

Abstract
Background
An inverse association between maternal smoking and preeclampsia has been frequently observed in epidemiologic studies for several decades. In the May 2015 issue of this journal, Lisonkova and Joseph described a simulation study suggesting that bias from left truncation might explain the inverse association. The simulations were based on strong assumptions regarding the underlying mechanisms through which bias might occur.
Methods
To examine the sensitivity of the previous authors’ conclusions to these assumptions, we constructed a new Monte Carlo simulation using published estimates to frame our data generating parameters. We estimated the association between smoking and preeclampsia across a range of scenarios that incorporated abnormal placentation and early pregnancy loss.
Results
Our results confirmed that the previous authors’ findings are highly dependent on assumptions regarding the strength of association between abnormal placentation and preeclampsia. Thus, the bias they described may be less pronounced than was suggested.
Conclusions
Under empirically-derived constraints of these critical assumptions, left truncation does not appear to fully explain the inverse association between smoking and preeclampsia. Further, when considering processes in which left truncation may result from the exposure, it is important to precisely describe the target population and parameter of interest before assessing potential bias. We comment on the specification of a meaningful target population when assessing maternal smoking and preeclampsia as a public health issue. We describe considerations for defining a target population in studies of perinatal exposures when those exposures cause competing events (e.g., early pregnancy loss) for primary outcomes of interest.
For several decades, epidemiologic evidence has consistently shown an inverse association between maternal smoking during pregnancy and the occurrence of preeclampsia.1,2 In a simulation study published in the May 2015 issue of EPIDEMIOLOGY, Lisonkova and Joseph assessed whether differential left truncation from early pregnancy loss (henceforth, early loss) by maternal smoking status could explain this counterintuitive but frequently observed result.3 For a given outcome and a given time scale, when individuals come under study after the time scale origin and the outcome can occur prior to the observed time, those outcomes are referred to as ‘left truncated.’4 An analogous scenario arises when a competing event occurs prior to outcome follow-up, thus precluding observation of the event of interest. Lisonkova and Joseph also refer to this second process as left truncation; and we adopt their terminology here. In the context of this paper, left truncation can occur due to early loss (i.e., fetal death before 20 weeks of pregnancy), which results in some pregnancies being excluded from the study population. Left truncation can induce selection bias of exposure-outcome associations if the magnitude of left truncation differs by exposure.5
Under a number of assumptions including no direct effect of smoking on preeclampsia, Lisonkova and Joseph concluded that such differential left truncation caused a biased risk ratio (RR) of 0.85 (−0.16 on the natural log scale, with 95% confidence interval, CI, −0.31, −0.02). We wholeheartedly agree with Lisonkova and Joseph that this puzzling association should be explored as a possible result of bias, and we commend their thoughtful insights into alternative mechanisms. However, the authors’ findings are dependent on three assumptions that merit additional consideration.
The simulation study assumed that: (1) All fetuses surviving past 20 weeks of pregnancy with abnormal placentation eventually progress to preeclampsia. (2) Zero fetuses surviving past 20 weeks of pregnancy without abnormal placentation progress to preeclampsia. (3) Maternal smoking has no effect on abnormal placentation. Regarding assumptions 1 and 2, pathological placental features are substantially more frequent in preeclamptic pregnancies as compared to non-preeclamptic pregnancies, but the two conditions are not deterministic.6–8 For example, Vinnars et al. reported that prevalence of pathological features (i.e., decidual arteriopathy, accelerated villous maturation, any placental infarction) ranged 4–20% in non-preeclamptic pregnancies and 35–80% in severely preeclamptic pregnancies.8 Regarding assumption 3, smoking during pregnancy has been associated with increased risk of impaired placental development.9,10
The first motivation for this study was to quantify the impact of these three prior assumptions on the conclusion that left truncation could explain the observed smoking-preeclampsia association. Second, a precise discussion of bias must pertain to a well-defined target population.11 Thus, we also assessed the impact of the choice of target population on the potential for bias due to left truncation. We conclude with a discussion of our findings in the context of public health interpretation.
METHODS
To address the impact of these three assumptions on the conclusion that left truncation could explain the observed smoking-preeclampsia association, we performed a Monte Carlo simulation study using published studies to frame our data generating parameters. We estimated bias of the natural-log transformed risk ratio (lnRR) for smoking and preeclampsia across a range of scenarios. In a preliminary scenario, we used the same parameters as the previous authors and replicated their published RR of 0.85, with similar precision (95% CI 0.72, 1.01). We provide a SAS program in eAppendix 1 (see SAS macro, Supplemental Digital Content 1) that allows the user to explore alternative ranges of assumptions that we considered.
Target population and effect of interest
Though not often stated, in studies of the effects of smoking on preeclampsia, the target population has been the hypothetical population of pregnant women, were it possible to intervene to prevent all smoking-related early loss without also affecting the risk of smoking-related preeclampsia or sources of early loss not related to smoking. That is, the causal effect of implicit interest often compares preeclampsia risk between smokers with non-smokers, while ensuring similar early loss in both groups. Assuming a time-fixed exposure and no heterogeneity of the risk ratio, the hypothetical interventions for this target population would correspond to ensuring that all mothers smoke from the earliest point in pregnancy versus ensuring that none of them smoke at all, while also ensuring against smoking-induced early loss. For comparability with the results of Lisonkova and Joseph who appear to have used this target population, we also use this target population and these hypothetical interventions in our simulations. In the discussion section, however, we illustrate an example using a target population comprising all conceptions.
Simulation setup
Figure 1 shows the causal diagram12 that served as the basis for our simulation. We first assigned a Bernoulli (zero-one) random variable for smoking with mean probability of 20%, which we consider more plausible than 50%.13,14 We note that smoking prevalence does not impact bias, but this alteration leads to more realistic confidence limits.
FIGURE 1.
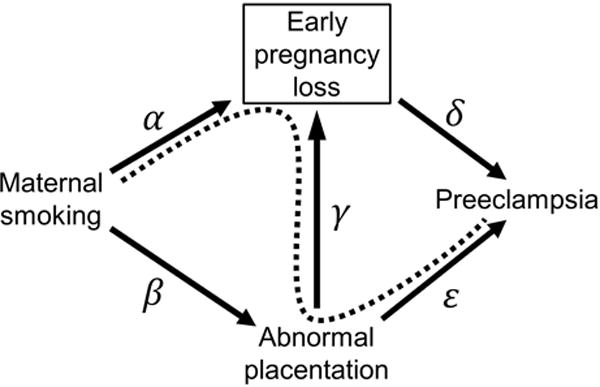
Causal diagram representing associations between maternal smoking, abnormal placentation, early pregnancy loss, and preeclampsia. Collider bias, denoted by the dashed line connecting maternal smoking and preeclampsia, arises in observational studies which implicitly restrict to pregnancies that survive past the point of early loss. Some simulation scenarios represent settings that assume null associations between smoking and early loss (edge α), smoking and abnormal placentation (edge β), or abnormal placentation and early loss (edge γ), such that arrows representing those associations would be absent from the figure. All simulations were based on the assumption that early loss precludes the occurrence of preeclampsia (edge δ).
To weaken the deterministic relation between abnormal placentation and preeclampsia (assumptions 1 and 2, represented in Figure 1 by edge ε), we examined a range of potential associations between abnormal placentation and preeclampsia based on data published by Vinnars et al.8 A detailed description of our approach to define data generating parameters using these published estimates is in eAppendix 2 (see Text and Tables, Supplemental Digital Content 2). Briefly, we considered two potential marginal risks of preeclampsia, 3% and 7%.2 Separately for each assumed marginal risk of preeclampsia, we used prior data8 to back-calculate risks of preeclampsia by abnormal placentation status across a range of scenarios. We selected data generating parameters for our simulations based on these results, so that our simulations would demonstrate how the strength of association between abnormal placentation and preeclampsia might impact potential left truncation bias. We selected two extreme scenarios (odds ratios, ORs, for abnormal placentation and preeclampsia of 2.2 and 96.0) and two middling scenarios (ORs of 7.3 and 21.0) that we believe are more plausible. Results pertaining to potential left truncation bias were similar for 3% and 7% marginal risk of preeclampsia; we therefore restrict our subsequent discussion to settings with marginal risk of preeclampsia of 3%. Given a marginal risk of preeclampsia of 3% and using the four ORs enumerated above, we modeled preeclampsia for pregnancies with abnormal placentation as a Bernoulli random variable with probabilities of 5.13%, 11.42%, 17.80%, and 38.22%; we modeled preeclampsia for pregnancies without abnormal placentation as a Bernoulli random variable with corresponding probabilities of 2.45%, 1.73%, 1.02%, and 0.64%. To compare our results with those derived from the previous authors’ assumptions, we also modeled preeclampsia as a Bernoulli random variable with probability of 99.99% for pregnancies with abnormal placentation (assumption 1) and 0.01% for pregnancies without abnormal placentation (assumption 2).
To explore the impact of differential abnormal placentation by maternal smoking (assumption 3, represented in Figure 1 by edge β), we modeled abnormal placentation as a Bernoulli random variable with probabilities uniformly distributed at levels between 5–10% and 10–15%; we included settings where abnormal placentation was similar by smoking status (5–10% in both groups), higher in smokers (10–15%) than non-smokers (5–10%), and lower in smokers (5–10%) than non-smokers (10–15%).
Among non-smokers without abnormal placentation, we modeled early loss as a Bernoulli random variable with mean probabilities of 10% and 20%. We modeled the direct effects on early loss of smoking (Figure 1, edge α) and abnormal placentation (Figure 1, edge γ) with risk ratios of 1.0, 1.5, 2.0, and 2.5. We assumed no departures from multiplicativity of the risk ratios for early loss, though such departures can be accommodated in our SAS program. Dropping impossible parameter combinations (e.g., 20% referent risk of early loss and risk ratios for smoking and abnormal placentation of 2.5 each), we generated data for 746 simulation scenarios. In each scenario, 1,000 cohorts were simulated, with 10,000 individuals per cohort. Simulations were conducted using SAS software version 9.3 (SAS Institute, Inc.; Cary, North Carolina USA).
RESULTS
Some of our results were similar to those reported by Lisonkova and Joseph. As expected, downward bias increased as risk ratios increased for the independent effects of smoking and abnormal placentation on early loss. We found that assumptions 1 and 2 in particular had notable impact on magnitude of bias. Figure 2 shows apparent risk ratios for the association between maternal smoking and preeclampsia across a range of simulation scenarios involving abnormal placentation and early loss, given a true risk ratio of 1.0.
FIGURE 2.
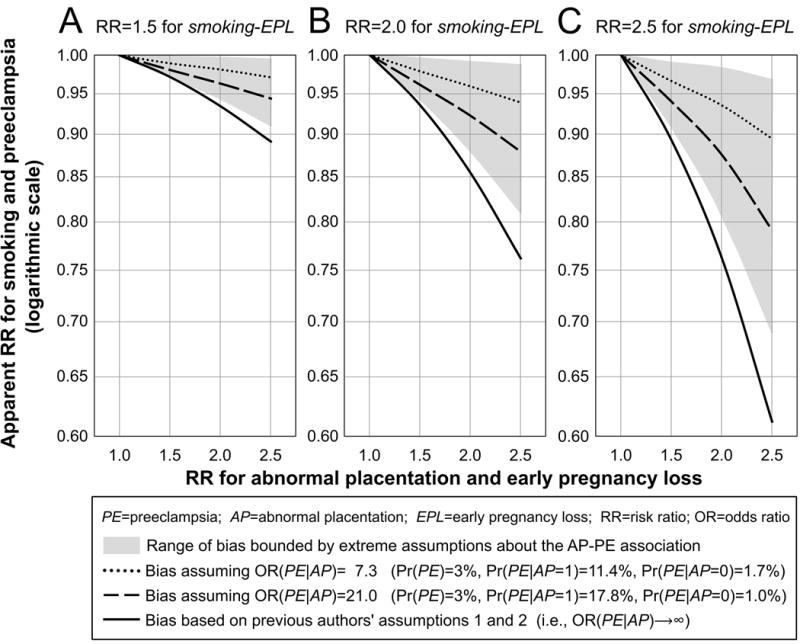
Apparent risk ratio (RR) estimates for the association between maternal smoking and preeclampsia (PE) across a range of simulation scenarios involving abnormal placentation (AP) and early pregnancy loss (EPL), given a true RR of 1.0. The RR for the association between smoking and early pregnancy loss was set to 1.5 (panel A), 2.0 (B), or 2.5 (C). The RR for smoking and abnormal placentation was set to 1.0 (assumption 3), the risk of early pregnancy loss was set to 10% for non-smokers without abnormal placentation, and the marginal risk of preeclampsia (Pr(PE)) was set to 3%. Line types and shading represent different assumptions regarding the relation between abnormal placentation and preeclampsia (assumptions 1 and 2). Solid lines depict bias estimated under the previous authors’ assumptions. Shaded areas cover the range of bias estimated within extreme empirically-derived assumptions about the odds ratio (OR) for abnormal placentation and preeclampsia; the weakest extreme was OR(PE|AP)=2.2 (Pr(PE)=3%, Pr(PE|AP=1)=5.1%, Pr(PE|AP=0)=2.5%) and the strongest extreme was OR(PE|AP)=96.0 (Pr(PE)=3%, Pr(PE|AP=1)=38.2%, Pr(PE|AP=0)=0.6%). Dashed and dotted lines depict bias estimated under middling empirically-derived assumptions about the OR for abnormal placentation and preeclampsia. Justification for our extreme and middling assumptions (shading, dashes, dots) is in eAppendix 2.
When we maintained the previous authors’ assumption of no causal relationship between smoking and preeclampsia, but relaxed the assumption of a deterministic relation between abnormal placentation and preeclampsia (assumptions 1 and 2), downward bias for the smoking-preeclampsia association was greatly attenuated. For example, the previous authors reported bias of the lnRR of −0.16 (RR(preeclampsia|smoking)=0.85) as shown in Figure 2B (solid line, RR(early-loss|AP)=2.0); this estimate is based on an assumption that all surviving pregnancies with abnormal placentation progress to preeclampsia, and zero surviving pregnancies without abnormal placentation progress to preeclampsia. When we increased the assumed risk of preeclampsia among those without abnormal placentation from 0% to 1.02% while maintaining a strong effect of abnormal placentation (AP) on preeclampsia (OR(preeclampsia|AP)=21.0), bias of the lnRR was reduced by half, from −0.16 to −0.08 (RR(preeclampsia|smoking)=0.92, Figure 2B, dashed-line). Reducing OR(preeclampsia|AP)) from 21.0 to 7.3 further decreased bias of the lnRR to −0.04 (RR(preeclampsia|smoking)=0.96, Figure 2B, dotted-line). The previous authors’ bias estimates ranged approximately 20–30% more extreme than the most extreme bias estimates from our simulations.
We also relaxed assumption 3 by allowing for a direct effect of smoking on abnormal placentation (Figure 1, edge β), which introduced a causal pathway from smoking to preeclampsia. Compared to when smoking was assumed to have no effect on abnormal placentation, downward bias was attenuated when smoking decreased risk of abnormal placentation, and was amplified when smoking increased risk of abnormal placentation. However, the relative impact of assumption 3 on bias was nominal compared to assumptions 1 and 2 (see eAppendix 3, Text and Figure, Supplemental Digital Content 3).
As expected, downward bias increased markedly with increasing baseline risk of early loss. For example, when baseline risk of early loss was 10%, OR(preeclampsia|AP)=21.0, RR(early-loss|smoking)=2.0, and RR(early-loss|AP) took values of 1.5, 2.0 and 2.5, the apparent RRs(preeclampsia|smoking) were 0.96, 0.92, and 0.88, respectively (Figure 2B, dashed line). Holding all other parameters constant, when baseline risk of early loss was increased to 20%, the corresponding RRs(preeclampsia|smoking) changed to 0.87, 0.72, and 0.54.
DISCUSSION
Lisonkova and Joseph posit that left truncation could fully explain the observed inverse smoking-preeclampsia association in the literature, based on their biased risk ratio estimate of 0.85. However, using published data on the relation between abnormal placentation and preeclampsia to inform parameters in our simulation setup, we show that their results are highly dependent on their assumptions regarding the strength of that association, and that the bias is likely less pronounced than Lisonkova and Joseph described. Summary measures in the literature for the relative risk for smoking and preeclampsia are near 0.70;1,2 for left truncation bias to explain the inverse association, we would expect the estimated bias in plausible simulation scenarios to be consistently stronger than 0.85. Under more empirically-derived constraints of these critical assumptions, left truncation does not appear to fully explain the inverse association between smoking and preeclampsia. Further, given the simulation implicitly contrasts all mothers smoking versus all mothers not smoking, we would expect an actual intervention to have a weaker effect since smoking prevalence of either 0% or 100% would be unrealistic.
In addition to abnormal placentation, other factors might introduce additional biasing pathways from smoking to preeclampsia. For illustrative purposes, we limited the causal structure in our simulations to include abnormal placentation as the key determinant of early loss that opened a potential biasing pathway. Because smoking is related to myriad health outcomes, it likely plays a key role in multiple etiologic pathways related to preeclampsia or early loss, leading to additional potential for bias.
Simulation studies and theoretical assessment of causal mechanisms are an essential tool in the field of perinatal epidemiology, where early loss limits our ability to directly assess many etiologic questions. Simulation studies provide the potential to evaluate hypotheses regarding unobservable but plausible phenomena. However, such approaches are strongly sensitive to assumptions underlying the simulated or causal mechanisms. Given frequent uncertainty regarding unobservable biologic processes in the field of perinatal epidemiology, simulation parameters should be faithful to the existing epidemiologic and clinical evidence, and incorporate the full range of possibilities – not just those with the highest likelihood.15
Specifying a meaningful target population
The specification of a meaningful target population extends on general concerns in perinatal epidemiology regarding the proper identification of the population-at-risk of an outcome. Perinatal epidemiologists often wish to estimate gestational-age-specific occurrence measures, which can often be handled using the fetuses-at-risk approach.16,17 In the setting of smoking and preeclampsia, however, our primary interest is not in estimating gestational-age-specific incidence of preeclampsia, but rather in estimating the extent to which smoking increases overall risk of preeclampsia. There is limited understanding of the complex mechanisms leading to early loss, which alters the population of mothers at risk for preeclampsia and is likely affected by maternal smoking. It is therefore in our interest to focus on a target population (e.g., all conceptions) which is not characterized by selection mechanisms that are exceedingly difficult for us to measure or control.
A risk ratio estimate that is internally valid for one population may be biased/invalid for another population, if the two populations differ in the distribution of modifiers on the risk ratio scale. Thus, a precise discussion of potential bias should include an explicit target population. We could imagine that the target population could include all conceptions (observed and unobserved pregnancies), all observed pregnancies, or all pregnancies that would be potentially at-risk for preeclampsia, whether the mother smoked or not.
Comparing expectations of the risk ratio in different target populations
For an illustrative example, we posit that a meaningful target of inference is the total (unobserved) population of conceptions. Under no confounding, the true risk ratio for all conceptions would be [(n preeclampsia events among smokers)/(N conceptions among smokers)]/[(n preeclampsia events among non-smokers)/(N conceptions among non-smokers)]. Note that the denominators refer to conceptions rather than to pregnancies surviving at least 20 weeks. Given 1,000 total conceptions, 20% smoking prevalence, and a true risk ratio of 1.0 (for illustrative purposes only to serve as a basis for easy assessment of upward and downward bias), let us assume the components of the true risk ratio to be (10/200)/(40/800).
Previous authors have supposed that in the observed data the number of pregnancies among smokers would be disproportionately lower than among non-smokers, because smoking causes increased risk of early loss. Under that assumption, let us suppose that 12.5% of conceptions among non-smokers experience early loss, and that smokers experience an absolute excess risk of early loss due to smoking of 12.5%, resulting in 25% risk of early loss among smokers. The observed data would comprise 150 pregnancies for smokers and 700 pregnancies for non-smokers, and the risk ratio using the observed (biased) data would then be (10/150)/(40/700)=1.17. Note that the numerator would not change since all preeclampsia events would have been observed in either scenario. In this scenario, where the only difference between the observed (biased) risk ratio and the unobserved (true) risk ratio is the exclusion of early losses, the true risk ratio is actually biased upward (1.17>1.0) for the target population of all conceptions, counter to the previous authors’ findings.
As mentioned above, the authors’ implicit target population appears to be the hypothetical population of pregnant mothers, had we been able to prevent all smoking-related early loss (the competing event) without also affecting the risk of smoking-related preeclampsia or other sources of early loss. This implication corresponds to removing edge α from the causal diagram in Figure 1. Shifting smokers’ risk of early loss from 25% to 12.5% (the risk among non-smokers) would result in an increase of smokers in the hypothetical population from 150 to 175. Based on the assumption that we were able to remove differential early loss without affecting the association between smoking and preeclampsia, we would suppose that the 25 additional smokers at risk of preeclampsia in the hypothetical population have the same risk of preeclampsia (10/150) as the smokers who did not experience early loss, resulting in (25×10)/150=1.7 additional preeclampsia events among smokers. The numerator would change in this scenario because we suppose that some early losses might have resulted in preeclampsia, had the early loss not occurred. The risk ratio in this hypothetical population would therefore be equal to (11.7/175)/(40/700)=1.17.
The unbiased risk ratio for this hypothetical target population is exactly equal to the biased risk ratio for the target population of all conceptions, which is not surprising since collider bias was prevented by the implicit removal of edge α from the causal diagram in Figure 1. Another way to prevent collider bias would be to assume we could prevent early loss altogether. Under the same assumptions as above, we would again arrive at a risk ratio of 1.17, still biased for the target population of all conceptions but unbiased for the target in which we removed the effects of smoking on early loss. The term ‘bias’ is therefore not useful without anchoring it to a specific estimand and target population.
The assumption that an intervention to reduce smoking-related early loss would not affect smoking-related preeclampsia would depend strongly on the specific intervention, so these results are tenuous in terms of describing realistic interventions. Neither analysis of these target populations identifies a biologic effect of smoking on preeclampsia because an intervention to eliminate smoking may affect many other mechanisms that influence preeclampsia. In fact, the analysis is predicated on the lack of a direct effect of smoking on preeclampsia. This example underscores the profound difficulty of intuiting biologic mechanisms from epidemiologic data when competing events are caused by the exposure.
Conditioning on the future: target populations in a public health context
An intervention to prevent or reduce maternal smoking throughout pregnancy would have to take place no later than conception or, alternatively, at the time when the pregnancy is recognized. Choosing to begin follow-up for preeclampsia at a later time (e.g., at 20 weeks of pregnancy) because that is when women become at risk of that outcome18 has several implications. First, it reduces the target population for the estimand to a subset of the target population for the exposure intervention: the women who remain alive and pregnant after conception or pregnancy recognition for at least 20 weeks. This restricted target population corresponds to the one used by Lisonkova and Joseph and in our simulations.
Second, given the effect of maternal smoking on early loss and (to a much lesser extent) on maternal mortality in that period, this restriction of the target population – if unintentional or unrecognized – creates potential for selection bias due to any condition or event that affects maternal mortality or early loss and preeclampsia (as shown by the dashed line in Figure 1).19–23 The upshot is that the effect being estimated in this restricted target population is a controlled direct effect: an effect of the smoking intervention through causal paths not mediated by survival of the mother and pregnancy to 20 weeks. Without assumptions24 that are often implausible, the estimation of such an effect requires special analytic methods.25,26
Finally, restricting the target population in this way would require a second intervention to keep every woman alive and pregnant between the time of the smoking intervention (at or before pregnancy initiation or recognition) and week 20 of the pregnancy, whether or not she smokes in that interval. This second intervention would be highly desirable, but because it is not feasible, the estimated effect has no public health relevance.
A second possible approach would be to estimate the effect of the smoking intervention in a different restricted target population: one composed of the women whose survival and pregnancy status through 20 weeks would not be affected by the smoking intervention. Specifically, this ‘principal stratum’ would consist of the women who would remain alive and pregnant to at least 20 weeks, whether they smoke or not.27–29 Although this restricted target population is unidentifiable and its corresponding effect measure estimates would require strong assumptions and special analytic methods, those estimates would have some value as measures of potential public health benefit. The estimated benefit would be only partial, however, as there is seldom, if ever, any good reason to assume that this subset of individuals who receive a public health intervention would be the only ones to benefit from it.
A third possibility would be to begin outcome follow-up at the time of the exposure intervention.22,27 In the present context, the target population would be defined as all women who would be subject to the smoking intervention, at or before the time of conception or pregnancy recognition. (We consider this target population to be closely related to the target population of all conceptions from the illustrative example.) This target population requires a study design that would enroll women who might become pregnant, who are trying to become pregnant, or who have just recognized that they are pregnant. We would have to accept that no cases of preeclampsia would occur in the first few months of follow-up. In return, the considerable benefit would be an avoidance of selection bias caused by conditioning on information from the future. Just as it would be unachievable in real life to require women to remain alive and pregnant between the smoking intervention and week 20 of pregnancy, the data analysis for this target population would not condition on such a requirement. It may be true that women are considered immune from preeclampsia until week 20 of pregnancy based on a commonly used demarcation of the risk period for preeclampsia; however, they are not immune during that period from outcomes that are affected by the smoking intervention and that also affect preeclampsia: specifically, early loss and maternal mortality. Finally, investigators wishing to account for smoking’s effects before pregnancy – such as its effect on fecundability30–32 – may need to shift the start of outcome follow-up further back in time to identify a relevant target population.
We conclude that in simulation studies that explore prenatal exposures associated with increased risk of early loss and other perinatal outcomes, investigators should implement plausible assumptions to estimate hypothetical intervention effects in well-defined target populations. Implicit to our discussion is the observation that in the case of maternal smoking and preeclampsia, the label ‘left truncation’ may not be precise; however, conceptualizing early loss as a competing event for preeclampsia clarifies the consequences of analytic decisions intended to address potential collider bias. Future research is needed to improve understanding of the causes of early loss and to further develop analytic methods29 for this setting. Without hypothesizing realistic, ethical interventions for well-defined target populations, however, it may be difficult to distinguish between statistical and public health measures of importance. Although investigators might have difficulty studying the separate effects of a smoking intervention on conception and implantation, early loss, and preeclampsia, they should maintain focus on estimating the population effects of realistic interventions (including interventions that do not eradicate smoking or early loss altogether).
Smoking may genuinely decrease preeclampsia among all observed pregnancies because women more prone to preeclampsia are at higher risk of smoking-related early loss, or through incidental effects of cigarette smoke components on placentation.33 Early maternal death or pregnancy loss, events that remove the mother from the population-at-risk for preeclampsia, are often tragic and should either be taken into account for the biases they can create or not omitted from the target population of inference.
As always, a reminder is required. Even if causal, the inverse association between maternal smoking and preeclampsia does not change the overall public health message about smoking. It is bad for human health in general, and for pregnant women and their pregnancies in particular.
Supplementary Material
SUPPLEMENTAL DIGITAL CONTENT 1. SAS macro to assess the impact of left truncation bias on the association between maternal smoking and preeclampsia. pdf.
SUPPLEMENTAL DIGITAL CONTENT 2. Text and tables that provide justification for our extreme and middling assumptions about the association between abnormal placentation and preeclampsia. pdf.
SUPPLEMENTAL DIGITAL CONTENT 3. Text and figure that provide data regarding our investigation of assumption 3, showing that it had negligible impact on bias compared to assumptions 1 and 2. pdf.
Acknowledgments
We wish to thank Quaker Harmon for helpful comments on early versions of this manuscript.
This research was supported in part by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (Grant no. NICHD 5T32HD052468) and the National Institute of Environmental Health Sciences (Grant no. NIEHS 5T32ES007018-38) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors report no conflicts of interest.
References
- 1.Conde-Agudelo A, Althabe F, Belizán JM, Kafury-Goeta AC. Cigarette smoking during pregnancy and risk of preeclampsia: a systematic review. Am J Obstet Gynecol. 1999;181(4):1026–1035. doi: 10.1016/s0002-9378(99)70341-8. [DOI] [PubMed] [Google Scholar]
- 2.Wei J, Liu C-X, Gong T-T, Wu Q, Wu L. Cigarette smoking during pregnancy and preeclampsia risk: a systematic review and meta-analysis of prospective studies. Oncotarget. 2015;6(41):43667–43678. doi: 10.18632/oncotarget.6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lisonkova S, Joseph KS. Left Truncation Bias as a Potential Explanation for the Protective Effect of Smoking on Preeclampsia. Epidemiology. 2015;26(3):436–440. doi: 10.1097/EDE.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howards PP, Hertz-Picciotto I, Poole C. Conditions for bias from differential left truncation. Am J Epidemiol. 2007;165(4):444–452. doi: 10.1093/aje/kwk027. [DOI] [PubMed] [Google Scholar]
- 5.Hernán MA, Hernández-Díaz S, Werler MM, Mitchell AA. Causal Knowledge as a Prerequisite for Confounding Evaluation: An Application to Birth Defects Epidemiology. Am J Epidemiol. 2002;155(2):176–184. doi: 10.1093/aje/155.2.176. [DOI] [PubMed] [Google Scholar]
- 6.Ghidini A, Salafia CM, Pezzullo JC. Placental vascular lesions and likelihood of diagnosis of preeclampsia. Obstet Gynecol. 1997;90(4 Pt 1):542–545. doi: 10.1016/s0029-7844(97)00360-8. [DOI] [PubMed] [Google Scholar]
- 7.Moldenhauer JS, Stanek J, Warshak C, Khoury J, Sibai B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am J Obstet Gynecol. 2003;189(4):1173–1177. doi: 10.1067/s0002-9378(03)00576-3. [DOI] [PubMed] [Google Scholar]
- 8.Vinnars M-T, Nasiell J, Ghazi S, Westgren M, Papadogiannakis N. The severity of clinical manifestations in preeclampsia correlates with the amount of placental infarction. Acta Obstet Gynecol Scand. 2011;90(1):19–25. doi: 10.1111/j.1600-0412.2010.01012.x. [DOI] [PubMed] [Google Scholar]
- 9.Dechanet C, Brunet C, Anahory T, Hamamah S, Hedon B, Dechaud H. Effects of cigarette smoking on embryo implantation and placentation and analysis of factors interfering with cigarette smoke effects (Part II) Gynecol Obstet Fertil. 2011;39(10):567–574. doi: 10.1016/j.gyobfe.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 10.Holloway A, Salomon A, Soares M, et al. Characterization of the adverse effects of nicotine on placental development: in vivo and in vitro studies. Am J Physiol Endocrinol Metab. 2014;306(4):E443–E456. doi: 10.1152/ajpendo.00478.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maldonado G, Greenland S. Estimating causal effects (with discussion) Int J Epidemiol. 2002;31:422–438. [PubMed] [Google Scholar]
- 12.Pearl J. Causal diagrams for empirical research. Biometrika. 1995;82(4):669–688. [Google Scholar]
- 13.Centers for Disease Control and Prevention. Smoking Prevalence Among Women of Reproductive Age — United States, 2006. MMWR Morb Mortal Wkly Rep. 2008;57(31):849–852. [PubMed] [Google Scholar]
- 14.Centers for Disease Control and Prevention. PRAMS and Smoking. 2012 http://www.cdc.gov/prams/tobaccoandprams.htm. Accessed October 6, 2015.
- 15.Maldonado G, Greenland S. The Importance of Critically Interpreting Simulation Studies. Epidemiology. 1997;8(4):453–456. [PubMed] [Google Scholar]
- 16.Yudkin PL, Wood L, Redman CW. Risk of unexplained stillbirth at different gestational ages. Lancet. 1987;1(8543):1192–1194. doi: 10.1016/s0140-6736(87)92154-4. [DOI] [PubMed] [Google Scholar]
- 17.Joseph KS. Theory of obstetrics: the fetuses-at-risk approach as a causal paradigm. J Obstet Gynaecol Canada. 2004;26(11):953–956. doi: 10.1016/s1701-2163(16)30414-5. [DOI] [PubMed] [Google Scholar]
- 18.Werler MM, Parker SE. Letter: Bias from conditioning on live birth in pregnancy cohorts: an illustration based on neurodevelopment in children after prenatal exposure to organic pollutants (Liew et al. 2015) Int J Epidemiol. 2015:1079–1080. doi: 10.1093/ije/dyv139. [DOI] [PubMed] [Google Scholar]
- 19.Hernández-Díaz S, Schisterman EF, Hernán MA. The birth weight “paradox” uncovered? Am J Epidemiol. 2006;164(11):1115–1120. doi: 10.1093/aje/kwj275. [DOI] [PubMed] [Google Scholar]
- 20.Banack HR, Kaufman JS. The obesity paradox: Understanding the effect of obesity on mortality among individuals with cardiovascular disease. Prev Med (Baltim) 2014;62:96–102. doi: 10.1016/j.ypmed.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Flanders WD, Eldridge RC, McClellan W. A nearly unavoidable mechanism for collider bias with index-event studies. Epidemiology. 2014;25(5):762–764. doi: 10.1097/EDE.0000000000000131. [DOI] [PubMed] [Google Scholar]
- 22.Liew Z, Olsen J, Cui X, Ritz B, Arah OA. Bias from conditioning on live birth in pregnancy cohorts: An illustration based on neurodevelopment in children after prenatal exposure to organic pollutants. Int J Epidemiol. 2015;44(1):345–354. doi: 10.1093/ije/dyu249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liew Z, Olsen J, Cui X, Ritz B, Arah OA. Response to Werler and Parker letter: Comment on live-birth bias in pregnancy cohorts. Int J Epidemiol. 2015:1080–1081. doi: 10.1093/ije/dyv140. [DOI] [PubMed] [Google Scholar]
- 24.Petersen ML, Sinisi SE, van der Laan MJ. Estimation of direct causal effects. Epidemiology. 2006;17(3):276–84. doi: 10.1097/01.ede.0000208475.99429.2d. [DOI] [PubMed] [Google Scholar]
- 25.VanderWeele TJ. Marginal structural models for the estimation of direct and indirect effects. Epidemiology. 2009;20(1):18–26. doi: 10.1097/EDE.0b013e31818f69ce. [DOI] [PubMed] [Google Scholar]
- 26.Vansteelandt S. Estimating direct effects in cohort and case-control studies. Epidemiology. 2009;20(6):851–860. doi: 10.1097/EDE.0b013e3181b6f4c9. [DOI] [PubMed] [Google Scholar]
- 27.Weuve J, Tchetgen Tchetgen EJ, Glymour MM, et al. Accounting for Bias Due to Selective Attrition: The Example of Smoking and Cognitive Decline. Epidemiology. 2012;23:119–128. doi: 10.1097/EDE.0b013e318230e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaix B, Evans D, Merlo J, Suzuki E. Commentary: Weighing up the Dead and Missing: Reflections on Inverse-probability Weighting and Principal Stratification to Address Truncation by Death. Epidemiology. 2012;23:129–131. doi: 10.1097/EDE.0b013e3182319159. [DOI] [PubMed] [Google Scholar]
- 29.Tchetgen Tchetgen EJ, Glymour MM, Shpitser I, Weuve J. Rejoinder: To Weight or Not to Weight?: On the Relation Between Inverse-probability Weighting and Principal Stratification for Truncation by Death. Epidemiology. 2012;23:132–137. [Google Scholar]
- 30.Baird DD, Wilcox AJ. Cigarette Smoking Associated With Delayed Conception. JAMA. 1985;253(20):2979–2983. [PubMed] [Google Scholar]
- 31.Weinberg CR, Wilcox AJ, Baird DD. Reduced fecundability in women with prenatal exposure to cigarette smoking. Am J Epidemiol. 1989;129(5):1072–1078. doi: 10.1093/oxfordjournals.aje.a115211. [DOI] [PubMed] [Google Scholar]
- 32.Curtis KM, Savitz DA, Arbuckle TE. Effects of cigarette smoking, caffeine consumption, and alcohol intake on fecundability. Am J Epidemiol. 1997;146(1):32–41. doi: 10.1093/oxfordjournals.aje.a009189. [DOI] [PubMed] [Google Scholar]
- 33.Bainbridge SA, Sidle EH, Smith GN. Direct placental effects of cigarette smoke protect women from pre-eclampsia: The specific roles of carbon monoxide and antioxidant systems in the placenta. Med Hypotheses. 2005;64(1):17–27. doi: 10.1016/j.mehy.2004.06.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL DIGITAL CONTENT 1. SAS macro to assess the impact of left truncation bias on the association between maternal smoking and preeclampsia. pdf.
SUPPLEMENTAL DIGITAL CONTENT 2. Text and tables that provide justification for our extreme and middling assumptions about the association between abnormal placentation and preeclampsia. pdf.
SUPPLEMENTAL DIGITAL CONTENT 3. Text and figure that provide data regarding our investigation of assumption 3, showing that it had negligible impact on bias compared to assumptions 1 and 2. pdf.