

Abstract
A significant proportion of breast cancer patients harbor clinically undetectable micrometastases at the time of diagnosis. If left untreated, these micrometastases may lead to disease relapse and possibly death. Hence, there is significant interest in the development of novel anti-metastatic agents that could also curb the growth of pre-established micrometastases. Like primary tumor, the growth of metastases also is driven by angiogenesis. Although the role of cysteine protease Cathepsin L (CTSL) in metastasis associated tumor cell functions such as migration and invasion is well recognized, its role in tumor angiogenesis remains less explored. The present study examines the contribution of CTSL to breast cancer angiogenesis and evaluates the anti-angiogenic efficacy of CTSL inhibitor KGP94. CTSL semi-quantitative RT-PCR analysis on breast tissue panels revealed significant upregulation of CTSL in breast cancer patients which strongly correlated with increased relapse and metastatic incidence and poor overall survival. Preclinically, CTSL ablation using shRNA or KGP94 treatment led to a significant reduction in MDA-MB-231 tumor cell induced angiogenesis in vivo. In-vitro assessments demonstrated a significant decrease in various angiogenic properties such as endothelial cell sprouting, migration, invasion, tube formation and proliferation in the presence of KGP94. Microarray analyses revealed a significant upregulation of cell cycle related genes by CTSL. Western blot analyses further confirmed upregulation of members of the cyclin family by CTSL. Collectively, these data indicate that CTSL is an important contributor to tumor angiogenesis and that the CTSL inhibition may have therapeutic utility in the treatment of breast cancer patients.
Keywords: Cathepsin L, Breast cancer, Angiogenesis, KGP94
Introduction
Breast cancer is the most prevalent cancer and the second leading cause of cancer deaths in women [1]. Metastatic disease continues to be the primary cause of treatment failure, high mortality and poor quality of life in breast cancer patients. This realization has led to significant interest in the development of agents that impair metastasis. While it is critical to disrupt tumor cell dissemination, it is equally important to retard the expansion of established micro-metastases.
Angiogenesis, the process of forming new blood vessels from pre-existing vasculature is an important hallmark of tumor progression [2]. To grow beyond a certain critical size, both primary tumors and distant metastases have to induce new vascular supply in order to meet their growing oxygen and nutrient demands. Moreover, tumor vasculature is immature and highly permeable due to inefficient mural cell coverage, discontinuous endothelial cell lining and the lack of a durable basement membrane [3]. These vascular anomalies provide an easy exit route to metastatic tumor cells attempting to escape from the primary tumor. In fact, Weidner et al., have demonstrated that tumor microvessel density strongly correlates with metastatic incidence in breast cancer patients [4]. Thus an effective anti-angiogenic agent would not only retard the growth of the primary tumor but could also exert significant anti-metastatic activity by impairing tumor cell dissemination and growth of metastatic lesions.
Recognition of the dependence of angiogenesis on proteolytic enzymes has raised interest in the evaluation of proteases as potential targets to disrupt tumor angiogenesis [5, 6]. While the role of cysteine proteases in tumor metastasis is well understood, their contribution to tumor angiogenesis remains less explored. Cysteine cathepsins, in particular cathepsin L (CTSL) is upregulated in a wide range of human cancers [7, 8]. In most normal cells, CTSL is mainly present within the lysosomes where it participates in degradative proteolysis of intracellular and endocytosed proteins [9]. However, transformation dependent CTSL overexpression and alterations in trafficking mechanism shunt this lysosomal protease into the secretory pathway [10–12]. Upon secretion, CTSL interacts with glycosaminoglycan component of proteoglycans present on the cell surface and extracellular matrix where it undergoes an activating conformational change [13, 14]. Secreted CTSL promotes tumor metastasis by degrading several components of the basement membrane and extracellular matrix and further amplifies the proteolytic cascade by activating latent pro-forms of key metastasis promoting proteases [15–19]. In fact, CTSL overexpression has been shown to switch poorly tumorigenic and non-metastatic human melanoma cells to a highly tumorigenic and aggressively metastatic phenotype [20]. Conversely, tumor progression studies in CTSL−/− mice revealed that CTSL deficiency significantly hampered the progression of benign encapsulated pancreatic tumors into highly invasive carcinomas [21]. Further, our previous findings have reported that pharmacological inhibition of CTSL using 3-bromophenyl-3-hydroxyphenyl-ketone thiosemicarbazone (KGP94) significantly impairs the invasive and metastatic capacities of human breast and prostate cancer cells [22–24]. While these studies are clearly indicative of the importance of CTSL in metastatic progression, they shed little light on the involvement of CTSL in the angiogenic process. Thus, the goal of the present studies was to assess the contribution of CTSL in breast cancer angiogenesis and to evaluate the anti-angiogenic efficacy of KGP94, a thiosemicarbazone based small molecule inhibitor that disrupts CTSL activity by blocking its active site [25].
Materials and methods
Cell culture
Human lung microvascular endothelial cells (HMVEC-L) and Human umbilical vein endothelial cells (HUVEC) were cultured in EGM2 MV media supplied by Lonza (CC-3202) and Ham’s F12 nutrient mixture (Kaighn’s modification) supplemented with 10 % fetal bovine serum, heparin (100 mg/L) and endothelial cell growth supplement (E2759, Sigma) respectively. All human and murine breast cancer cell lines were cultured in appropriate medium (MDA-MB-231 and 4T-07 in DMEM; 4T1 in RPMI; EMT6 in Waymouth medium) supplemented with 10 % fetal bovine serum. All cells were maintained at 37 °C in a humidified atmosphere of 5 % CO2 in air.
Breast qPCR array
CTSL expression levels in breast cancer patients were determined by performing semiquantitative RT-PCR analysis on TissueScan Breast cancer panel (Origene BCRT102) as per manufacturer’s instructions. CTSL mRNA levels were normalized to β-actin and are shown as relative levels normalized to normal tissue pools.
Clinical data analysis
For survival analyses on clinical datasets, KM plotter database was used. The program integrates gene expression information from TCGA, EGA and GEO microarray databases with clinical outcome [26]. For progression free, distant metastases free and overall survival analyses, patients were stratified into below median and above median expression groups and tested for statistical significance using log-rank test.
CTSL secretion analysis
Breast cancer cells and endothelial cells were cultured in 10 cm dishes. When the cells reached ~60 % confluency, cell culture media was replaced with serum free media. Cell conditioned media were harvested 24 h later, centrifuged at 1000 rpm for 10 min to remove cell debris and stored at −20 °C until analysis. Secreted CTSL levels were determined by performing enzyme linked immune sorbent assay on cell conditioned media using Cathepsin L kit (human—R&D systems, DY952; mouse—My Biosource, MBS280424) as per manufacturers’ instructions.
CTSL knockdown
6.25 × 105 MDA-MB-231 cells were seeded in a 6 well dish. At 70–80 % confluency, cells were transfected with CTSL shRNA plasmids (Origene TG305172) using Lipofectamine LTX plus reagent (Invitrogen) as per manufacturer’s instructions. Transfected clones were selected in the presence of puromycin and expanded. CTSL knockdown efficiency was tested by performing western blot on whole cell lysates and ELISA on cell culture supernatants. Clones exhibiting >80 % knockdown efficiency were used for in vivo and in vitro studies.
Reagent
KGP94 was kindly provided by Dr. Kevin Pinney (Baylor University). For in vitro evaluations, KGP94 was dissolved in sterile DMSO to obtain a stock concentration of 25 mM and stored at −20 °C. The stock was then diluted in cell culture media to achieve a working concentration of 25 μM. For in vivo assays, KGP94 was sonicated in 10 % tween 80 solution for 30 min and then diluted in 1 M HEPES buffer to desired concentrations. The drug was then filter sterilized and stored at 4 °C.
Intradermal assay
All in vivo experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Florida. Induction of angiogenesis by breast cancer MDA-MB-231 cells and the ability of KGP94 to inhibit tumor angiogenesis in vivo was measured by performing intradermal assay as described previously [27]. 5 × 105 parental or CTSL knockdown MDA-MB-231 cells were injected intradermally on the ventral surface of athymic NCR female nu/nu mice at four sites. To facilitate easy location of the site of tumor cell inoculation, one drop of trypan blue solution was added to impart a light blue color to the cell suspension. 10 or 20 mg/kg KGP94 was administered intra-peritoneally on a daily basis. 3 days later, the mice were euthanized and their skin flaps were removed and promptly analyzed. Tumor angiogenesis was evaluated by counting the number of blood vessels growing into the tumor nodule using a Zeiss Stemi SV 6 dissecting microscope. Tumor nodule images were captured using a Leica MZ 16 F camera and Leica Application Suite software.
Migration assay
Migration assays were performed using BD falcon cell culture inserts (353097). The bottom of these inserts is a polyethylene terephthalate membrane with 8 μm pores. 1 × 103 HMVEC-L cells were seeded into cell culture inserts in the presence of desired concentrations of purified human CTSL (Calbiochem 219402) and allowed to migrate through the pores in the membrane. 24 h later, non-migrated cells in the top chamber were scraped off using cotton swabs. Cells that had migrated to the other side of the membrane were stained with crystal violet and quantified under a light microscope.
Invasion assay
For invasion assays, BD falcon cell culture inserts (353097) were coated with matrigel as per manufacturer’s instructions and seeded with 5 × 103 HMVEC-L cells in serum free media. Cells were incubated in the presence of indicated concentrations of purified CTSL or KGP94 or tumor conditioned media. 24 h later, non-invaded cells were scraped off; invaded cells were stained with crystal violet and counted.
Sprouting assay
750 HMVEC-L cells suspended in 4.2 % methylcellulose solution (Sigma, viscosity 4000 cP) were seeded per well of a 96 well plate to form endothelial spheres. 24 h later the spheres were harvested using a wide bore pipette and centrifuged at 1300 rpm for 3 min. Approximately 48 spheres were suspended in a 2 mg/mL collagen solution and added to each well of a 24 well plate pre-coated with 4.2 % methylcellulose-and 2 mg/mL collagen mixture. The spheres were incubated at 37 °C for 30 min to solidify and embed the spheres within the collagen methyl cellulose matrix. 30 min later, 200 μL media containing desired concentrations of purified CTSL or KGP94 or tumor conditioned media was added. The spheres were visualized and imaged 8 h later using a Zeiss Axioplan 2 microscope.
Tube formation assay
200 μL matrigel was added to each well of a 24 well plate and solidified at 37 °C for 30 min. 4 × 104 HMVEC-L or 1 × 105 HUVEC cells were seeded on solidified matrigel and in the presence of desired concentrations of purified CTSL or KGP94 or tumor conditioned media and incubated at 37 °C for 8 h. Endothelial tubes were quantified and imaged using Zeiss Axioplan 2 microscope.
Proliferation assays
2.5 × 103 HMVEC-L or HUVEC cells were seeded in a 24 well dish. 48 h later, the cells were treated with desired concentrations of purified CTSL or KGP94. At each time point, cell culture media was replaced with WST-1 reagent (Dojindo) in phenol red free media. 4 h later viability was determined by measuring the amount of formazan dye formation at 450 nm using the Spectramax M5 (Molecular Devices) spectrophotometer.
BrdU incorporation assay was performed using BrdU cell proliferation assay kit (Cell Signaling, 6813) as per manufacturer’s instructions. Briefly, 1 × 103 HMVEC-L cells were seeded in a 96 well plate. 48 h later, the cells were treated with desired concentrations of purified CTSL and 1× BrdU. 24 h later, the cells were fixed, incubated with detection antibody for 1 h, washed followed by incubation with HRP conjugated secondary antibody for 1 h. 3,3′,5,5′-Tetramethylbenzidine substrate was added and the amount of BrdU incorporation was measured spectrophotometrically at 450 nm using a Spectramax M5 plate reader.
Microarray analysis
HMVEC-L cells were stimulated with 10 ng/mL purified CTSL. 4 h later, total RNA was extracted using RNeasy plus kit (Qiagen) as per manufacturer’s instructions and assessed using Agilent Bioanalyzer 2000. All samples had a RNA integrity number of 9.9 or higher. 200 ng of total RNA was labeled using Affymetrix Whole Transcriptome Plus Expression kit, following manufacturer’s instructions and 5.5 μg cDNA was hybridized onto HTA 2.0 Transcriptome Array (Affymetrix).
Data were normalized using RMA as implemented in Partek and later analyzed with the R software (http://www.r-project.org). Microarray data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE75744.
Limma package was employed to perform statistical analysis applying the empirical Bayes method proposed by Smyth [28], which produces stable estimates when the number of replicates is small. Genes were considered differentially expressed with p-values of <0.05. For the significant genes, correlations between samples were evaluated by hierarchical clustering using the open software Cluster 3.0 [29]. Data were adjusted by centering the gene expressions by the median. Gene expression and microarray samples were clustered according to centroid linkage after similarity measurement by centered correlation. Gene ontology analysis was performed with the Gene Set Enrichment Analysis (GSEA) software. This method first considers the gene expression profiles from two classes to rank genes according to the strength of the correlation with a class. Then, given a priori defined sets of genes, the software determine if genes (belonging to the same set) are randomly distributed throughout the ranked list or significantly overexpressed in one or other class [30]. We defined the gene sets as genes sharing the same biological process. A significant biological process was defined by a nominal p value <0.05, after 1000 permutations. If enriched biological processes were redundant, a leading edge analysis was performed. This analysis determines common genes in the leading-edge subsets between similar biological processes. Cytoscape software [31] and the GeneMania plugin [32] were used to infer a network between common significant genes. In the network, genes (nodes) were connected if they had physical interaction (protein–protein interaction, red edges), genetic interaction (green edges), participate in the same reaction within a pathway (light blue edges), shared protein domains (brown edges) or they were predicted to have functional relationship (orange edges). The thicker the edge between two genes, the higher was the Pearson’s correlation coefficient across multiple conditions in an experiment.
Quantitative polymerase chain reaction
Gene expression analysis was performed at 1, 2, 4 and 12 h after CTSL addition to HMVEC-L cells. Cells were then rinsed with PBS, lysed with TRIzol reagent (ThermoFisher Scientific) and processed for RNA extraction. Briefly, chloroform was added to the cell homogenate to separate out DNA and proteins, and RNA was eluted using 100 % isopropyl alcohol. Following RNA quantification using a ND-1000 spectrophotometer, samples were reverse transcribed using Taqman reverse transcription reagents (ThermoFisher Scientific) as per manufacturer’s instructions. The list of primers used to amplify cDNA of interest is shown in Supplementary Table 1. Relative gene expression was determined by performing quantitative PCR using StepOne Real-Time PCR system (ThermoFisher Scientific).
Results
CTSL expression level serves as a prognosticator of clinical outcome of breast cancer patients
A significant upregulation in CTSL expression levels was observed in breast cancer patients compared to normal individuals (Fig. 1a). In order to determine the significance of CTSL upregulation in disease progression and survival of breast cancer patients, CTSL expression profiles were examined in a microarray compendium of primary tumors from 1809 breast cancer patients with known clinical outcome [26]. We observed that patients that expressed high levels of CTSL were at a significantly higher risk of relapse, developing metastatic disease, and death (Fig. 1b–d). CTSL upregulation results in an increase in its secretion as demonstrated by the increased serum and urinary CTSL levels in various cancer settings [33–36]. In order to examine the role of CTSL in tumor angiogenesis, CTSL secretion by endothelial cells was evaluated. Compared to the level of CTSL secreted by breast cancer cells, CTSL secretion by human microvascular endothelial cells (HMVEC-L) was undetectable (Fig. 1e).
Fig. 1.
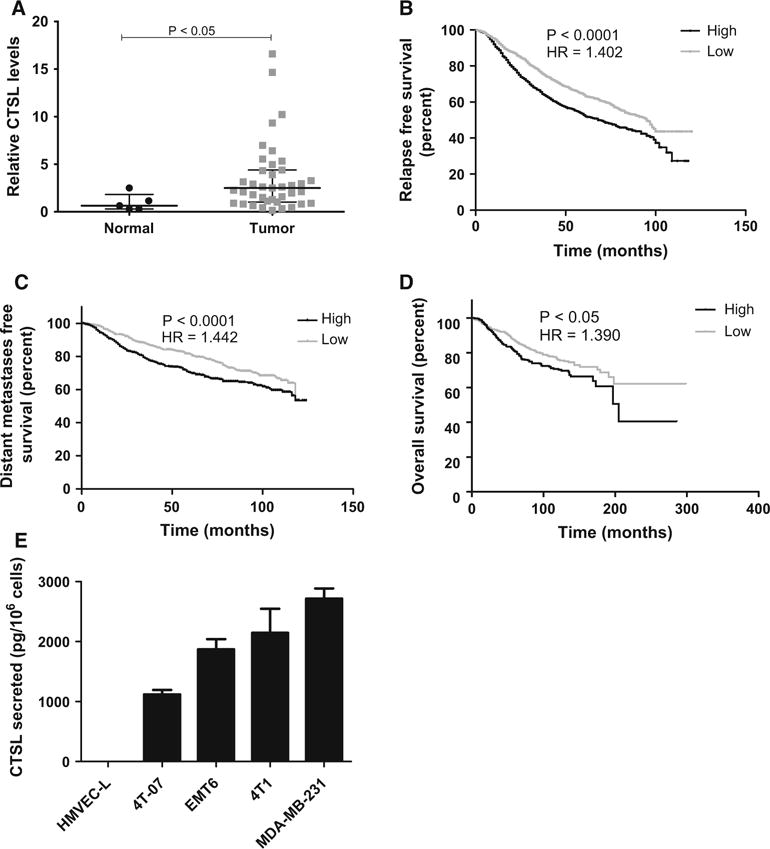
CTSL upregulation in breast cancer. a CTSL semiquantitative PCR testing CTSL expression in breast cancer patients using Tissues can breast tissue panels. CTSL mRNA levels were normalized to β-actin. Line, median; whiskers, values at 25th and 75th percentiles; Mann–Whitney test. b–d Kaplan–Meier plots of relapse free, distant metastases free and overall survival using the KM plotter meta-analysis database. Breast cancer patients were stratified on the basis of CTSL expression level. Statistical significance was determined using log rank test. e Quantification of CTSL secretion by ELISA on 24 h conditioned media. Bars represent mean and s.e.m. from three independent experiments
CTSL ablation suppresses tumor angiogenesis in vivo
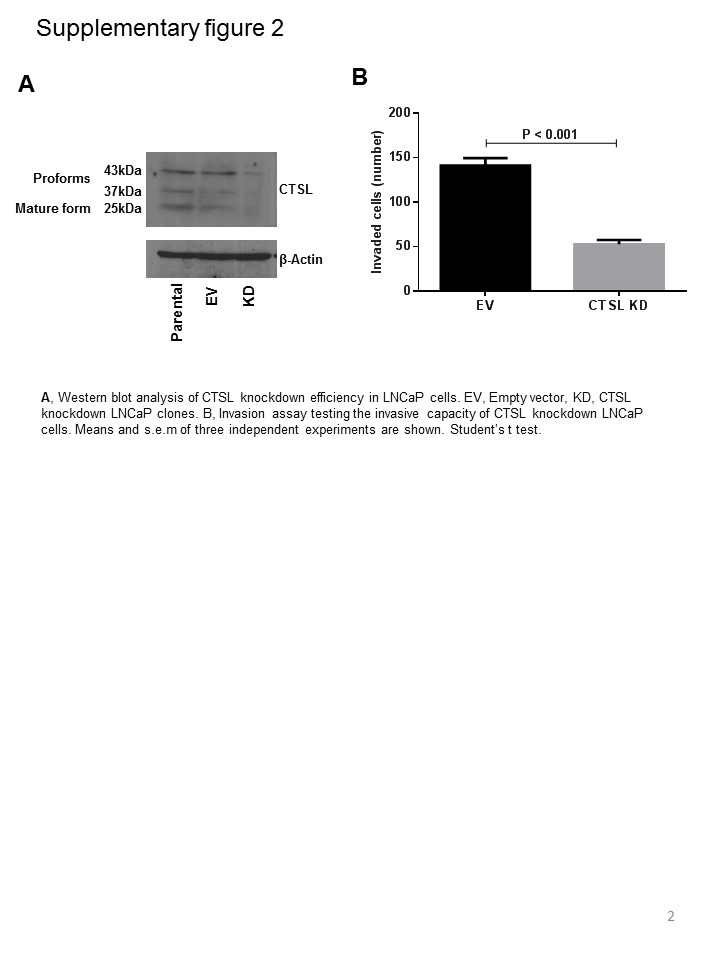
To test whether CTSL plays a role in tumor angiogenesis, CTSL knockdown MDA-MB-231 cells were generated and clone exhibiting >80 % knockdown efficiency (Supplementary Fig. 1a, b) was selected for testing the effect of CTSL deficiency on angiogenesis. Empty vector transfected as well as CTSL knockdown MDA-MB-231 breast cancer cells were intradermally inoculated into the ventral skin flaps of female nude mice (Fig. 2a, b). Compared to the empty vector control, tumor nodules in mice inoculated with CTSL knockdown tumor cells showed a significant decrease in their angiogenic capacity thus suggesting that CTSL is a major contributor to breast tumor cell initiated angiogenesis. Viability assay confirmed that the decrease in blood vessel formation was not due to an effect on cell proliferation (Supplementary Fig. 1c). Similarly, compared to untreated controls, tumor nodules in mice that were treated with 10 or 20 mg/kg KGP94 showed a significant reduction in their ability to induce blood vessel formation.
Fig. 2.
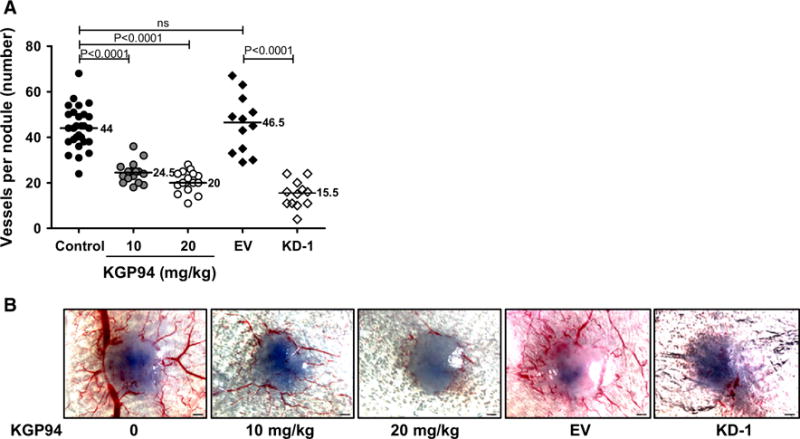
CTSL inhibition suppresses tumor angiogenesis in vivo. a Intradermal assay testing the effect of genetic or pharmacological ablation of CTSL on MDA-MB-231 tumor angiogenesis; n = 16. EV empty vector, KD-1, CTSL knockdown MDA-MB-231 tumor nodules; line, median; Mann–Whitney test. b Representative images of tumor nodules from a
CTSL promotes in vitro angiogenic properties of endothelial cells
Even though endothelial cells do not secrete CTSL, they are exposed to tumor cell secreted CTSL within the tumor microenvironment. In order to elucidate whether tumor cell secreted CTSL can act upon endothelial cells in a paracrine manner to elicit an angiogenic response, we tested the effect of purified CTSL on various angiogenesis associated endothelial cell functions. In the presence of CTSL, endothelial cells exhibited a dose dependent increase in their ability to migrate and invade through matrigel (Fig. 3a, b). While KGP94 had no effect on the invasive capacity of naïve endothelial cells, it reduced CTSL stimulated endothelial cell invasion to near baseline levels. In an attempt to closely mimic the tumor microenvironmental conditions, endothelial cell invasiveness in the presence of tumor conditioned media was evaluated. While conditioned media harvested from parental MDA-MB-231 cells led a marked increase in endothelial cell invasion, endothelial cells that were incubated either with KGP94 or with conditioned media derived from CTSL knockdown MDA-MB-231 cells showed a significant reduction in their invasive capacity (Fig. 3c). These results collectively indicate that tumor derived CTSL could significantly enhance endothelial cell migratory and invasive capacities. Certain proteases have been shown to facilitate sprouting by degrading the obstructive extracellular matrix as these sprouts penetrate into the interstitium to form new vessels [37]. Hence, we tested the effect of CTSL on the sprouting of endothelial spheres embedded within a collagen matrix. Both purified CTSL and tumor conditioned media led to a marked increase in endothelial sphere sprouting in a dose dependent fashion (Fig. 3d). In order to determine the contribution of CTSL to tumor conditioned media stimulated sprouting, endothelial spheres were incubated in presence of conditioned media harvested from CTSL knockdown MDA-MB-231 cells. Both CTSL deficiency and KGP94 treatment led to a significant reduction in tumor conditioned media or purified CTSL stimulated endothelial sprouting.
Fig. 3.
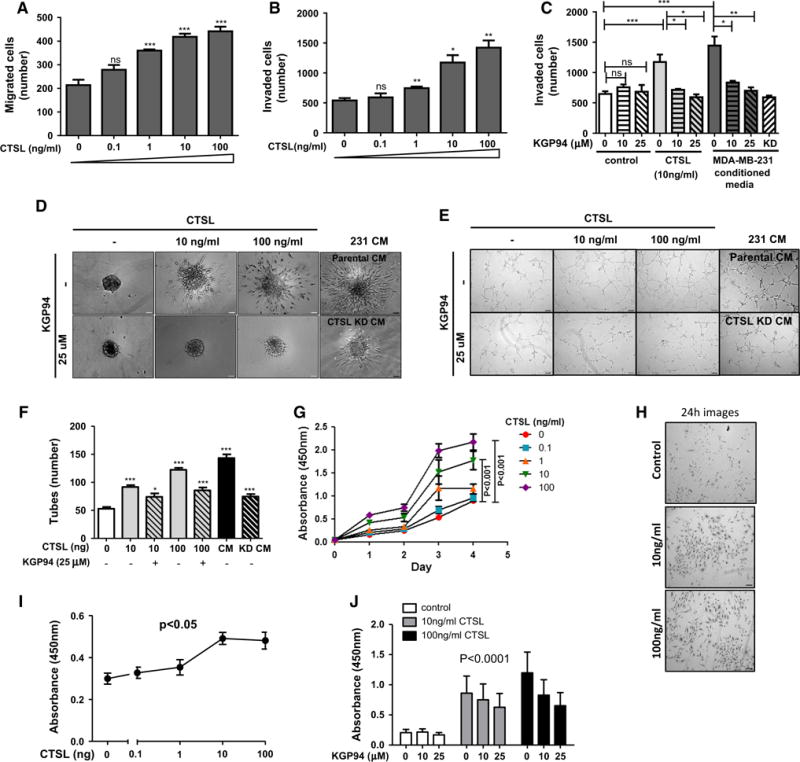
CTSL promotes endothelial cell pro-angiogenic functions. a and b Human microvascular endothelial cell (HMVEC-L) transwell migration and invasion assays in presence of indicated concentrations of purified CTSL. Means and s.e.m. from three independent experiments are shown. c CTSL or tumor conditioned media stimulated HMVEC-L invasion assay with or without KGP94. Bars represent means and s.e.m from three independent experiments. *p < 0.05; **p < 0.005; ***p < 0.0005; Student’s t test. d Images of endothelial sphere sprouts upon incubation with indicated doses of purified CTSL or tumor conditioned media with or without KGP94. ×20 magnification. e Images of tubes formed by CTSL or tumor conditioned media stimulated endothelial cells treated with or without KGP94. ×5 magnification. f Quantification of total number of tubes formed. *p < 0.05; ***p < 0.0001; Student’s t test. g Measurement of proliferation of endothelial cells exposed to increasing concentrations of purified CTSL. Results are mean ± s.e.m; analysis of variance. h Representative images of endothelial cells stimulated with indicated concentrations of CTSL taken 24 h after initial exposure, ×5 magnification. i BrdU incorporation assay to quantify DNA synthesis upon 24 h exposure to various concentrations of purified CTSL. j Measurement of the effect of KGP94 on CTSL stimulated endothelial cell proliferation. Results are mean ± s.e.m; Student’s t test
Newly formed vascular sprouts elongate, branch out and connect with neighboring sprouts to form an extensive capillary network. This step of the angiogenesis process can be evaluated in vitro using the tube forming assay [38]. In the presence of extracellular matrix such as Matrigel, endothelial cells form a capillary like network that resembles the capillary bed in vivo (Fig. 3e, f; Supplementary Fig. 2a, b). Endothelial cells that were incubated with either purified CTSL or tumor conditioned media showed a striking increase in their tube forming capacity. Both pharmacological and genetic ablation of CTSL led to a dramatic decrease in the extent of tube formation.
Increased endothelial cell proliferation comprises an important pro-angiogenic response. Thus, endothelial cells were exposed to various concentrations of purified CTSL and their proliferation was assessed over a period of 96 h (Fig. 3g, h; Supplementary Fig. 2c). Endothelial cells that were incubated in the presence of CTSL showed a marked increase in viability in a dose dependent fashion. In order to confirm that the increase in viability was indeed due to increased cell proliferation, we quantified the extent of DNA synthesis. Endothelial cells incubated in the presence of CTSL showed a significant increase in bromo-deoxyuridine uptake indicating an increase in DNA synthesis and thus, cell proliferation (Fig. 3i). These results collectively suggest that CTSL facilitates various aspects of the angiogenic process including endothelial cell sprouting, migration and invasion through extracellular matrix, capillary like tube formation and proliferation.
CTSL promotes expression of cell cycle genes in endothelial cells
Transcriptome profiling of HMVEC-L cells exposed to purified CTSL revealed significant alteration in the gene expression pattern of cells exposed to purified CTSL (upregulated—1636; downregulated—1323 genes). The heat map of significantly altered genes showed a strong correlation between HMVEC-L cell samples (Fig. 4a). Gene set enrichment analysis of the 2959 genes that were differentially expressed in response to CTSL stimulation, revealed strong activation of cell cycle processes (Fig. 4b–d).
Fig. 4.
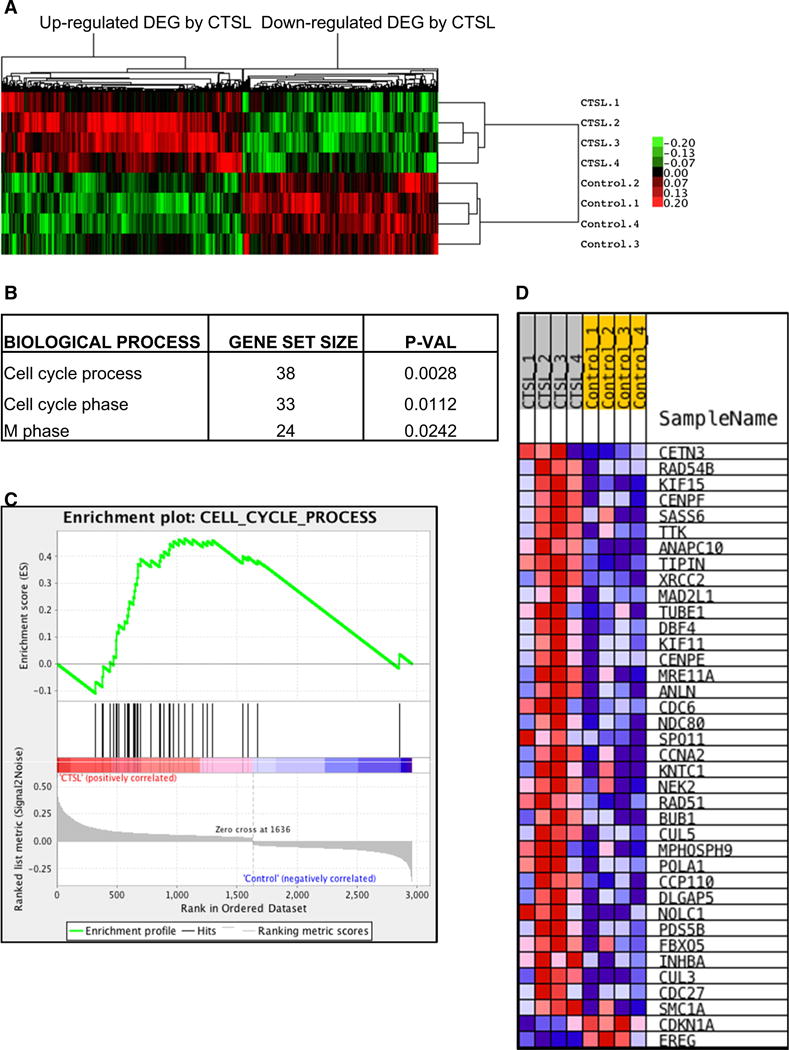
CTSL promotes expression of cell cycle genes. a Heat map of differentially expressed genes, based on microarray analysis of endothelial cells stimulated with or without purified CTSL; n = 4. b Main enriched gene sets sharing same biological process activated in response to CTSL stimulation of endothelial cells. c and d, Enrichment plot and heat map for the gene set related with cell cycle process
Since several enriched biological processes were related to cell cycle, a leading edge analysis was performed (Table 1). Relatedness between common genes in those leading edges was confirmed by inferring a network, visualized with Cytoscape (Fig. 5a). Quantitative PCR validation of select common genes from those leading edges showed significant upregulation of mitotic genes such as centrosome associated protein E (CENPE), kinesin like protein 15 (KIF15) and cyclin A (Fig. 5b–d). Increased protein levels of cyclin D and cyclins involved in G2 to mitotic phase transition such as cyclins A and B further confirmed stimulation of cell cycle in response to CTSL exposure (Fig. 5e).
Table 1.
Common genes in the cell cycle leading edges
| Symbol | Gene name |
|---|---|
| RAD54B | NA |
| KIF15 | Kinesin family member 15 |
| CENPF | Centromere protein F, 350/400 kDa (mitosin) |
| TTK | TTK protein kinase |
| ANAPC10 | Anaphase promoting complex subunit 10 |
| TIPIN | TIMELESS interacting protein |
| XRCC2 | X-ray repair complementing defective repair in Chinese hamster cells 2 |
| MAD2L1 | MAD2 mitotic arrest deficient-like 1 (yeast) |
| DBF4 | DBF4 homolog (S. cerevisiae) |
| KIF11 | Kinesin family member 11 |
| CENPE | Centromere protein E, 312 kDa |
| MRE11A | MRE11 meiotic recombination 11 homolog A (S. cerevisiae) |
| ANLN | Anillin, actin binding protein |
| CDC6 | Cell division cycle 6 homolog (S. cerevisiae) |
| NDC80 | NDC80 kinetochore complex component homolog (S. cerevisiae) |
| SPO11 | SPO11 meiotic protein covalently bound to DSB homolog (S. cerevisiae) |
| CCNA2 | Cyclin A2 |
| KNTC1 | Kinetochore associated 1 |
| NEK2 | NIMA (never in mitosis gene a)-related kinase 2 |
| RAD51 | RAD51 homolog (S. cerevisiae) |
| BUB1 | Budding uninhibited by benzimidazoles 1 homolog (yeast) |
| CUL5 | Cullin 5 |
| MPHOSPH9 | M-phase phosphoprotein 9 |
| POLA1 | Polymerase (DNA directed), alpha 1, catalytic subunit |
Fig. 5.
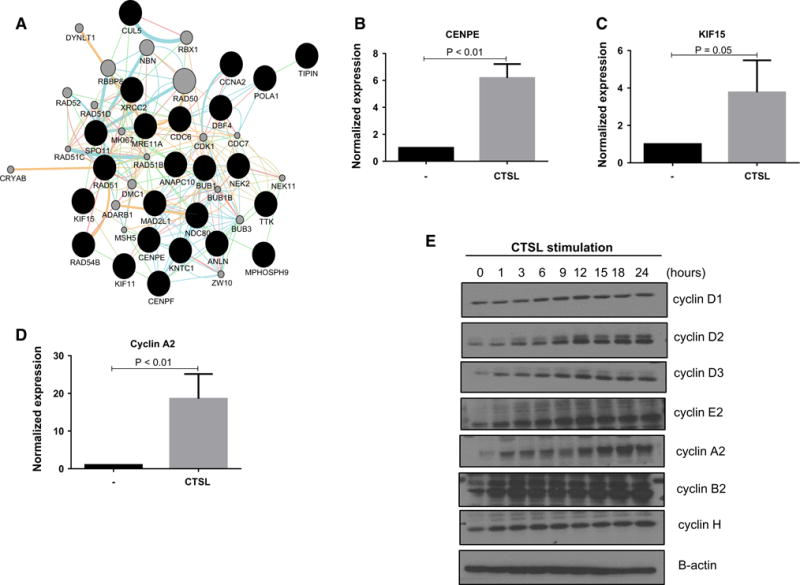
a Cytoscape analysis of common genes in the leading edges of biological processes related to cell cycle. Black nodes represent genes input by the user, grey nodes are genes inferred by the GeneMania plugin. Edge color indicates the type of interaction between two nodes: Red edges protein–protein interaction; green edges genetic interaction; light blue edges same reaction within a pathway; brown edges shared protein domain; orange edges predicted to have functional relationship. The thicker the edge between two nodes, the higher the Pearson’s correlation coefficient across multiple conditions in an experiment. b–d Gene expression analysis of select genes from a upon stimulation with 10 ng/mL purified CTSL. e Western blot analysis of cyclin expression in response to CTSL stimulation of HMVEC-L cells
Discussion
During the course of tumor progression, expression profiles of several genes are altered significantly. While some of these alterations are key to malignant progression, a majority of them represent bystander effect and thus do not contribute to tumorigenicity. Hence, we first evaluated the relationship between CTSL expression status and survival and metastatic incidence in breast cancer patients. The strong association between CTSL upregulation and disease relapse, metastatic incidence and overall survival (Fig. 1) suggested that CTSL over-expression is not a bystander effect but a key factor driving breast cancer progression and metastatic aggressiveness. While several genetic and pharmacological CTSL intervention approaches have demonstrated that CTSL targeting holds significant anti-metastatic potential, it remains unknown whether CTSL targeting would also yield anti-tumor effects in a breast cancer setting. Although the role of CTSL in tumor angiogenesis remains less explored, several clinical and experimental findings are strongly suggestive of its involvement in the angiogenic process. In patients with coronary heart diseases, the formation of collateral coronary vessels as an alternative source of blood supply aids the recovery of the heart from ischemic insult. In these patients, plasma CTSL level serves as an important biomarker for rich collateral vessel formation [39]. CTSL is also highly expressed in human abdominal aortic aneurysm lesions and gene knockout studies have revealed that CTSL plays an important role in aneurysm development by promoting pathological angiogenesis, inflammatory cells recruitment, and aortic wall matrix degradation [40]. Further, key proangiogenic factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) have been shown to induce CTSL expression. Keerthivasan et al. identified a 47 base pair VEGF responsive element in CTSL promoter region and demonstrated that VEGF stimulation induces CTSL transcription in glioblastoma cells [41]. A strong positive correlation between VEGF and CTSL expression status has been reported in adult chronic myeloid leukemia and pediatric acute myeloid leukemia patients [42, 43]. In fact, CTSL upregulation was associated with inferior event free and overall survival of these AML patients [42]. Similarly, bFGF has been reported to promote cysteine cathepsin expression in various pathological conditions involving angiogenesis such as intraocular angiogenesis and ischemic diseases [44, 45]. Development and progression of glomerulosclerosis is characterized by podocyte detachment from the glomerular basement membrane. Both bFGF and PDGF have been shown to promote podocyte secretion of CTSL to facilitate degradation of glomerular basement membrane [46].
Angiogenesis is a complex and dynamic process that progresses through several tightly controlled events including vascular sprouting, endothelial cell migration and invasion through the extracellular matrix, tube formation and proliferation. Quiescent endothelial cells are held together through tight homotypic vascular E-cadherin interactions. However, in the presence of proangiogenic factors, endothelial cells dissolve their VE-cadherin contacts to form new vascular sprouts in the direction of the pro-angiogenic stimulus [47]. Certain proteases have been shown to facilitate sprouting by degrading the obstructive extracellular matrix as these sprouts penetrate into the interstitium to form new vessels [37]. In agreement with these observations, endothelial cells exhibited a dramatic increase in their sprouting and tube forming capacity in the presence of purified or tumor derived CTSL (Fig. 3d–f). Once activated, endothelial cells depend on proteases to degrade the basement membrane and extracellular matrix as they migrate and invade through the interstitium. Our present study shows that both purified and tumor derived CTSL results in a significant enhancement of endothelial cell migration and invasion (Fig. 3a–c). Conversely, CTSL ablation using KGP94 or knockdown approach resulted in a significant impairment of tumor angiogenic properties such as endothelial cell sprouting, invasion, tube formation and proliferation. These results are in agreement with observations made in previous melanoma studies in which intra-tumoral gene delivery of single chain variable fragment CTSL neutralizing antibody resulted in a marked reduction in tumorigenicity, growth and angiogenesis of human melanoma xenografts [48]. In contrast to these findings, CTSL deficiency had no effect on microvascular density in different mouse models of pancreatic endocrine tumors [21]. Similarly, contrary to its pro-tumorigenic function in various tumor types including prostate, breast, pancreatic, ovarian, gastric and colorectal cancers, CTSL has been shown to suppress tumor progression in mouse epidermis [8, 49]. These discrepancies in certain models suggest that its tumorigenic function may vary with certain tumor types.
In addition to angiogenesis, CTSL has also been implicated to participate in alternative mechanisms of vascularization such as vasculogenesis. During the process of vasculogenesis, bone marrow derived endothelial progenitor cells are recruited to the hypoxic tissue to form denovo blood vessels. Gene expression analysis of endothelial progenitor cells in ischemic disease models revealed that their pro-angiogenic effects are primarily mediated by CTSL [50]. CTSL was critical for the integration of circulating endothelial progenitor cells into hypoxic tissues and both pharmacological and genetic ablation of CTSL impaired neovascularization of ischemic tissues.
Previous studies with CTSL and matrix metalloprotease (MMP) inhibitors have shown that inactivation of CTSL and MMP proteolytic function severely impairs the extracellular matrix degradative capacity of endothelial cells [51, 52]. CTSL mediated augmentation of endothelial cell sprouting, tube formation and invasion through collagen or matrigel may therefore be attributed to its proteolytic effects on extracellular matrix components. While our studies clearly demonstrate upregulation of cell cycle genes in response to CTSL (Figs. 4 and 5), the mechanism behind CTSL stimulated endothelial cell proliferation remains obscure. A recent study in ischemic disease model may perhaps shed some light on this unanticipated CTSL function. Proteomic analysis identified CTSL to be the most significant mediator of bFGF stimulated angiogenesis [45]. bFGF over-expression in skeletal muscle cells enhanced CTSL secretion which in turn triggered endothelial cell migration by activating JNK signaling pathway. MMPs have also been reported to activate transforming growth factor β pathway and epidermal growth factor receptor signaling to promote tumor angiogenesis [53, 54]. CTSL stimulated endothelial cell proliferation could thus potentially be a downstream effect of activation of cell signaling pathways.
While peptide based CTSL inhibitor NSITC demonstrated similar anti-angiogenic effects, it is cell permeable and exerts serious cytotoxic effects on endothelial cells [52]. On the contrary, due to its cell impermeable nature KGP94 may exert its anti-angiogenic property by targeting tumor secreted CTSL without having any deleterious side effects on normal lysosomal CTSL function.
In conclusion, CTSL upregulation is associated with poor clinical outcomes of breast cancer patients. Our study has demonstrated that CTSL plays a key role in tumor angiogenesis by activating various angiogenesis associated endothelial cell functions such as migration, invasion, sprouting, tube formation and proliferation. Treatment with KGP94, a small molecule CTSL inhibitor previously shown to impair tumor cell metastasis, led to a significant suppression of CTSL stimulated angiogenic properties of endothelial cells and also inhibited tumor angiogenesis in vivo. While there exists a concern that impairment of angiogenesis might increase metastasis [55] based on the possibility that such anti-angiogenic agent therapy may induce tumor hypoxia, a known enhancer of tumor cell dissemination [56, 57], the generality of this conclusion is controversial in light of studies showing that the impact of such therapy on the tumor microenvironment is highly variable, leading to improvements, reductions, or no change in tumor oxygenation (reviewed in [58, 59]). Taken together the present results demonstrate that targeting CTSL to suppress both the invasive and pro-angiogenic functions of tumor cells could be of significant benefit in the treatment of metastatic cancer patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Dr. Kevin Pinney of Baylor University and OXiGENE Inc. for providing KGP94. These studies were supported in part by a grant from the National Cancer Institute (US Public Health Service Grant R01 CA169300).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10585-016-9790-1) contains supplementary material, which is available to authorized users.
Conflicts of interest The authors have no conflict of interest to disclose.
References
- 1.Siegel R, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat Rev. 2011;37(1):63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weidner N, et al. Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N Engl J Med. 1991;324(1):1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 5.Burden RE, et al. Antibody-mediated inhibition of cathepsin S blocks colorectal tumor invasion and angiogenesis. Clin Cancer Res. 2009;15(19):6042–6051. doi: 10.1158/1078-0432.CCR-09-1262. [DOI] [PubMed] [Google Scholar]
- 6.Lakka SS, et al. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene. 2004;23(27):4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- 7.Chauhan SS, Goldstein LJ, Gottesman MM. Expression of cathepsin L in human tumors. Cancer Res. 1991;51(5):1478–1481. [PubMed] [Google Scholar]
- 8.Sudhan DR, Siemann DW. Cathepsin L targeting in cancer treatment. Pharmacol Ther. 2015;155:105–116. doi: 10.1016/j.pharmthera.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman HA, Riese RJ, Shi GP. Emerging roles for cysteine proteases in human biology. Annu Rev Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- 10.Gottesman MM, Sobel ME. Tumor promoters and Kirsten sarcoma virus increase synthesis of a secreted glycoprotein by regulating levels of translatable mRNA. Cell. 1980;19(2):449–455. doi: 10.1016/0092-8674(80)90519-x. [DOI] [PubMed] [Google Scholar]
- 11.Rabin MS, Doherty PJ, Gottesman MM. The tumor promoter phorbol 12-myristate 13-acetate induces a program of altered gene expression similar to that induced by platelet-derived growth factor and transforming oncogenes. Proc Natl Acad Sci USA. 1986;83(2):357–360. doi: 10.1073/pnas.83.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denhardt DT, et al. Close relationship of the major excreted protein of transformed murine fibroblasts to thiol-dependent cathepsins. Cancer Res. 1986;46(9):4590–4593. [PubMed] [Google Scholar]
- 13.Jordans S, et al. Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological pH and redox conditions. BMC Biochem. 2009;10:23. doi: 10.1186/1471-2091-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novinec M, Lenarcic B, Turk B. Cysteine cathepsin activity regulation by glycosaminoglycans. Biomed Res Int. 2014;2014:309718. doi: 10.1155/2014/309718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everts V, et al. Osteoclastic bone degradation and the role of different cysteine proteinases and matrix metalloproteinases: differences between calvaria and long bone. J Bone Miner Res. 2006;21(9):1399–1408. doi: 10.1359/jbmr.060614. [DOI] [PubMed] [Google Scholar]
- 16.Goretzki L, et al. Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett. 1992;297(1–2):112–118. doi: 10.1016/0014-5793(92)80339-i. [DOI] [PubMed] [Google Scholar]
- 17.Ishidoh K, Kominami E. Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem Biophys Res Commun. 1995;217(2):624–631. doi: 10.1006/bbrc.1995.2820. [DOI] [PubMed] [Google Scholar]
- 18.Laurent-Matha V, et al. Processing of human cathepsin D is independent of its catalytic function and auto-activation: involvement of cathepsins L and B. J Biochem. 2006;139(3):363–371. doi: 10.1093/jb/mvj037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason RW, et al. Elastinolytic activity of human cathepsin L. Biochem J. 1986;233(3):925–927. doi: 10.1042/bj2330925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frade R, et al. Procathepsin-L, a proteinase that cleaves human C3 (the third component of complement), confers high tumorigenic and metastatic properties to human melanoma cells. Cancer Res. 1998;58(13):2733–2736. [PubMed] [Google Scholar]
- 21.Gocheva V, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20(5):543–556. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavarria GE, et al. Initial evaluation of the antitumour activity of KGP94, a functionalized benzophenone thiosemicarbazone inhibitor of cathepsin L. Eur J Med Chem. 2012;58:568–572. doi: 10.1016/j.ejmech.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 23.Sudhan DR, Siemann DW. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin Exp Metastasis. 2013;30(7):891–902. doi: 10.1007/s10585-013-9590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sudhan DR, et al. Cathepsin L inactivation leads to multimodal inhibition of prostate cancer cell dissemination in a preclinical bone metastasis model. Int J Cancer. 2016;138:2665–2677. doi: 10.1002/ijc.29992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar GD, et al. Functionalized benzophenone, thiophene, pyridine, and fluorene thiosemicarbazone derivatives as inhibitors of cathepsin L. Bioorg Med Chem Lett. 2010;20(22):6610–6615. doi: 10.1016/j.bmcl.2010.09.026. [DOI] [PubMed] [Google Scholar]
- 26.Gyorffy B, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 27.Siemann DW, et al. Impact of tumor cell VEGF expression on the in vivo efficacy of vandetanib (ZACTIMA; ZD6474) Anticancer Res. 2009;29(6):1987–1992. [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth GK. Limma: linear models for microarray data. 397–420. Springer; New York: 2005. [Google Scholar]
- 29.de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinform Appl Notes. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montojo J, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26(22):2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishida Y, et al. Increased cathepsin L levels in serum in some patients with ovarian cancer: comparison with CA125 and CA72-4. Gynecol Oncol. 1995;56(3):357–361. doi: 10.1006/gyno.1995.1063. [DOI] [PubMed] [Google Scholar]
- 34.Siewinski M, et al. Determination of cysteine peptidases-like activity and their inhibitors in the serum of patients with ovarian cancer treated by conventional chemotherapy and vitamin E. J Exp Ther Oncol. 2004;4(3):189–193. [PubMed] [Google Scholar]
- 35.Svatek RS, et al. Role of urinary cathepsin B and L in the detection of bladder urothelial cell carcinoma. J Urol. 2008;179(2):478–484. doi: 10.1016/j.juro.2007.09.037. discussion 84. [DOI] [PubMed] [Google Scholar]
- 36.Tumminello FM, et al. Cathepsin D, B and L circulating levels as prognostic markers of malignant progression. Anticancer Res. 1996;16(4B):2315–2319. [PubMed] [Google Scholar]
- 37.van Hinsbergh VW, Koolwijk P. Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc Res. 2008;78(2):203–212. doi: 10.1093/cvr/cvm102. [DOI] [PubMed] [Google Scholar]
- 38.Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628–635. doi: 10.1038/nprot.2010.6. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, et al. Plasma cathepsin L and its related pro/antiangiogenic factors play useful roles in predicting rich coronary collaterals in patients with coronary heart disease. J Int Med Res. 2010;38(4):1389–1403. doi: 10.1177/147323001003800421. [DOI] [PubMed] [Google Scholar]
- 40.Sun J, et al. Cathepsin L activity is essential to elastase perfusion-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2500–2508. doi: 10.1161/ATVBAHA.111.230201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keerthivasan S, et al. Transcriptional upregulation of human cathepsin L by VEGF in glioblastoma cells. Gene. 2007;399(2):129–136. doi: 10.1016/j.gene.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 42.Jain M, et al. Cathepsins B and L in peripheral blood mononuclear cells of pediatric acute myeloid leukemia: potential poor prognostic markers. Ann Hematol. 2010;89(12):1223–1232. doi: 10.1007/s00277-010-1012-3. [DOI] [PubMed] [Google Scholar]
- 43.Samaiya M, et al. Epigenetic regulation of cathepsin L expression in chronic myeloid leukaemia. J Cell Mol Med. 2011;15(10):2189–2199. doi: 10.1111/j.1582-4934.2010.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamada Y, et al. Involvement of cysteine proteases in bFGF-induced angiogenesis in guinea pig and rat cornea. J Ocul Pharmacol Ther. 2000;16(3):271–283. doi: 10.1089/jop.2000.16.271. [DOI] [PubMed] [Google Scholar]
- 45.Chung JH, et al. Cathepsin L derived from skeletal muscle cells transfected with bFGF promotes endothelial cell migration. Exp Mol Med. 2011;43(4):179–188. doi: 10.3858/emm.2011.43.4.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asanuma K, et al. Selective modulation of the secretion of proteinases and their inhibitors by growth factors in cultured differentiated podocytes. Kidney Int. 2002;62(3):822–831. doi: 10.1046/j.1523-1755.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- 47.Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol. 2010;22(5):617–625. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 48.Frade R, Rousselet N, Jean D. Intratumoral gene delivery of anti-cathepsin L single-chain variable fragment by lentiviral vector inhibits tumor progression induced by human melanoma cells. Cancer Gene Ther. 2008;15(9):591–604. doi: 10.1038/cgt.2008.51. [DOI] [PubMed] [Google Scholar]
- 49.Dennemarker J, et al. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene. 2010;29(11):1611–1621. doi: 10.1038/onc.2009.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urbich C, et al. Cathepsin L is required for endothelial progenitor cell-induced neovascularization. Nat Med. 2005;11(2):206–213. doi: 10.1038/nm1182. [DOI] [PubMed] [Google Scholar]
- 51.Handsley MM, Edwards DR. Metalloproteinases and their inhibitors in tumor angiogenesis. Int J Cancer. 2005;115(6):849–860. doi: 10.1002/ijc.20945. [DOI] [PubMed] [Google Scholar]
- 52.Rebbaa A, et al. The anti-angiogenic activity of NSITC, a specific cathepsin L inhibitor. Anticancer Res. 2009;29(11):4473–4481. [PubMed] [Google Scholar]
- 53.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–176. [PMC free article] [PubMed] [Google Scholar]
- 54.Yu WH, et al. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev. 2002;16(3):307–323. doi: 10.1101/gad.925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paez-Ribes M, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cairns RA, Hill RP. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 2004;64(6):2054–2061. doi: 10.1158/0008-5472.can-03-3196. [DOI] [PubMed] [Google Scholar]
- 57.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129(3):465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006;66(24):11520–11539. doi: 10.1158/0008-5472.CAN-06-2848. [DOI] [PubMed] [Google Scholar]
- 59.Siemann DW, Horsman MR. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol Ther. 2015;153:107–124. doi: 10.1016/j.pharmthera.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.