do not support further studies in this arena. Surprisingly, however, DFO-PN(4.3) accumulated in regions of incision generated inflammation and additional studies of this PN in inflammatory models seem warranted.
Experimental Procedures
Synthesis and radiolabeling of DFO-PNs
An outline of the syntheses is given in results along with Scheme 1. Details of synthesis are given in Supplementary materials.
Determination of DFO-PN Volumes
Volume was determined by FPLC using an ÄKTA Purifier 10 and Superdex™ 200 10/300GL column (GE Healthcare) with a running buffer of 0.05 M sodium phosphate, 0.15 M NaCl (0.1% Tween, pH 7.2) and flow rate of 0.8 ml/min. Standards (GE Healthcare) were Ferritin, Ribonuclease A, Carbonic Anhydrase, and Conalbumin and Blue Dextran 2000. AMW (apparent molecular weight based on size exclusion retention) was plotted versus Kav. Kav = (Ve−Vo)/(Vt−Vo), Vt = total volume, Ve = elution volume, Vo = void volume. Globular protein equivalent molecular volumes (in kDa) were converted to DFO-PN diameters in nm using the relationship: Radius in nm = 0.066*M1/3 where M is the molecular weight of a globular protein expressed in daltons 26.
DFO-PN Pharmacokinetics
All animal studies were approved by the Institutional Review Committee of the Massachusetts General Hospital. All studies of DFO-PN pharmacokinetics used female nude mice (25–30 g; 6–8 weeks old; nu/nu) and employed tail vein injections. (All mouse studies used nu/nu mice.) All pharmacokinetic studies used an anesthesia of 2% isoflurane in oxygen and flow rate 2L/min, and all animals were sacrificed with CO2. For PK (n=6 per compound), non-tumor bearing animals received 10 nmoles of DFO-PN(4.3) or DFO-PN(10). Some 50 μl of blood was collected by tail snip at multiple time points for each animal. Approximately 20 blood samples were collected per compound over 30 h (Figure 2). Blood was diluted (25 μl, 700 μl PBS, 10 mM EDTA), with fluorescence measured in a Cary Eclipse Fluorescence Spectrophotometer, excitation at 675 nm and emission from 695 nm.
Serum fluorescence was fit with two-phase decay curve using Graphpad Prism software: cp = A • e−αt + B • e−βt. The microconstants were then obtained: , kprem = α + β − kvas,return − kelim. Half lives were calculated from: .
Mouse tumor model
mCherry/HT-29 cells were a gift from Dr. Darshini Kuruppu. Cells were grown as described by ATCC for HT-29 cells, detached, pelleted and 200 μl of cell suspension containing 106 cells in Matrigel (BD Bioscience) was injected subcutaneously into right and left shoulders of anesthetized nu/nu mice. Tumors were allowed to grow 5–7 days. A total of 10 animals were used, 6 for biodistribution, 2 for PET/CT, 2 for fluorescence imaging and 3 for whole body elimination.
DFO-PN biodistribution and whole body elimination
150 μl of either [89Zr]:DFO-PN(10) or [89Zr]:DFO-PN(4.3) (400 μCi, ~2 nmole) was injected into tumor bearing mice (n=6). Some 24 h or 48 h later, animals were sacrificed with carbon dioxide and tissues collected. Radioactivity was measured with Perkin Elmer, Wizard2 2480. For determination of whole body radioactivity, mice (n=3 per compound) were injected with 200 μCi of the indicated agent, anesthetized with ketamine/xylazine (90/10 mg/kg ip), and lowered into the well Capintec cr-25 dose calibrator. Mice were then returned to their cages and employed for additional determinations of whole body radioactivity.
Tumor surface fluorescence imaging
Tumor bearing animals (2) were injected with 2 nmoles DFO-PN(10), n=2. After 48 h, a Kodak FX multispectral imaging system (Carestream Molecular Imaging, Rochester, NY) was used with excitation was at 540, 580, 620, 650, 690, 710, 720, 730, 750 and 760 nm, and emission at 700 nm for Cy5.5. Excitation at multiple wavelengths (450, 470, 510, 520, 530, 540, 550, 570, and 590 nm) with the emission at 700 nm was used for mCherry. Manufacturer’s software was used to determine (unmix) the Cy5.5 and mCherry fluorescence. Animals (n=2) were anesthetized as above for fluorescent and then X-ray images.
Tumor model for PET/CT
Mice (n-2) bearing xenografts were injected with approximately 10 MBq of [89Zr]:DFO-PN(10). Under anesthesia, animals were imaged on the eXplore VISTA microPET/CT scanner (General Electric Healthcare, Fairfield, CT) at 1 h, 24 h, and 48 h post injection. PET emission data were collected for 10 minutes at the 1 h post-injection time point and for 20 minutes at the 24 h and 48 h time points. Immediately following each PET acquisitions, the animal also underwent x-ray CT (40 kVp, 300 μA) for anatomical reference. PET data were sorted into sinograms using Fourier rebinning 42 then reconstructed using an ordered subsets expectation maximization algorithm 43 with 16 subsets and 2 iterations producing images with approximately 1.5 mm spatial resolution 44. PET images were calibrated to yield units of standardized uptake value, that is, radioactivity concentration normalized by injected dose and body weight. PET SUV volumes were aligned to the CT images for anatomical reference. Images presented are slices from 3D volumes.
Transient ischemia model
Rats (n=5, 350–400 g, male Wistar) were used. Pre-emptive buprenorphine (0.08mg/kg SC) was given in each animal. Rats were anesthetized with 2% isoflurane. Temperature was monitored and kept between 36–36.5 degrees centegrade using a temperature control system (FHC, ME, USA). A transient right middle cerebral artery (MCA) occlusion was performed under laser Doppler monitoring (Perimed, Sweden) using a burned tip 4-0 nylon filament (Ethicon Inc.) inserted from the external carotid artery to the internal carotid artery circulation. MCA occlusion was maintained 50–55 minutes.
Tumor model PET/CT
Mice (n=2) were injected with approximately 20 MBq [89Zr]:DFO-PN(4.3) 24 h after stroke onset. At an additional 24 h (n=3) or 48 h (n=2) later, rats were anesthetized and positioned on the scanning bed for PET/CT acquisition. PET and CT data acquisition, reconstruction, and image processing were performed as described above. After imaging, animals were euthanized and ischemia was visualized using a 2% triphenyl tetrazolium chloride (TTC) staining on two millimeter thick slices of the excised brain.
Whole body radioactivity determination
Mice (n=3) were anesthetized with ketamine/xylazine 80/10 mg/kg i.p, and then injected with 89Zr-DFO-PNs (15 mBq, IV, tail vein). Animals were placed in padded holder and momentarily placed in a Capitec CRC 25PE dose calibrator. Three animals were used for [89Zr]:DFO-PN4.3) and for [89Zr]:DFO-PN(10).
Supplementary Material
Acknowledgments
Funding sources: This work was supported by R01 EB 117699 (LJ), R01 EB 009691(LJ) and T32 EB013180 (support for MQW and DWW).
Footnotes
The authors declare no competing financial interests.
Supporting information available includes detailed synthetic procedures. This material is available free of charge at http://pubs.acs.org.
References
- 1.Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, Richardson DR. Iron chelators for the treatment of cancer. Current medicinal chemistry. 2012;19:2689–702. doi: 10.2174/092986712800609706. [DOI] [PubMed] [Google Scholar]
- 2.Lui GY, Obeidy P, Ford SJ, Tselepis C, Sharp DM, Jansson PJ, Kalinowski DS, Kovacevic Z, Lovejoy DB, Richardson DR. The iron chelator, deferasirox, as a novel strategy for cancer treatment: oral activity against human lung tumor xenografts and molecular mechanism of action. Molecular pharmacology. 2013;83:179–90. doi: 10.1124/mol.112.081893. [DOI] [PubMed] [Google Scholar]
- 3.Richardson D, Ponka P, Baker E. The effect of the iron(III) chelator, desferrioxamine, on iron and transferrin uptake by the human malignant melanoma cell. Cancer research. 1994;54:685–9. [PubMed] [Google Scholar]
- 4.Kovacevic Z, Kalinowski DS, Lovejoy DB, Quach P, Wong J, Richardson DR. Iron Chelators: Development of Novel Compounds with High and Selective Anti-Tumour Activity. Current drug delivery. 2010 May 27; doi: 10.2174/156720110791560991. Epub. [DOI] [PubMed] [Google Scholar]
- 5.Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochimica et biophysica acta. 2009;1790:702–17. doi: 10.1016/j.bbagen.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Torti SV, Torti FM. Ironing out cancer. Cancer research. 2011;71:1511–4. doi: 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nature reviews Cancer. 2013;13:342–55. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan W, Taylor AJ, Ellims AH, Lefkovits L, Wong C, Kingwell BA, Natoli A, Croft KD, Mori T, Kaye DM, et al. Effect of iron chelation on myocardial infarct size and oxidative stress in ST-elevation-myocardial infarction. Circulation Cardiovascular interventions. 2012;5:270–8. doi: 10.1161/CIRCINTERVENTIONS.111.966226. [DOI] [PubMed] [Google Scholar]
- 9.Liachenko S, Tang P, Xu Y. Deferoxamine improves early postresuscitation reperfusion after prolonged cardiac arrest in rats. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:574–81. doi: 10.1097/01.WCB.0000057742.00152.3F. [DOI] [PubMed] [Google Scholar]
- 10.Marin PC, Im MJ, Girotto JA, Borschel G, Bickel KD. Effects of hydroxyethyl-starch-bound deferoxamine on ischemia/reperfusion injury in chronic nerve compression. Journal of reconstructive microsurgery. 1998;14:485–90. doi: 10.1055/s-2007-1000211. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Hu R, Li F, Meng H, Lin J, Zhu G, Wang X, Feng H. Deferoxamine attenuates traumatic brain injury in rats. Di-San Junyi Daxue Xuebao. 2012;34:2349–2352. [Google Scholar]
- 12.Fudman EJ, Till GO, Fox IH. Deferoxamine induced decreases of lipid peroxides in rheumatoid arthritis. The Journal of rheumatology. 1987;14:686–91. [PubMed] [Google Scholar]
- 13.Salvarani C, Baricchi R, Lasagni D, Boiardi L, Piccinini R, Brunati C, Macchioni P, Portioli I. Effects of desferrioxamine therapy on chronic disease anemia associated with rheumatoid arthritis. Rheumatology international. 1996;16:45–8. doi: 10.1007/BF01816434. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y, Rempe DA. Prophylactic neuroprotection against stroke: low-dose, prolonged treatment with deferoxamine or deferasirox establishes prolonged neuroprotection independent of HIF-1 function. Journal of cerebral blood flow and metabolism. 2011;31:1412–23. doi: 10.1038/jcbfm.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanson LR, Roeytenberg A, Martinez PM, Coppes VG, Sweet DC, Rao RJ, Marti DL, Hoekman JD, Matthews RB, Frey WH, 2nd, et al. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. The Journal of pharmacology and experimental therapeutics. 2009;330:679–86. doi: 10.1124/jpet.108.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selim M, Yeatts S, Goldstein JN, Gomes J, Greenberg S, Morgenstern LB, Schlaug G, Torbey M, Waldman B, Xi G, Palesch Y. Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke. 2011;42:3067–74. doi: 10.1161/STROKEAHA.111.617589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domingo JL. The use of chelating agents in the treatment of aluminum overload. Journal of toxicology Clinical toxicology. 1989;27:355–67. doi: 10.3109/15563658909000356. [DOI] [PubMed] [Google Scholar]
- 18.Volf V, Burgada R, Raymond KN, Durbin PW. Early chelation therapy for injected Pu-238 and Am-241 in the rat: comparison of 3,4,3-LIHOPO, DFO-HOPO, DTPA-DX, DTPA and DFOA. International journal of radiation biology. 1993;63:785–93. doi: 10.1080/09553009314552191. [DOI] [PubMed] [Google Scholar]
- 19.White DL, Durbin PW, Jeung N, Raymond KN. Specific sequestering agents for the actinides. 16. Synthesis and initial biological testing of polydentate oxohydroxypyridinecarboxylate ligands. Journal of medicinal chemistry. 1988;31:11–8. doi: 10.1021/jm00396a005. [DOI] [PubMed] [Google Scholar]
- 20.Guo Y, Yuan H, Claudio NM, Kura S, Shakerdge N, Mempel TR, Bacskai BJ, Josephson L. PEG-like nanoprobes: multimodal, pharmacokinetically and optically tunable nanomaterials. PloS one. 2014;9:e95406. doi: 10.1371/journal.pone.0095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Y, Yuan H, Rice WL, Kumar AT, Goergen CJ, Jokivarsi K, Josephson L. The PEG-Fluorochrome Shielding Approach for Targeted Probe Design. J Am Chem Soc. 2012;134:19338–41. doi: 10.1021/ja309085b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duncan R. The dawning era of polymer therapeutics. Nature reviews Drug discovery. 2003;2:347–60. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 23.Kratz F, Muller IA, Ryppa C, Warnecke A. Prodrug strategies in anticancer chemotherapy. ChemMedChem. 2008;3:20–53. doi: 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]
- 24.Govindan SV, Michel RB, Griffiths GL, Goldenberg DM, Mattes MJ. Deferoxamine as a chelator for 67Ga in the preparation of antibody conjugates. Nuclear medicine and biology. 2005;32:513–9. doi: 10.1016/j.nucmedbio.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Ruggiero A, Holland JP, Hudolin T, Shenker L, Koulova A, Bander NH, Lewis JS, Grimm J. Targeting the internal epitope of prostate-specific membrane antigen with 89Zr-7E11 immuno-PET. Journal of nuclear medicine. 2011;52:1608–15. doi: 10.2967/jnumed.111.092098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erickson HP. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biological Procedures Online. 2009;11:32–50. doi: 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum SE. Basic Pharmacokinetics and Pharmacodynamics: An Integrated Textbook and Computer Simulations. John Wiley and Sons; Hoboken, NJ: 2011. [Google Scholar]
- 28.Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, Stefani C, Lovejoy DB, Sharpe PC, Bernhardt PV, Richardson DR. Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors. Journal of medicinal chemistry. 2009;52:5271–94. doi: 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- 29.Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, Stefani C, Basha MT, Sharpe PC, Jansson PJ, Kalinowski DS, et al. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. Journal of medicinal chemistry. 2012;55:7230–44. doi: 10.1021/jm300768u. [DOI] [PubMed] [Google Scholar]
- 30.Merlot AM, Kalinowski DS, Richardson DR. Novel chelators for cancer treatment: where are we now? Antioxidants & redox signaling. 2013;18:973–1006. doi: 10.1089/ars.2012.4540. [DOI] [PubMed] [Google Scholar]
- 31.Choi HS, Gibbs SL, Lee JH, Kim SH, Ashitate Y, Liu F, Hyun H, Park G, Xie Y, Bae S, et al. Targeted zwitterionic near-infrared fluorophores for improved optical imaging. Nature biotechnology. 2013;31:148–53. doi: 10.1038/nbt.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pauli J, Licha K, Berkemeyer J, Grabolle M, Spieles M, Wegner N, Welker P, Resch-Genger U. New Fluorescent Labels with Tunable Hydrophilicity for the Rational Design of Bright Optical Probes for Molecular Imaging. Bioconjugate chemistry. 2013;24(7):1174–85. doi: 10.1021/bc4000349. [DOI] [PubMed] [Google Scholar]
- 33.Ebert B, Riefke B, Sukowski U, Licha K. Cyanine dyes as contrast agents for near-infrared imaging in vivo: acute tolerance, pharmacokinetics, and fluorescence imaging. Journal of biomedical optics. 2011;16:066003. doi: 10.1117/1.3585678. [DOI] [PubMed] [Google Scholar]
- 34.Gotthardt M, van Eerd-Vismale J, Oyen WJ, de Jong M, Zhang H, Rolleman E, Maecke HR, Behe M, Boerman O. Indication for different mechanisms of kidney uptake of radiolabeled peptides. Journal of nuclear medicine. 2007;48:596–601. doi: 10.2967/jnumed.106.036020. [DOI] [PubMed] [Google Scholar]
- 35.Melis M, Vegt E, Konijnenberg MW, de Visser M, Bijster M, Vermeij M, Krenning EP, Boerman OC, de Jong M. Nephrotoxicity in mice after repeated imaging using 111In-labeled peptides. Journal of nuclear medicine. 2010;51:973–7. doi: 10.2967/jnumed.109.074310. [DOI] [PubMed] [Google Scholar]
- 36.Behe M, Kluge G, Becker W, Gotthardt M, Behr TM. Use of polyglutamic acids to reduce uptake of radiometal-labeled minigastrin in the kidneys. Journal of nuclear medicine. 2005;46:1012–5. [PubMed] [Google Scholar]
- 37.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halaschek-Wiener J, Wacheck V, Schlagbauer-Wadl H, Wolff K, Kloog Y, Jansen B. A novel Ras antagonist regulates both oncogenic Ras and the tumor suppressor p53 in colon cancer cells. Molecular medicine (Cambridge, Mass ) 2000;6:693–704. [PMC free article] [PubMed] [Google Scholar]
- 39.Chen B, Friedman B, Cheng Q, Tsai P, Schim E, Kleinfeld D, Lyden PD. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke. 2009;40:e666–74. doi: 10.1161/STROKEAHA.109.551341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann A, Bredno J, Wendland M, Derugin N, Ohara P, Wintermark M. High and Low Molecular Weight Fluorescein Isothiocyanate (FITC)-Dextrans to Assess Blood-Brain Barrier Disruption: Technical Considerations. Translational stroke research. 2011;2:106–11. doi: 10.1007/s12975-010-0049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez-Lopez D, Faustino J, Daneman R, Zhou L, Lee SY, Derugin N, Wendland MF, Vexler ZS. Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. The Journal of neuroscience. 2012;32:9588–600. doi: 10.1523/JNEUROSCI.5977-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Defrise M, Kinahan PE, Townsend DW, Michel C, Sibomana M, Newport DF. Exact and approximate rebinning algorithms for 3-D PET data. IEEE transactions on medical imaging. 1997;16:145–58. doi: 10.1109/42.563660. [DOI] [PubMed] [Google Scholar]
- 43.Hudson HM, Larkin RS. Accelerated image reconstruction using ordered subsets of projection data. IEEE transactions on medical imaging. 1994;13:601–9. doi: 10.1109/42.363108. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Seidel J, Tsui BM, Vaquero JJ, Pomper MG. Performance evaluation of the GE healthcare eXplore VISTA dual-ring small-animal PET scanner. Journal of nuclear medicine. 2006;47:1891–900. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.