

Abstract
In Alzheimer disease (AD), the accumulation of amyloid beta (Aβ) begins decades before cognitive symptoms and progresses from intraneuronal material to extracellular plaques. To date, however, the precise mechanism by which the early buildup of Aβ peptides leads to cognitive dysfunction remains unknown. Here, we investigate the impact of the early Aβ accumulation on temporal and frontal lobe dysfunction. We compared the performance of McGill-R-Thy1-APP transgenic AD rats with wild-type littermate controls on a visual discrimination task using a touchscreen operant platform. Subsequently, we conducted studies to establish the biochemical and molecular basis for the behavioral alterations. It was found that the presence of intraneuronal Aβ caused a severe associative learning deficit in the AD rats. This coincided with reduced nuclear translocation and genomic occupancy of the CREB co-activator, CRTC1, and decreased production of synaptic plasticity-associated transcripts Arc , c-fos , Egr1 , and Bdnf . Thus, blockade of CRTC1-dependent gene expression in the early, preplaque phase of AD-like pathology provides a molecular basis for the cognitive deficits that figure so prominently in early AD.
Keywords: Alzheimer disease, amyloid beta, CRTC1, learning and memory, touchscreen operant platform
Introduction
Alzheimer disease (AD) is the leading cause of dementia worldwide and is characterized by a progressive decline in cognitive function ( Selkoe 2001 ). A consistent pathological feature of this neurodegenerative disease is the accumulation of extracellular amyloid beta (Aβ) plaques in brain regions important for learning and memory. Emerging evidence from transgenic animal models and human patients indicates that Aβ also accumulates intraneuronally and contributes to disease progression ( LaFerla et al. 2007 ; Cuello et al. 2012 ).
In this regard, the progressive and predictable evolution of pathology in transgenic animal models facilitates the study of early AD. Previously, we have shown that McGill-R-Thy1-APP transgenic rats display full AD-like amyloid pathology, with appearance of Aβ plaques matching progression of plaque appearance in human patients ( Leon et al. 2010 ). These rats develop the first Aβ plaques at 6–9 months of age in the subiculum, with subsequent plaques appearing in the hippocampus and entorhinal cortex. As is the case with AD patients, these rats show an intraneuronal Aβ accumulation of the toxic oligomeric soluble form occurring in the hippocampus and neocortex, which precedes plaque formation ( Iulita et al. 2014 ). This early accumulation of Aβ alters synaptic plasticity through persistent inhibition of long-term potentiation in the CA1 area of the hippocampus ( Qi et al. 2014 ).
While memory deficits are a consistent feature of AD, they overlap with cognitive impairments associated with frontal dysfunction, which can be difficult to differentiate in humans, especially in the early phases of the disease. Since the rat brain matures at a rate similar to humans ( Whishaw et al. 2001 ) and because rats perform well in tests of prefrontal-executive function ( Chudasama and Robbins 2006 ; Bussey et al. 2012 ), the McGill transgenic rat model provides a unique opportunity to identify the precise molecular mechanisms associated with prefrontal cognitive decline. Using an automated touchscreen platform, we examined performance of transgenic AD rats on a complex task that assessed visual discrimination, associative learning, and behavioral control ( Schoenbaum et al. 2002 ; Chudasama and Robbins 2003 ). Similar touchscreen platforms are used in identifying cognitive decline in clinical AD patients ( Blackwell et al. 2004 ). Here, we show that transgenic AD rats were severely impaired in learning the visual stimulus–reward association, as they committed many errors and showed long reaction times. Importantly, this impairment was evident during the early stages of the disease, when the Aβ is predominantly intraneuronal.
Changes in gene expression affect cognition during pathological aging and, in AD, altered cAMP/Ca 2+ response element-binding protein (CREB)-mediated transcription is associated with memory decline ( Pugazhenthi et al. 2011 ). Gene transcription mediated by CREB requires the CREB-regulated transcription coactivator 1 (CRTC1) ( Conkright et al. 2003 ; Iourgenko et al. 2003 ), which is widely expressed in the hippocampus ( Zhou et al. 2006 ; Watts et al. 2011 ) where it contributes to control of gene expression necessary for memory ( Sekeres et al. 2012 ). In this study, we show that transgenic AD rats experience a blockade in nuclear CRTC1 translocation in the hippocampus and a related reduction in the expression of memory function gene transcripts including Arc , c-fos , Egr1 , and Bdnf . Together, these results expand our understanding as to the manner in which intraneuronal Aβ accumulation contributes to disease progression in the early stages of the AD-like amyloid pathology, before the appearance of Aβ plaques.
Materials and Methods
Animals
McGill-R-Thy1-APP transgenic rats express the Swedish double and Indiana genetic mutations in the human amyloid precursor protein gene, hAβPP (Fig. 1A ). Male and female McGill-R-Thy1-APP transgenic rats homozygous for the mutated hAβPP transgene and their wild-type littermates were obtained from our animal colony. The animals were 4 months of age at the beginning of the experiments and were sacrificed before the appearance of amyloid plaques in the transgenic animals, at 6 months of age. Animals were housed in humidity and temperature controlled rooms under 12-hour light cycle and had access to water ad libitum . Food pellets were restricted to maintain animals at ∼90% of their free-food weight during behavioral testing. All experiments were carried out with approval from institutional Animal Care Committee and under strict adherence to the guidelines set out by the Canadian Council on Animal Care.
Figure 1.

The McGill-R-Thy1-APP rat transgenic model of the Alzheimer amyloid pathology exhibits a preplaque phase preceding the appearance of extracellular plaques where Aβ is primarily accumulated intraneuronally in the cerebral cortex and hippocampus. ( A ) DNA construct used for expression of transgenic human APP with Swedish double (Swe**) and Indiana (Ind*) mutations. Thy1: the neuron-specific murine thy1.2 promoter. ( B ) Expression of Aβ peptides following transgenesis. Aβ-burdened neurons (arrows, lower left panel) were present in the hippocampus and cerebral cortex (CC) of 6-month-old Alzheimer transgenic rats. Extracellular Aβ plaques were widespread across the hippocampus and in the cerebral cortex in 15-month-old Alzheimer transgenic rats. Higher magnification micrographs illustrate Aβ-burdened neurons of lamina V of the cerebral cortex at the preplaque stages and the occurrence of Aβ-immunoreactive plaques at the postplaque stages, respectively.
Touchscreen Operant Platform
Behavior testing was conducted in automated, operant touchscreen chambers (Lafayette Instruments, IN, USA). Each chamber was equipped with: 1) a houselight, 2) a food magazine fitted with a light-emitting diode and photocells to detect food collection entries, 3) a pellet dispenser that delivered 45-mg dustless precision sucrose pellets (Ren's Pet Depot, Ontario, Canada) and 4) a 12″ × 12″ touch sensitive monitor (Elo Touch Solutions, USA). Two computer graphic stimuli were presented on the left and right side of the touchscreen (Fig. 2A,B ). A black Plexiglas mask was attached to the front of the screen ∼0.6″ from the surface of the display to restrict the rats' access to the visual stimuli through a left and right response window (2.05″ × 2.05″). The apparatus and online data collection for each chamber were controlled using the Whisker control system ( Cardinal and Aitken 2010 ).
Figure 2.

Alzheimer transgenic rats at the preplaque stage were severely impaired in associative learning. ( A ) Photograph showing touchscreen operant platform and rat making nose-poke touch response to visual stimulus. ( B ) Schematic illustration of stimulus pair used in the visual discrimination task and the reward contingencies. ( C, D ) Individual learning curves for wild-type and Alzheimer transgenic rats. The blue line represents criterion performance at 70% accuracy on noncorrection trials over 2 consecutive training sessions. ( E ) Proportion of total correction and noncorrection trials for the first 35 sessions only. Wild-type rats commit more noncorrection trials and fewer correction trials with time, whereas the Alzheimer rats fail to show such improvement. ( F ) Preplaque Alzheimer transgenic rats made many noncorrection trial errors and ( G ), correction trial errors, while learning the stimulus–reward association and never reached criterion performance, indicating that they never learned the stimulus–reward association. ( H ) Alzheimer transgenic rats were very slow in making a response. ( I ) The Alzheimer transgenic rats were also slow in collecting food rewards when they made a correct response. Data represent mean ± SEM. *** P < 0.001.
Behavioral Procedure
Pretraining
Rats were first habituated to the apparatus and then trained to make a nose-poke touch response to a white square (2″ × 2″) that was presented on the left or right side of the screen. A nose-poke touch response to the white square was rewarded with a single sucrose pellet. Animals transitioned to the acquisition phase of the visual discrimination task when their performance reached 50 reward pellets delivered within a 20-min session.
Visual Discrimination Task
Each session began with the illumination of the house light and the food magazine light. After a 5-s intertrial interval, the rat initiated the trial by making a food magazine entry. This resulted in simultaneous presentation of 2 stimuli on the screen; one was associated with a sucrose pellet (the A+ stimulus) and the second was not (the B− stimulus). The rewarded stimulus was counterbalanced across subjects. The same pair of stimuli was presented on every trial, and the left/right positions of the stimuli (i.e., which stimulus was on the left and which was on the right) were determined pseudorandomly. The rat was required to make a nose-poke touch response to one of the stimuli. The stimuli remained on the screen until the rat made a nose-poke touch response to either stimulus. A correct response to A+ was followed by the disappearance of the stimuli and the delivery of a sucrose pellet concomitant with illumination of the food magazine. The next trial was initiated by a food magazine entry after a 5-s intertrial interval. An incorrect response to B− resulted in the disappearance of the stimuli from the screen and a 5-s timeout period during which all of the lights were extinguished. Consequently, rats were presented with correction trials such that after an incorrect response, the same stimulus configuration (i.e., the A+ and B− stimuli remained in the same left/right positions) was presented over successive trials until the rat responded correctly. Each session comprised a maximum of 60 noncorrection trials, with up to an infinite number of correction trials. Rats were given up to 1 h to complete the session. Rats were required to learn to respond to the correct, reinforced stimulus to an average criterion of 70% accuracy for 2 consecutive days, for noncorrection trials only. Rats were given a total of 50 sessions to acquire the visual discrimination task.
Locomotor Activity
After behavioral testing, locomotor activity was assessed using 4 standard home cage activity frames. Each home cage was a clear polycarbonate tub (61 cm wide × 37 cm long × 20.5 cm high) lined with sawdust and covered with a barrier filter lid (Ancare, NY, USA). Each home cage was placed within a Cage Rack SmartFrame™ (58 cm wide × 60.33 cm long × 2.11 cm high) equipped with infrared photobeams located on the interior perimeter of the frame (Lafayette Instruments). The rat was placed in the activity cage for 2 h. The total number of horizontal beam breaks was recorded using MotorMonitor™ software, version 5.05 and transmitted to a Dell Optiplex 745 computer.
Immunoenzymatic Reactions
Preparation of tissue was as previously described ( Hanzel et al. 2014 ). To reveal amyloid neuropathology, tissue sections were incubated overnight in anti-McSA1 (MediMabs, QC, Canada), a mouse monoclonal antibody raised against synthetic peptide of Aβ corresponding to amino acids 1–12 of human APP ( Grant et al. 2000 ). Tissue sections were then incubated in goat anti-mouse antibody (MP Biochemicals), followed by a mouse anti-peroxidase monoclonal antibody complex (MAP/HRP complex, MediMabs) and developed using DAB as the chromogen (Vector Laboratories, Inc.). Sections were then processed through a graded alcohol series, defatted and cleared in xylenes, and coverslipped using Permount mounting medium (Fisher Scientific). Aβ-immunostained sections were imaged using a Zeiss Microimaging desk scanner (Zeiss Microimaging).
Immunofluorescence
Briefly, sections were blocked with 10% normal serum and incubated at 4°C for 48 h in a PBST with 5% serum and rabbit monoclonal anti-CRTC1 (Cell Signaling Technology, 2587) antibody solution. Sections were washed and incubated with cross-preabsorbed secondary antibodies, which minimizes the potential for cross-reactivity, conjugated to Alexa 594 (Jackson Immunoresearch Laboratories, Inc., 112-585-167). Finally, sections were washed with PBS, counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and coverslipped with Aqua Polymount (Polysciences).
Image Analysis
For CRTC1 nuclear translocation analysis, 3 distinct cell populations from the anterior aspect of the dorsal hippocampus were sampled: 1) the granule cell layer of the lateral blade of the dentate gyrus (DG), 2) the superficial pyramidal layer of CA1, and 3) the superficial pyramidal layer of CA3. Five regions on up to 3 sections were sampled under guidance of the DAPI channel for each CA1, CA3, and DG using a Zeiss LSM 510 confocal microscope (Carl Zeiss Canada). Confocal images were saved with 16-bit depth and were processed using NIH ImageJ software (National Institutes of Health, USA). For the analysis of signals, an ad hoc, automated macro using NIH ImageJ software was applied. Five fields spanning 73.12 × 73.12 µm centered over the appropriate cellular layer were imaged on up to 3 sections per animal. Sampling in this manner yielded counts of ∼275 neurons in CA1, 180 neurons in CA3, and 500 neurons in the DG for each animal. Nuclear CRTC1 was defined as that signal which overlapped with the DAPI staining for nuclear heterochromatin. The optical slice of the confocal microscopy was <0.9 µm, giving sufficient resolution to show overlapping signals. The macro was programmed to automatically select the DAPI images and apply background removal, Gaussian blur, thresholding according to mean filter, and finally watershedding to separate touching nuclei. This macro was programmed to then automatically define the extent of the DAPI-labeled nuclei and to use those defined regions to measure, in the second channel, CRTC1 signal. Nuclear CRTC1 signal was measured as the mean signal intensity of CRTC1 overlapping DAPI signal, while total CRTC1 was represented as the mean signal intensity of the field.
Subcellular Fractionation
Subcellular fractionation of frozen hippocampal tissue was completed using the Biovision, Inc. Nuclear/Cytosol Fractionation Kit (Cat: K266) according to manufacturers’ instructions. Protein concentrations were determined according to the Lowry method. Normalized loads of each extract were analyzed by western blotting. Fraction purity was confirmed using antibodies toward nuclear-specific histone H3 (Cell Signaling Technology, 9715) and cytosolic cyclophilin A (Abcam, ab41684). Nuclear and cytosolic levels of CRTC1 were revealed using the rabbit monoclonal anti-CRTC1 antibody (Cell Signaling Technology). Primary antibodies were detected using HRP-conjugated goat anti-rabbit secondary antibody (Jackson Immunoresearch Laboratories, Inc., 111-035-144). Membranes were processed according to directions using the Enhanced Chemiluminescent System (GE Healthcare) and were exposed to Hyblot CL films (Denville). Relative integrated optical density was determined using the CLIQS 1D gel analysis software (Totallab Ltd).
Chromatin Immunoprecipitation (ChIP) Assay
Cortical tissue was cross-linked in formaldehyde solution (50 m m HEPES-KOH [pH 7.5], 100 m m NaCl, 1 m m EDTA, 0.5 m m EGTA, 1% formaldehyde, pH 8.0). After quenching the formaldehyde reaction by adding 2.5 m glycine solution, tissue was homogenized in lysis buffer (10 m m Tris–HCl [pH 8.0], 0.1% sodium deoxycholate, 0.5% N-laurylsarcosine), supplemented with Complete™ protease inhibitors (Roche). DNA was sheared to 100–500-bp fragments ( Supplementary Fig. 1 ) using a Bioruptor Plus UCD-300 sonicator (Diagenode). After sonication, 10% Triton X-100 was added to the tissue lysate and the mix was cleared by centrifugation at 15 000× g for 15 min (4°C). Protein concentration was determined using the Bradford assay (BioRad).
Tissue lysate was first precleared by incubation with PureProteome protein A magnetic beads (EMD Millipore). Cleared tissue lysate was then added to PureProteome protein A magnetic beads previously saturated with antibodies, and immunoprecipitation was carried overnight at 4°C. The antibodies used were CREB, CRTC1 rabbit monoclonal antibodies (1:250, Cell Signaling Technology), and anti-rat IgG rabbit polyclonal antibody as a negative control (1:250, Sigma–Aldrich). Beads were washed with RIPA wash buffer (50 m m HEPES–KOH [pH 7.6], 500 m m LiCl, 1 m m EDTA, 1% Nonidet P-40, and 0.7% Na-deoxycholate) followed by one wash with TE buffer supplemented with 50 m m NaCl. DNA–protein complexes were eluted from the beads using elution buffer (50 m m Tris–HCl [pH 8.0], 10 m m EDTA, 1% SDS) and by heating the samples at 65°C for 15 min with frequent vortexing. Contaminating RNAs were digested using RNaseA (Sigma–Aldrich) followed by a proteinase K (Roche) treatment to digest remaining proteins. The immunoprecipitated DNA fragments were purified using the Qiaquick PCR purification kit (Qiagen) and were eluted in 30 μL of 10 m m Tris–HCl (pH 8.0). Quantification of immunoprecipitated DNA was assessed by qPCR with EvaGreen ® (MBI EVOlution EvaGreen qPCR Mix, Montreal Biotech, Inc.) using the Illumina Eco Instrument and Software (Illumina, Inc.). For a list of primers used, see Supplementary Table 1 . ChIP data were normalized to input DNA from each sample.
Gene Expression Analysis
Total RNA was extracted from rat cortex using the RNeasy Mini Kit (Qiagen), following manufacturer instructions. Residual DNA was removed by on-column DNase digestion using the RNase-Free DNase Set (Qiagen). To generate cDNA, total RNA was retro-transcribed using an oligo-dT primer with the Omniscript RT Kit (Qiagen). Quantification of transcript expression was assessed by qRT-PCR with EvaGreen ® (MBI EVOlution EvaGreen qPCR Mix, Montreal Biotech, Inc.) using the Illumina Eco Instrument and Software (Illumina, Inc.). Expression of each gene was normalized to the housekeeping gene for β-actin. For the list of primers used, see Supplementary Table 2 .
Data Analysis
For the behavioral results, data for each variable were subjected to an independent samples t test or repeated-measures ANOVA using SPSS statistical software, version 20 (SPSS, Inc.). Statistical significance was set at P < 0.05. Mann–Whitney nonparametric tests were performed when normal distribution data could not be assumed. The brains of all Alzheimer transgenic and wild-type rats were analyzed to make direct comparisons with CRTC1 expression. Subsequent tissue processing for western blot, ChIP, and qPCR was performed on a randomly selected subset of behaviorally tested animals. This number secured sufficient statistical power and is specified in the results section for each application.
Results
Intraneuronal Aβ Accumulation Precedes Plaque Deposition
McGill-R-Thy1-APP transgenic rats express the human APP gene with Swedish double and Indiana mutations under control of the neuron-specific murine Thy1.2 promoter (Fig. 1A ). These mutations are associated with a rapidly progressing early-onset form of AD. Using a highly specific antibody that targets amino acids 1–12 of human Aβ ( Grant et al. 2000 ), immunohistochemical analysis revealed that extracellular Aβ plaques were present across the hippocampus and cerebral cortex in 15-month-old Alzheimer transgenic rats (Fig. 1B ). Importantly, although extracellular Aβ plaques had yet to deposit, the preplaque phase of the Aβ pathology was characterized by heavy burden of intraneuronal Aβ in the hippocampus and neocortex of 6-month-old rats that underwent behavioral testing.
Intraneuronal Aβ Impairs Associative Learning in AD Rats
We tested preplaque transgenic Alzheimer rats and their wild-type controls because we were interested in determining the effect of intraneuronal Aβ on associative learning. Rats were presented with a pair of visual stimuli on a touchscreen (Fig. 2A,B ), and a nose-poke touch response to the correct stimulus was rewarded with a sucrose pellet. For each trial, the left/right position of the correct stimulus was pseudorandom. When the rat made an incorrect response, the same trial with the same left/right stimulus configuration was repeated (i.e., correction trials) until the rat responded correctly. Therefore, correction trial errors indicate the animal was repeating incorrect responses to the same side in successive trials whereas noncorrection trial errors indicate the animal was repeating errors to the same stimulus.
The learning curves for individual wild-type ( n = 5) and Alzheimer transgenic rats ( n = 10) are shown in Figure 2C and D , respectively. Rats were trained until they reached a criterion of 70% accuracy for noncorrection trials on 2 consecutive sessions. Rats were given a maximum of 50 sessions to acquire the task. All wild-type rats quickly learned that only one stimulus was positively associated with reward, and they reached criterion performance in an average of 15 sessions, committing few errors, displaying high motivation and fast decision times in this task. In contrast, only one rat from the Alzheimer transgenic group learned the stimulus–reward relationship after 31 sessions, which was, twice as many sessions as the control wild type. The remaining 9 rats failed to reach criterion, even with 50 sessions of training (mean sessions ± S.E.M.: WT, 14.60 ± 2.42; TG, 48.80 ± 2.04; t13 = 10.141; P < 0.001). We plotted the proportion of total trials within a session that were noncorrection or correction trials to gauge some sense of how the trial types were distributed (Fig. 2E ). This analysis confirmed that the wild-type rats made a higher proportion of noncorrection trials as training progressed while reducing the need for repeat trials to correct their errors. In contrast, the Alzheimer rats performed a lower proportion of noncorrection trials and an almost equivalent proportion of correction trials within a session, reflecting no obvious pattern in learning. In fact, when we look at the error types alone, the Alzheimer rats made many noncorrection trial errors: ( t13 = 5.652; P < 0.001; Figure 2F ) as well as correction errors ( t13 = 4.508; P < 0.001; Figure 2G ) as if they were choosing randomly.
Another notable impairment was in their speed of response. The Alzheimer rats took over 10 s to make their response suggesting that the animals were severely compromised in their capacity to make a choice ( P < 0.001, Mann–Whitney U test; Fig. 2H ), and they were slower than the wild types in collecting their reward ( t13 = 9.191; P < 0.001; Figure 2I ). However, their long latencies cannot be attributed entirely to a motor deficit. First, although the Alzheimer rats were less active than the wild types in their locomotor behavior ( F1,12 = 25.74; P < 0.0001), both groups showed a general decline in activity with time ( F7,84 = 35.40; P < 0.0001) and were no different from each other for the last 45 min of locomotor testing. Second, while Alzheimer rats took over 10 s to make their choice, they took only 3 s to collect their food suggesting they are not slow in all aspects of behavior. Moreover, the rats consumed all their reward pellets indicating that they were not demotivated. Thus, the preplaque transgenic Alzheimer rats had a significant cognitive impairment in this early phase of the amyloid pathology, where Aβ peptides are primarily accumulated intraneuronally.
Intraneuronal Aβ Blocks CRTC1 Nuclear Translocation
To investigate molecular changes associated with the earliest stages of the AD-like amyloid pathology (akin to the human preclinical AD), we analyzed the brains of the young transgenic rats at the preplaque stage after behavioral testing. For instance, it is well accepted that glutamatergic synaptic transmission promotes nuclear translocation of CRTC1 in a calcium- and calcineurin-dependent manner ( Screaton et al. 2004 ; Li et al. 2009 ; Ch'ng et al. 2012 ), leading to a transient stabilization of the CREB transcription complex, and activation of CRE-regulated gene expression ( Bittinger et al. 2004 ). Upon phosphorylation by salt-inducible kinase, CRTC1 is shuttled back to the cytoplasm ( Takemori and Okamoto 2008 ). Therefore, CRTC1-dependent gene expression is tightly regulated by its subcellular localization and Aβ has been shown to negatively affect CRTC1-dependent gene transcription ( Espana et al. 2010 ). This led us to investigate whether the accumulation of intraneuronal Aβ is sufficient to impede CRTC1 nuclear translocation in this rat model.
The brains of wild type ( n = 5) and AD transgenic rats ( n = 10) were immunohistochemically processed (Fig. 3A–C ), and nuclear CRTC1 was defined as that signal which overlapped with DAPI staining for heterochromatin. We found that AD rats showed a significant reduction in nuclear CRTC1 in the dentate gyrus (DG) ( t13 = 2.824; P < 0.01), and in the pyramidal neurons of CA1 ( t13 = 4.572; P < 0.001). Not surprisingly, the most striking nuclear accumulation occurred in CA3 ( t13 = 3.002; P < 0.01), a region known to have the most excitatory connections. A reduction in total CRTC1 expression (cytoplasmic and nuclear) was observed only in area CA1 ( t13 = 1.841; P < 0.05; Figure 3D ) indicating that reduced nuclear CRTC1 was primarily due to reduced cytoplasmic-nuclear translocation, rather than to a global reduction of available CRTC1.
Figure 3.

Confocal imaging reveals reduced nuclear accumulation of CRTC1 in neurons of the hippocampal formation before Aβ plaque deposition. ( A ) Brain diagram showing the hippocampal region and method of confocal image analysis ( B-D ) Immunofluorescent staining revealed CRTC1 expression in CA1, the dentate gyrus (DG), and CA3 regions of the hippocampus in wild-type and Alzheimer transgenic rats. A significant reduction in nuclear CRTC1 was observed in CA1, the DG, and CA3 regions of the hippocampus in Alzheimer transgenic compared with wild-type rats when examined at 6 months of age. Immunofluorescent staining revealed a significant reduction in total CRTC1 in AD transgenic rats only in the CA1 region, indicating that the reduction of nuclear CRTC1 was not due to an overall reduction of available CRTC1. These results indicate that reduced nuclear CRTC1 in neurons across the hippocampal formation is an early event in the Aβ pathology preceding the appearance of amyloid plaques. Data represent the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001. Scale bar A = 5 µm; B–D = 20 µm.
We next prepared cytosolic and nuclear subcellular fractions to confirm reduced nuclear CRTC1 in Alzheimer rats. Hippocampal tissue from a randomly selected subset of behaviorally tested wild-type ( n = 4) and Alzheimer transgenic ( n = 4) rats was used for this analysis. We found that CRTC1 was highly expressed in the cytosolic fraction and to a lesser extent in the nuclear fraction (Fig. 4A ). We found no significant difference in CRTC1 expression in the cytosolic fractions between wild-type and Alzheimer transgenic rats (Fig. 4B ). Consistent with the confocal imaging results (Fig. 3B–D ), nuclear CRTC1 was significantly reduced in Alzheimer transgenic rats (Fig. 4C; t6 = 2.174; P < 0.05). Together, these results indicate that intraneuronal Aβ impaired CRTC1 nuclear translocation at the early stages of AD-like amyloid pathology.
Figure 4.

Subcellular fractionation confirms impaired CRTC1 nuclear translocation in hippocampus at preplaque AD stages. ( A ) CRTC1-immunoreactive bands were detected in cytosolic and nuclear fractions of hippocampal tissue from wild-type and Alzheimer transgenic rats. ( B, C ) While no significant difference in CRTC1 levels was observed in the cytosolic fractions, a significant reduction in nuclear CRTC1 occurred in the Alzheimer transgenic rats. Cyclophilin A and histone H3 were used as cytosolic and nuclear markers, respectively. Data represent the mean ± SEM. * P < 0.05.
Intraneuronal Aβ Accumulation Results in Reduced CRTC1 Genomic Occupancy and Neuroplasticity-Related Gene Expression
Learning involves Hebbian and homeostatic forms of synaptic plasticity that require production of key plasticity-related proteins and is highly dependent on CRTC1 for gene transcription. To test whether CRTC1 promoter occupancy is altered during the early stages of the Aβ pathology, we performed a chromatin co-immunoprecipitation (ChIP) assay. The results revealed decreased binding of CRTC1 to genetic regions examined in the Alzheimer transgenic rats. Figure 5A shows a 2-fold reduction of the immediate early genes Arc ( t8 = 3.790; P < 0.01), c-fos ( t8 = 2.054; P < 0.05), and Egr1 ( t8 = 2.314; P < 0.05) , as well as the growth factor Bdnf ( t8 = 2.354; P < 0.05) in the Alzheimer transgenic rats ( n = 6) compared with wild-type rats ( n = 4). Previous studies have shown that inactive CREB binds to gene promoters and co-factors such as CRTC1 are required for gene expression ( Mayr and Montminy 2001 ; Kornhauser et al. 2002 ; Conkright et al. 2003 ). Accordingly, ChIP analyses did not reveal significant differences in CREB promoter binding between the wild-type and Alzheimer transgenic rats (Fig. 5B ). A decrease in CRTC1 binding to promoter elements is expected to result in a concomitant reduction in CRTC1-dependent gene expression. To test whether the lower binding of CRTC1 in AD rats was consistent with this scenario, we assessed the expression of those genes under CRTC1 regulatory control. We found that the Aβ-induced decreased binding of CRTC1 to gene promoters was directly reflected by diminished transcript production for Arc ( t8 = 2.857; P < 0.05), c-fos ( t8 = 2.219; P < 0.05), Egr1 ( t8 = 3.621; P < 0.01), and Bdnf ( t8 = 2.063; P < 0.05) in AD rats compared with wild-type (Fig. 5C ).
Figure 5.
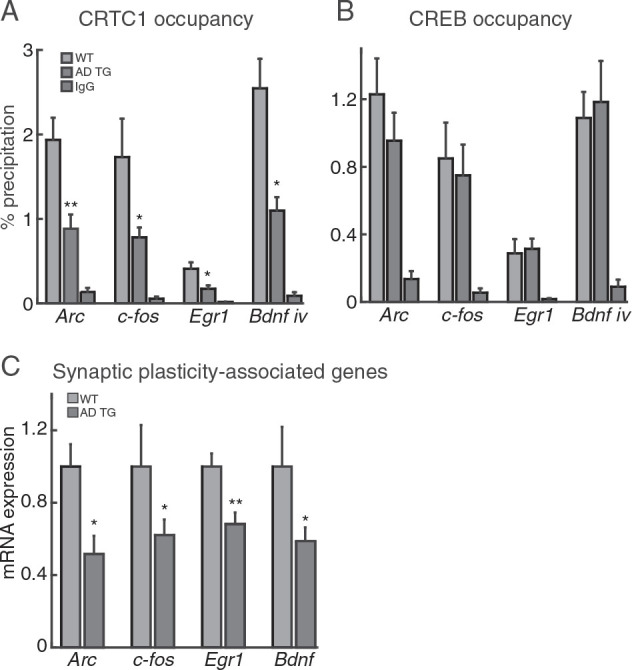
CRTC1 promoter occupancy and CRTC1-dependent gene expression are reduced at preplaque AD stages. ( A ) ChIP assays demonstrate recruitment of CRTC1 to CRE-responsive Arc , c-fos , Egr1 , and Bdnf promoters. For CRTC1, we observed a 2-fold reduction in AD transgenic rats of recruitment to Arc, c-fos, Egr1, and Bdnf promoters relative to the same promoters in wild-type rats. ( B ) Recruitment of CREB to CRE-responsive promoters was not different in Alzheimer transgenic rats. IgG indicates immunoprecipitation with an irrelevant antibody and confirms specific binding of CRTC1 and CREB antibodies. ( C ) Gene expression analysis of CREB-dependent genes in Alzheimer transgenic rats compared with wild-type animals revealed reduced production of transcripts for Arc , c-fos , Egr1 , and Bdnf . Data represent the mean ± SEM. * P < 0.05, ** P < 0.01.
Discussion
Biomarker evidence suggests that pathological processes underlying AD begin decades prior to the overt manifestation of cognitive symptoms ( Jack et al. 2010 ). To intervene at preclinical stages of the disease, however, further investigation is needed to understand the mechanisms by which an early buildup of Aβ peptides contributes to disease progression ( Donohue et al. 2014 ). In this study, we found that abnormal levels of intraneuronal Aβ caused a severe associative learning deficit in AD transgenic rats. Impaired cognition coincided with reduced nuclear translocation and genomic occupancy of the CREB co-activator, CRTC1, and decreased production of synaptic plasticity-associated transcripts Arc , c-fos , Egr1 , and Bdnf .
We provide the first evidence of an impairment in associative learning in Alzheimer transgenic rats using an automated touchscreen behavior platform. The operant touchscreen platform for behavioral testing holds translational value given its use in humans ( Chudasama and Robbins 2006 ). Just like patients at early stages of AD, our preplaque Alzheimer transgenic rats were slow learners, unable to correct their errors ( Blackwell et al. 2004 ). While a previous study using a mouse Alzheimer model failed to demonstrate an acquisition or learning impairment on the visual discrimination task ( Romberg et al. 2013 ), acquisition deficits are known to be among the major contributors of cognitive decline in AD patients ( Becker et al. 1987 ; Germano and Kinsella 2005 ). Pasquier and colleagues showed an acquisition deficit in early AD ( Pasquier et al. 2001 ); AD patients (Mini Mental State Examination score of 23) acquired less information in a list-learning test compared with healthy counterparts. It is feasible that a difficulty to learn in early AD can lead to incomplete storage and retention of information, leading to poor recall or retrieval of information at a later stage. This might explain why the preplaque transgenic AD rats needed several repeat correction trials; they never learned the stimulus–reward association. In support of this, when differences in initial learning were controlled, Alzheimer patients showed the same rate of forgetting on a picture recognition test administered at intervals over the course of a week when compared with healthy controls. Thus, it is feasible that the anterograde amnesic deficit observed in AD might be related to an initial learning deficit ( Kopelman 1985 ).
The Alzheimer transgenic rats demonstrated rigidity and inflexibility in their behavior, as shown by the increased number of errors to the incorrect stimulus, and the need for repeat trials to help them correct their errors. The errors resulted in timeout and no reward, but these animals were unable to use this negative feedback to guide their response and continued to respond incorrectly. This type of inflexibility is typically associated with prefrontal dysfunction and can be experimentally produced through lesions to the orbitofrontal cortex in rats ( Chudasama and Robbins 2003 ) and monkeys ( Dias et al. 1996 ). It is also observed in patients with frontotemporal dementia, who have a more selective ventral orbital prefrontal pathology ( Rahman et al. 1999 ). It is possible, therefore, that the executive impairment of inflexibility and disinhibition shown by Alzheimer rats was due to pathology extending beyond the temporal lobe into the orbitofrontal cortex and was compounded by pathology to other prefrontal regions ( Bussey et al. 1997 ; Chudasama and Robbins 2003 ). However, the learning difficulty observed in the preplaque Alzheimer transgenic rats cannot be attributed to prefrontal dysfunction alone, because rats with selective prefrontal or orbitofrontal lesions do not show an acquisition deficit when tested on the same task ( Bussey et al. 1997 ; Chudasama et al. 2001 ; Chudasama and Robbins 2003 ).
Notably, the Alzheimer rats took over 10 s to make a decision to respond to the touchscreen and showed very long reward collection latencies. Since the rats collected all their reward pellets, we can rule out demotivation as the primary cause of the learning impairment. Likewise, we can reject the possibility that AD rats were visually impaired since their wild-type littermates improved their accuracy for noncorrection trials and were therefore able to track the correct stimulus to some extent. Although the Alzheimer rats were less active than the wild types, it was not the case that they were motorically incapacitated; the Alzheimer rats were substantially faster to collect their rewards (i.e., within 3 s on average) than they were to make a choice response. One possibility is that the Alzheimer rats were easily distracted or became disengaged while in the process of making a response. This might hinder the animals' ability to actively monitor or sequence their actions or effectively use working memory to guide their responses. Thus, their long reaction times may reflect a general difficulty in scheduling goal directed actions. This hypothesis needs to be tested directly but is in keeping with human case reports of AD patients showing that cognitive decline is accompanied by slow decision-making and motor actions ( Hebert et al. 2010 ; Buchman and Bennett 2011 ; Bennett et al. 2012 ).
CRTC1 is implicated in dendrite arborization of developing cortical neurons ( Li et al. 2009 ; Finsterwald et al. 2010 ), neuronal survival in response to ischemia ( Sasaki et al. 2011 ), addiction mediated through brain reward circuits ( Hollander et al. 2010 ; Dietrich et al. 2011 ), circadian clock entrainment ( Jagannath et al. 2013 ; Sakamoto et al. 2013 ), mood regulation ( Breuillaud et al. 2012 ), as well as hippocampal L-LTP and memory ( Zhou et al. 2006 ; Kovacs et al. 2007 ; Sekeres et al. 2012 ). Our study provides new insight into how CRTC1-dependent gene expression is coincident with dysfunction on associative learning tasks at the early stages of Aβ pathology. It is likely that intraneuronal Aβ disrupts the normal tracking of synaptic glutamatergic activity that is achieved through CRTC1 nuclear translocation ( Ch'ng et al. 2012 ). Consistent with this idea, we observed decreased gene transcription of Bdnf, Arc, Egr1 , and c-fos . Our data build on the work of others that has shown a similar hippocampal reduction in nuclear CRTC1 in a mouse model of AD and decreased CRTC1 levels in human brain at intermediate Braak III–VI pathological stages ( Parra-Damas et al. 2014 ). We extend these results by documenting the behavioral impact of dysregulated CRTC1 in a robust rat model of Aβ pathology, using an advanced cognitive testing platform.
Certain benefits exist in using rats as a model for AD. For example, the rat is physiologically, genetically, and morphologically closer to humans than mice, has a complex CNS, and, like humans, has postnatal brain development ( Whishaw et al. 2001 ). In addition, rats are behaviorally well characterized and demonstrate complex cognitive behaviors ( Whishaw et al. 2001 ; Chudasama and Robbins 2006 ). Rats are increasingly used to mimic key pathological hallmarks of neurodegenerative diseases including AD, and it has been reported that some transgenic rat models offer more accurate representations of the human disease compared with mice bearing the same transgene ( Do Carmo and Cuello 2013 ). Finally, a key benefit of using a rat model in studies of cognition is that dysfunction can be more readily detected as rats have fewer available mechanisms for neural compensation. Conceptually, cognitive reserve results from underlying neural mechanisms including neural reserve, the resilience of pre-existing cognitive networks, and neural compensation, which allows the use of compensatory neural resources ( Stern et al. 2005 ). BDNF, which we found to be reduced at the transcript level in Alzheimer rats, enhances neural reserve in humans by increasing efficiency in cognitive networks underlying executive control ( van Praag et al. 2000 ). Cognitive reserve is enhanced in individuals with greater educational and occupational attainment and through factors such as aerobic physical exercise and life-long learning (reviewed in Stern [2012 ]). The consequence is that individuals with higher cognitive reserve will present AD symptoms at a later stage of the pathology ( Hanyu et al. 2008 ). In this regard, the sensitivity of the touchscreen platform could prove useful in detecting subtle cognitive decline in individuals at early stages of AD, and in individuals with high cognitive reserve. For instance, it is worth noting that a paired associate learning touchscreen task in humans has been used to detect early cognitive impairments, which were correlated with alterations in biomarkers of CNS synaptic plasticity ( Kiddle et al. 2015 ).
In summary, intraneuronal Aβ accumulation is sufficient to disrupt CRTC1-dependent gene expression and cognitive function at the early stages of the AD-like amyloid pathology, before Aβ plaques appear. These results provide greater understanding relating to the early progression of Aβ pathology and identify CRTC1-dependent gene transcription as target for therapeutic intervention at the early AD stages.
Supplementary Material
Supplementary material can be found here.
Funding
This work was supported by grants from the Canadian Institutes of Health Research awarded to Y.C. (grant number: 102507) and A.C.C. (grant number: 102752) and Centres of Excellence in Neurodegeneration (grant number: 01074) to A.C.C. S.D.C. is the holder of the Charles E. Frosst/Merck Research Associate Position. A.C.C. is the holder of the McGill University Charles E. Frosst/Merck Chair in Pharmacology. We wish to thank Dr. A. Frosst, the Frosst family, and Merck Canada for their continuing support.
Supplementary Material
Notes
A.R.A. is now at the Centre for Addiction and Mental Health (CAMH), Toronto, ON, Canada. We thank Vanessa Knight for her help with behavioral testing. Conflict of Interest : None declared.
References
- Becker JT , Boller F , Saxton J , McGonigle-Gibson KL . 1987. . Normal rates of forgetting of verbal and non-verbal material in Alzheimer's disease . Cortex . 23 : 59 – 72 . [DOI] [PubMed] [Google Scholar]
- Bennett DA , Schneider JA , Buchman AS , Barnes LL , Boyle PA , Wilson RS . 2012. . Overview and findings from the rush Memory and Aging Project . Curr Alzheimer Res . 9 : 646 – 663 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA , McWhinnie E , Meltzer J , Iourgenko V , Latario B , Liu X , Chen CH , Song C , Garza D , Labow M . 2004. . Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins . Curr Biol . 14 : 2156 – 2161 . [DOI] [PubMed] [Google Scholar]
- Blackwell AD , Sahakian BJ , Vesey R , Semple JM , Robbins TW , Hodges JR . 2004. . Detecting dementia: novel neuropsychological markers of preclinical Alzheimer's disease . Dement Geriatr Cogn Disord . 17 : 42 – 48 . [DOI] [PubMed] [Google Scholar]
- Breuillaud L , Rossetti C , Meylan EM , Merinat C , Halfon O , Magistretti PJ , Cardinaux JR . 2012. . Deletion of CREB-regulated transcription coactivator 1 induces pathological aggression, depression-related behaviors, and neuroplasticity genes dysregulation in mice . Biol Psychiatry . 72 : 528 – 536 . [DOI] [PubMed] [Google Scholar]
- Buchman AS , Bennett DA . 2011. . Loss of motor function in preclinical Alzheimer's disease . Expert Rev Neurother . 11 : 665 – 676 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey TJ , Holmes A , Lyon L , Mar AC , McAllister KA , Nithianantharajah J , Oomen CA , Saksida LM . 2012. . New translational assays for preclinical modelling of cognition in schizophrenia: the touchscreen testing method for mice and rats . Neuropharmacology . 62 : 1191 – 1203 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey TJ , Muir JL , Everitt BJ , Robbins TW . 1997. . Triple dissociation of anterior cingulate, posterior cingulate, and medial frontal cortices on visual discrimination tasks using a touchscreen testing procedure for the rat . Behav Neurosci . 111 : 920 – 936 . [DOI] [PubMed] [Google Scholar]
- Cardinal RN , Aitken MR . 2010. . Whisker: a client-server high-performance multimedia research control system . Behavior Res Meth . 42 : 1059 – 1071 . [DOI] [PubMed] [Google Scholar]
- Ch'ng TH , Uzgil B , Lin P , Avliyakulov NK , O'Dell TJ , Martin KC . 2012. . Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus . Cell . 150 : 207 – 221 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y , Bussey TJ , Muir JL . 2001. . Effects of selective thalamic and prelimbic cortex lesions on two types of visual discrimination and reversal learning . Eur J Neurosci . 14 : 1009 – 1020 . [DOI] [PubMed] [Google Scholar]
- Chudasama Y , Robbins TW . 2003. . Dissociable contributions of the orbitofrontal and infralimbic cortex to pavlovian autoshaping and discrimination reversal learning: further evidence for the functional heterogeneity of the rodent frontal cortex . J Neurosci . 23 : 8771 – 8780 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y , Robbins TW . 2006. . Functions of frontostriatal systems in cognition: comparative neuropsychopharmacological studies in rats, monkeys and humans . Biol Psychol . 73 : 19 – 38 . [DOI] [PubMed] [Google Scholar]
- Conkright MD , Canettieri G , Screaton R , Guzman E , Miraglia L , Hogenesch JB , Montminy M . 2003. . TORCs: transducers of regulated CREB activity . Mol Cell . 12 : 413 – 423 . [DOI] [PubMed] [Google Scholar]
- Cuello AC , Allard S , Ferretti MT . 2012. . Evidence for the accumulation of Abeta immunoreactive material in the human brain and in transgenic animal models . Life Sci . 91 : 1141 – 1147 . [DOI] [PubMed] [Google Scholar]
- Dias R , Robbins TW , Roberts AC . 1996. . Dissociation in prefrontal cortex of affective and attentional shifts . Nature . 380 : 69 – 72 . [DOI] [PubMed] [Google Scholar]
- Dietrich JB , Takemori H , Grosch-Dirrig S , Bertorello A , Zwiller J . 2011. . Cocaine induces the expression of MEF2C transcription factor in rat striatum through activation of SIK1 and phosphorylation of the histone deacetylase HDAC5 . Synapse . 66 : 61 – 70 . [DOI] [PubMed] [Google Scholar]
- Do Carmo S , Cuello AC . 2013. . Modeling Alzheimer's disease in transgenic rats . Mol Neurodegener . 8 : 37 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue MC , Sperling RA , Salmon DP , Rentz DM , Raman R , Thomas RG , Weiner M , Aisen PS . 2014. . The preclinical Alzheimer cognitive composite: measuring amyloid-related decline . JAMA Neurol . 71(8) : 961 – 970 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espana J , Valero J , Minano-Molina AJ , Masgrau R , Martin E , Guardia-Laguarta C , Lleo A , Gimenez-Llort L , Rodriguez-Alvarez J , Saura CA . 2010. . {beta}-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1 . J Neurosci . 30 : 9402 – 9410 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterwald C , Fiumelli H , Cardinaux JR , Martin JL . 2010. . Regulation of dendritic development by BDNF requires activation of CRTC1 by glutamate . J Biol Chem . 285 : 28587 – 28595 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano C , Kinsella GJ . 2005. . Working memory and learning in early Alzheimer's disease . Neuropsychol Rev . 15 : 1 – 10 . [DOI] [PubMed] [Google Scholar]
- Grant SM , Ducatenzeiler A , Szyf M , Cuello AC . 2000. . Abeta immunoreactive material is present in several intracellular compartments in transfected, neuronally differentiated, P19 cells expressing the human amyloid beta-protein precursor . J Alzheimers Dis . 2 : 207 – 222 . [DOI] [PubMed] [Google Scholar]
- Hanyu H , Sato T , Shimizu S , Kanetaka H , Iwamoto T , Koizumi K . 2008. . The effect of education on rCBF changes in Alzheimer's disease: a longitudinal SPECT study . Eur J Nucl Med Mol Imaging . 35 : 2182 – 2190 . [DOI] [PubMed] [Google Scholar]
- Hanzel CE , Pichet-Binette A , Pimentel LS , Iulita MF , Allard S , Ducatenzeiler A , Do Carmo S , Cuello AC . 2014. . Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer's disease . Neurobiol Aging . 35 : 2249 – 2262 . [DOI] [PubMed] [Google Scholar]
- Hebert LE , Bienias JL , McCann JJ , Scherr PA , Wilson RS , Evans DA . 2010. . Upper and lower extremity motor performance and functional impairment in Alzheimer's disease . Am J Alzheimers Dis Other Demen . 25 : 425 – 431 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA , Im HI , Amelio AL , Kocerha J , Bali P , Lu Q , Willoughby D , Wahlestedt C , Conkright MD , Kenny PJ . 2010. . Striatal microRNA controls cocaine intake through CREB signalling . Nature . 466 : 197 – 202 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourgenko V , Zhang W , Mickanin C , Daly I , Jiang C , Hexham JM , Orth AP , Miraglia L , Meltzer J , Garza D et al. . 2003. . Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells . Proc Natl Acad Sci USA . 100 : 12147 – 12152 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iulita MF , Allard S , Richter L , Munter LM , Ducatenzeiler A , Weise C , Carmo SD , Klein WL , Multhaup G , Cuello AC . 2014. . Intracellular Abeta pathology and early cognitive impairments in a transgenic rat model overexpressing human amyloid precursor protein: a multidimensional study . Acta Neuropatho Commun . 2 : 61 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr , Knopman DS , Jagust WJ , Shaw LM , Aisen PS , Weiner MW , Petersen RC , Trojanowski JQ . 2010. . Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade . Lancet Neurol . 9 : 119 – 128 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannath A , Butler R , Godinho SI , Couch Y , Brown LA , Vasudevan SR , Flanagan KC , Anthony D , Churchill GC , Wood MJ et al. . 2013. . The CRTC1-SIK1 pathway regulates entrainment of the circadian clock . Cell . 154 : 1100 – 1111 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiddle SJ , Steves CJ , Mehta M , Simmons A , Xu X , Newhouse S , Sattlecker M , Ashton NJ , Bazenet C , Killick R et al. . 2015. . Plasma protein biomarkers of Alzheimer's disease endophenotypes in asymptomatic older twins: early cognitive decline and regional brain volumes . Transl Psychiatry . 5 : e584 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman MD . 1985. . Rates of forgetting in Alzheimer-type dementia and Korsakoff's syndrome . Neuropsychologia . 23 : 623 – 638 . [DOI] [PubMed] [Google Scholar]
- Kornhauser JM , Cowan CW , Shaywitz AJ , Dolmetsch RE , Griffith EC , Hu LS , Haddad C , Xia Z , Greenberg ME . 2002. . CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events . Neuron . 34 : 221 – 233 . [DOI] [PubMed] [Google Scholar]
- Kovacs KA , Steullet P , Steinmann M , Do KQ , Magistretti PJ , Halfon O , Cardinaux JR . 2007. . TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity . Proc Natl Acad Sci USA . 104 : 4700 – 4705 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM , Green KN , Oddo S . 2007. . Intracellular amyloid-beta in Alzheimer's disease . Nat Rev Neurosci . 8 : 499 – 509 . [DOI] [PubMed] [Google Scholar]
- Leon WC , Canneva F , Partridge V , Allard S , Ferretti MT , DeWilde A , Vercauteren F , Atifeh R , Ducatenzeiler A , Klein W et al. . 2010. . A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment . J Alzheimers Dis . 20 : 113 – 126 . [DOI] [PubMed] [Google Scholar]
- Li S , Zhang C , Takemori H , Zhou Y , Xiong ZQ . 2009. . TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons . J Neurosci . 29 : 2334 – 2343 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B , Montminy M . 2001. . Transcriptional regulation by the phosphorylation-dependent factor CREB . Nat Rev Mol Cell Biol . 2 : 599 – 609 . [DOI] [PubMed] [Google Scholar]
- Parra-Damas A , Valero J , Chen M , Espana J , Martin E , Ferrer I , Rodriguez-Alvarez J , Saura CA . 2014. . Crtc1 activates a transcriptional program deregulated at early Alzheimer's disease-related stages . J Neurosci . 34 : 5776 – 5787 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier F , Grymonprez L , Lebert F , Van der Linden M . 2001. . Memory impairment differs in frontotemporal dementia and Alzheimer's disease . Neurocase . 7 : 161 – 171 . [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S , Wang M , Pham S , Sze CI , Eckman CB . 2011. . Downregulation of CREB expression in Alzheimer's brain and in Abeta-treated rat hippocampal neurons . Mol Neurodegener . 6 : 60 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y , Klyubin I , Harney SC , Hu N , Cullen WK , Grant MK , Steffen J , Wilson EN , Do Carmo S , Remy S et al. . 2014. . Longitudinal testing of hippocampal plasticity reveals the onset and maintenance of endogenous human Aβ-induced synaptic dysfunction in individual freely behaving pre-plaque transgenic rats: rapid reversal by anti-Aβ agents . Acta Neuropathologica Commun . 2 : 175 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S , Sahakian BJ , Hodges JR , Rogers RD , Robbins TW . 1999. . Specific cognitive deficits in mild frontal variant frontotemporal dementia . Brain . 122(8) : 1469 – 1493 . [DOI] [PubMed] [Google Scholar]
- Romberg C , Horner AE , Bussey TJ , Saksida LM . 2013. . A touch screen-automated cognitive test battery reveals impaired attention, memory abnormalities, and increased response inhibition in the TgCRND8 mouse model of Alzheimer's disease . Neurobiol Aging . 34 : 731 – 744 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K , Norona FE , Alzate-Correa D , Scarberry D , Hoyt KR , Obrietan K . 2013. . Clock and light regulation of the CREB coactivator CRTC1 in the suprachiasmatic circadian clock . J Neurosci . 33 : 9021 – 9027 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T , Takemori H , Yagita Y , Terasaki Y , Uebi T , Horike N , Takagi H , Susumu T , Teraoka H , Kusano K et al. . 2011. . SIK2 Is a Key Regulator for Neuronal Survival after Ischemia via TORC1-CREB . Neuron . 69 : 106 – 119 . [DOI] [PubMed] [Google Scholar]
- Schoenbaum G , Nugent S , Saddoris MP , Gallagher M . 2002. . Teaching old rats new tricks: age-related impairments in olfactory reversal learning . Neurobiol Aging . 23 : 555 – 564 . [DOI] [PubMed] [Google Scholar]
- Screaton RA , Conkright MD , Katoh Y , Best JL , Canettieri G , Jeffries S , Guzman E , Niessen S , Yates JR 3rd , Takemori H et al. . 2004. . The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector . Cell . 119 : 61 – 74 . [DOI] [PubMed] [Google Scholar]
- Sekeres MJ , Mercaldo V , Richards B , Sargin D , Mahadevan V , Woodin MA , Frankland PW , Josselyn SA . 2012. . Increasing CRTC1 function in the dentate gyrus during memory formation or reactivation increases memory strength without compromising memory quality . J Neurosci . 32 : 17857 – 17868 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ . 2001. . Alzheimer's disease: genes, proteins, and therapy . Physiol Rev . 81 : 741 – 766 . [DOI] [PubMed] [Google Scholar]
- Stern Y . 2012. . Cognitive reserve in ageing and Alzheimer's disease . Lancet Neurol . 11 : 1006 – 1012 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y , Habeck C , Moeller J , Scarmeas N , Anderson KE , Hilton HJ , Flynn J , Sackeim H , van Heertum R . 2005. . Brain networks associated with cognitive reserve in healthy young and old adults . Cereb Cortex . 15 : 394 – 402 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori H , Okamoto M . 2008. . Regulation of CREB-mediated gene expression by salt inducible kinase . J Steroid Biochem Mol Biol . 108 : 287 – 291 . [DOI] [PubMed] [Google Scholar]
- van Praag H , Kempermann G , Gage FH . 2000. . Neural consequences of environmental enrichment . Nat Rev Neurosci . 1 : 191 – 198 . [DOI] [PubMed] [Google Scholar]
- Watts AG , Sanchez-Watts G , Liu Y , Aguilera G . 2011. . The distribution of messenger RNAs encoding the three isoforms of the transducer of regulated cAMP responsive element binding protein activity in the rat forebrain . J Neuroendocrinol . 23 : 754 – 766 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whishaw IQ , Metz GA , Kolb B , Pellis SM . 2001. . Accelerated nervous system development contributes to behavioral efficiency in the laboratory mouse: a behavioral review and theoretical proposal . Dev Psychobiol . 39 : 151 – 170 . [DOI] [PubMed] [Google Scholar]
- Zhou Y , Wu H , Li S , Chen Q , Cheng XW , Zheng J , Takemori H , Xiong ZQ . 2006. . Requirement of TORC1 for late-phase long-term potentiation in the hippocampus . PLoS One . 1 : e16 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.