

Abstract
Purpose
Aberrant activation of the NF-κB transcription factors underlies the aggressive behavior and poor outcome of pancreatic ductal adenocarcinoma (PDAC). However, clinically effective and safe NF-κB inhibitors are not yet available. Because NF-κB transcription factors can be activated by the Interleukin-1 Receptor-Associated Kinase (IRAK) downstream of the Toll-like receptors (TLRs), but has not been explored in PDAC, we sought to investigate the role of IRAK in the pathobiology of PDAC.
Experimental Design
We examined the phosphorylation status of IRAK4 (p-IRAK4), the master regulator of TLR signaling, in PDAC cell lines, in surgical samples and commercial tissue microarray. We then performed functional studies using small molecule IRAK1/4 inhibitor, RNA-interference and CRISPR/Cas9n techniques to delineate the role of IRAK4 in NF-κB activity, chemoresistance, cytokine production and growth of PDAC cells in vitro and in vivo.
Results
p-IRAK4 staining was detectable in the majority of PDAC lines and about 60% of human PDAC samples. Presence of p-IRAK4 strongly correlated with phospho-NF-κB/p65 staining in PDAC samples and is predictive of postoperative relapse and poor overall survival. Inhibition of IRAK4 potently reduced NF-κB activity, anchorage-independent growth, chemoresistance and secretion of pro-inflammatory cytokines from PDAC cells. Both pharmacologic suppression and genetic ablation of IRAK4 greatly abolished PDAC growth in mice and augmented the therapeutic effect of gemcitabine by promoting apoptosis, reducing tumor cell proliferation and tumor fibrosis.
Conclusions
Our data established IRAK4 as a novel therapeutic target for PDAC treatment. Development of potent IRAK4 inhibitors is needed for clinical testing.
INTRODUCTION
To date, the prognosis of pancreatic ductal adenocarcinoma (PDAC) remains dismal with an overall 5-year-survival rate of about 8%, which has not changed in the last four decades(1, 2). In addition to the universal presence of strong oncogenic events such as activating mutation of KRas, the constitutive activation of the NF-κB transcription factors is another major mechanism that drives the aggressive behavior of PDAC(3–6). Understanding how NF-κB is activated in PDAC is clinically important and has been an area of intense interest(5). For instance, expression of oncogenic KRas upregulates IL-1α and GSK3α production to activate the NF-κB pathway in a feedforward manner(7, 8). High miR-301a level suppresses translation of NF-κB repressing factor protein, thereby activating NF-κB, which in turn enhances miR-301a expression to create a positive feedforward loop(9). However, direct inhibitors of NF-κB or microRNAs have yet to be successful in clinic, indicating the need to explore other strategies to curb this pathway.
Another element that contributes to the recalcitrant nature of PDAC, in addition to its strong intrinsic survival signaling, is the uniquely inflamed and immunosuppressive extrinsic microenvironment, which is believed to hamper cytotoxic T lymphocyte infiltration and drug delivery(10, 11). In normal tissue, inflammation is triggered by the engagement of the Toll-like receptors (TLRs), which activate the innate immune response through the NF-κB and p38/MAPK signaling cascades(12). Several lines of evidence indicate that stimulation of the TLRs could drive NF-κB activity in PDAC. For instance, ligation of TLR4 enhances the invasiveness of PDAC cells in an NF-κB-dependent manner(13). In genetically-engineerd mouse models, ligation of TLR7 or TLR9 accelerates stromal inflammation and progression of PDAC activation of the NF-κB and MAPK pathways(14, 15). However, the intrinsic role of TLR signaling in neoplastic PDAC cells and how it affects the extrinsic tumor microenvironemt have not been thoroughly described.
In this study, we demonstrate for the first time, that the Interleukin-1 Receptor-Associated Kinase 4 (IRAK4), the master kinase that relays signaling downstream of TLRs(16, 17), is constitutively activated in PDAC cell lines, patient-derived cell lines and tumor samples. Activated IRAK4 staining positively correlates with activated NF-κB in human PDAC samples, and importantly, portends high postoperative relapse and poor patient survival. We showed that pharmacological blockade or silencing of IRAK4 potently suppressed NF-κB activity, abolished the tumorigenic potential of human and murine PDAC cells, and greatly sensitized PDAC cells to various chemotherapeutics in vivo and in vitro. Our data lay a solid foundation for testing IRAK4 inhibitors as a novel class of therapeutic agents for PDAC treatment.
MATERIAL AND METHODS
(Further details included in supplementary data)
PDAC lines
Human PDAC lines AsPC-1, BxPC-3, Capan-1, Capan-2, CFPAC-1, HPAF-II, HPAC, MIA Paca-2, PANC-1, SW1990, and HPNE cells were purchased from ATCC, which performs authentication on its own cell lines. All cells were used for fewer than 6 months after receipt or resuscitation from cryopreservation. HPDE, MPanc96, Hs766T were kind gifts from Dr. David Linehan (University of Rochester, NY). KRas-Ink4a cells were a kind gift from Dr. David DeNardo (Washington University, MO) and were published(18). Patient-derived PDAC cell lines (PDCLs) Pa01c, Pa02c and Pa14c were kind gifts from Dr. Channing Der (University of North Carolina, NC) and published(19, 20).
Cancer-associated fibroblasts (CAFs)
Human CAFs were isolated from the PDAC specimen of a patient who underwent Whipple procedure and expanded using a published method(21). CAFs were confirmed to be α-SMA-positive and pan-cytokeratin-negative by immunofluorescence.
Human PDAC samples, tissue microarray and survival analysis
FFPE PDAC and matched adjacent normal pancreatic tissues diagnosed between year 2000 and 2010 were retrieved from the Department of Pathology and Immunology at WUSTL under IRB-approved protocol (#201404143). All selected patients had documented follow up to 6 years after surgery. None received neoadjuvant treatment prior to surgery but all received adjuvant treatment containing either 5-FU or gemcitabine. Pancreatic tissue array (catalog#PA961B) was purchased from US Biomax.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
IHC were performed using p-IRAK4 (T345S) (A8A8, ABNOVA, 1:200), p-NF-κB/p65 (S536) (Abcam ab86299, 1:200), p-TAK1 (S184/187) (Bioss bs-3439R, 1:200). IHC scores were determined by the sum of Intensity (0: none, 1: faint, 2: weak, 3: moderate, 4: strong) and Distribution (0: none, 1:<25%, 2:25–50%, 3:50–75%, 4:>75%) sub-scores. All samples were scored two times in a blinded manner. Further details provided in supplementary data.
Plasmids and creation of stable cell lines
Stable knockdowns were generated using pSuper-Retro-Puro or neo/GFP plasmids encoding scramble sequence, or indicated shRNAs as described(22). Further details included in supplementary data.
Drugs and reagents
5-FU, gemcitabine and paclitaxel were purchased from the Siteman Cancer Center Pharmacy. The IRAK1/4 inhibitor, N-(2-Morpholinylethyl)-2-(3-nitrobenzoylamido)-benzimidazole, was purchased from Sigma (I5409). IMD-0354 was purchased from Selleckchem.
In vitro cell viability assay, synergism analysis and soft agar assay
Viability was assayed using Resazurin colorimetric analysis; synergism studies were performed as described(23); soft agar assay was performed as described(24). Further details provided in supplementary data.
In Vitro CAF Migration/Invasion Assay
CAFs were added to rehydrated 8 µm pore, 24-well cell culture inserts (with or without collagen, BD Bioscience). 200uL of serum-free conditioned media from PANC-1 or Capan-1 cells were added to lower chambers. After 24 hours, uninvaded CAFs were removed from the upper chamber and invaded cells were fixed with 4% paraformaldehyde, stained with toluidine blue overnight and counted under microscope.
Immunoblotting and quantitative real-time PCR
Details provided in supplementary data.
Cytokine Array Analysis
Human Cytokine Antibody Array C3 (cat# AAH-CYT-3) was purchased from Raybio. Membranes were incubated with serum-free conditioned media collected from 70–80% confluent Capan-1 cells treated with DMSO or 10µM of IRAK1/4i overnight (about 16 hours) and processed according to manufacturer’s protocol. Identical results were obtained from a repeat experiment using different sets of Capan-1 conditioned media.
NF-κB reporter assay
NF-κB reporter activity was performed using Dual-Glo® Luciferase Assay System and read with Synergy H4 Hybrid Multi-Mode Microplate Reader. All experiments were done three times in triplicate and data represented as means ± S.E.M.
Xenograft tumorigenesis assay and in vivo bioluminescence imaging
All animal experiments were conducted according to IACUC protocol (#20130191). Details provided in supplementary data.
Statistical Analysis
Normal distributions were compared by 2-tailed Student’s t test or ANOVA; tumor growth curves were compared using Mann-Whitney U tests; survivals were analyzed using Kaplan-Meier method and Log-rank test. Statistical analyses were performed with Graphpad Prism v6.0. P-values <0.05 were considered as statistically significant.
RESULTS
IRAK4 is constitutively phosphorylated in human PDAC cell lines, surgical samples and is associated with poor prognosis
To explore the intrinsic role of TLR signaling in PDAC, we first investigated the phosphorylation/activation status of Interleukin-1 Receptor-Associated kinases (IRAK)(25, 26), particularly IRAK1 and IRAK4 in a panel of well-characterized human PDAC lines. We found IRAK1 to be constitutively phosphorylated in nine, and IRAK4 in eleven, of twelve lines in the absence of TLR ligand stimulation. In contrast, IRAK4 phosphorylation was barely detected in both, whereas IRAK1 was weakly phosphorylated in one, of two non-transformed pancreatic ductal cell lines, HPNE and HPDE (Fig. 1A). Consistent with published literature, the majority of PDAC lines also showed constitutive phosphorylation of NF-κB/p65 and iκB(5, 6). Similarly, p-IRAK4 is detectable in four early-passaged patient-derived cell lines (PDCLs), along with increased p-NF-κB/p65 and p-iκBα (Supplementary Fig. S1A).
Figure 1. IRAK4 is constitutively phosphorylated in PDAC cell lines and surgical samples.

A, western blots showing phosphorylation status of IRAK1, IRAK4, NF-κB and iκBα in twelve PDAC lines versus two non-transformed pancreatic ductal lines (HPDE and HPNE). B, representative IHC images showing p-IRAK4 staining in PDAC surgical samples (N=103), but not in the adjacent normal pancreatic tissue, of two patients. Total IRAK4 staining is present in both normal and PDAC tissues (scale bar: 200µm). C, scatter plot showing quantification of p-IRAK4 IHC score on matched normal and PDAC samples (N=30 each), and all PDAC cases (N=103) (ns: not significant). D, representative IHC images showing overlap of p-IRAK4 and p-NF-κB/p65 staining in PDAC tissues of two patients. E, correlation analysis of p-IRAK4 and p-NF-κB/p65 IHC intensity of 85 PDAC samples from tissue microarray. Shown here is best-fit line with 95% confidence interval. F, representative H&E and IHC images of the pancreas of KPC mice during progression from pre-neoplastic PanIN lesions to PDAC. G, Relapse-free survival (RFS) and overall survival (OS) by Kaplan-Meier analysis of PDAC patients who underwent Whipple resection as stratified by presence or absence of p-IRAK4 staining (N=73).
We next investigated the phosphorylation status of IRAK4 (p-IRAK4) in 103 resected human PDAC samples by immunohistochemistry using a phospho-antibody that was previously reported in melanoma(27). We found p-IRAK4 to be present and significantly stronger in the ductal epithelia of 20 out of 30 (or 66.7%) PDAC samples (Fig. 1B and 1C), compared to their matched normal panceratic tissues. In all 103 PDAC samples analyzed, p-IRAK4 expression is present in 58.8% of cases at various intensities and extent in the neoplastic ductal cells (Fig. 1C), which is significantly higher than normal pancreatic tissue (p=0.0002). As a control, total IRAK4 protein is detected in both normal pancreatic and PDAC tissues (Fig. 1B). We next validated this finding using a commercially available pancreatic tissue microarray, and found p-IRAK4 IHC stainig to be present at various intensities in 63.5% of PDAC samples, which is comparable to our findings in surgical samples (Supplementary Fig. S1B and S1C).
To gain an initial insight of whether p-IRAK4 may contribute to NF-κB activaty in PDAC, we next determined the correlation between the staining intensities of p-IRAK4 and p-NF-κB/p65. We found complete overlap of p-IRAK4 and p-NF-κB/p65 IHC staining in the neoplastic epithelia of surgical PDAC samples (Fig. 1D). IHC analysis of 85 PDAC samples from tissue microarray showed various degrees of p-NF-κB /p65 staining intensities in 87% of samples (Supplementary Fig. S1B, S1D) and importantly, we noted strong correlation between the staining intensities of p-IRAK4 and p-NF-κB/p65 (Spearmann R 0.87, p<0.0001; Fig. 1E). In a genetically-engineered p48-Cre;p53Flox/WT;LSL-KRasG12D (or KPC) mouse model of PDAC, both p-IRAK4 and p-NF-κB/p65 stainings are present at very low level starting from early Pancreatic Intraepithelial Neoplasia (PanIN-1) and become increasingly stronger during progression to higher grade PanIN-2, PanIN-3 and PDAC (Fig. 1F), indicating a potential role of IRAK4 and NF-κB activity during PDAC development.
Since p-IRAK4 staining is detectable in the majority but not all PDAC samples, we determined whether p-IRAK4 staining is associated with prognosis. Remarkably, we found that patients with positive p-IRAK4 PDAC had significantly higher chance of postsurgical relapse and worse overall survival despite adjuvant treatment, compared to patients with negative p-IRAK4 tumors (6-year median relapse-free survival: 12.56 vs. 32.95 months, log-rank p=0.0122; 6-year median overall survival: not reached vs. 19.05 months, log-rank p=0.0065). We observed no difference in tumor size, nodal status, tumor grade, age and gender distribution between p-IRAK4-positive versus -negative tumor (not shown), establishing IRAK4 activation as an independent prognostic factor in PDAC. Together, our findings conclude that IRAK4 is frequently activated and associated with aggressive behaviour of PDAC, and provide a strong rationale to investigate the pathogenic role of IRAK4 in PDAC.
IRAK1/4 drives NF-κB activity and anchorage-independent growth of PDAC cells
To determine whether IRAK4 contributes to NF-κB activity in PDAC cells, we treated PDAC lines with a widely-described dual IRAK1/4 inhibitor(28). We found dose-dependent suppression of phosphorylated IRAK1, IRAK4, iκBα and NF-κB/p65 in all four PDAC lines tested (Fig. 2A). Consistently, IRAK1/4i significantly suppressed NF-κB-driven luciferase activity in five out of six PDAC lines, but not in BxPC-3 which has barely detectable p-IRAK1 and p-IRAK4 (Fig. 2B). Furthermore, IRAK1/4i dose-dependently suppressed the anchorage-independent (AI) growth of eight out of ten established PDAC lines in soft agar, and all three PDCLs tested, but had no significant suppression on the viability of HPNE, HPDE and most PDAC cells in monolayer culture except for mild effect in PANC-1 (Fig. 2C, Supplementary Fig. S2A, S2B, S2C). To exclude the non-specific effect of IRAK1/4i, ectopic expression of a constitutively active IKKβ mutant (S177E/S181E) rendered IRAK1/4i ineffective in suppressing the NF-κB-driven reporter activity and AI growth of PANC-1 cells (Fig. 2D, 2E). As a complementary experiment, although the NF-κB activity of BxPc-3 line is independent of IRAK1/4 inhibition and can be suppressed by the IKKβ inhibitor IMD-0354, overexpression of wild-type, but not kinase-dead IRAK4 significantly increased p-NF-κB, AI growth and rendered BxPC-3 cells sensitive to IRAK1/4i (Fig. 2F and 2G). These results suggest that the NF-κB activity of PDAC is dependent on IRAK1/4.
Figure 2. IRAK4 kinase contributes to NF-κB activity and anchorage-independent (AI) growth of PDAC cells.

A, western blots showing suppression of p-IRAK1, p-IRAK4, p-NF-κB/p65 and p-iκBα in the indicated PDAC lines following overnight treatment with IRAK1/4 inhibitor at two different concentrations. B, NF-κB luciferase reporter assay showing dose-dependent suppression of relative NF-κB activity (Firefly luciferase activity/Renilla) following overnight treatment of IRAK1/4 inhibitor in six PDAC lines stably expressing NF-κB-driven firefly luciferase and Renilla; means ± S.E.M. C, AI growth of PDAC lines seeded in DMSO or two different concentrations of IRAK1/4i for 3–4 weeks. D, relative NF-κB luciferase reporter activity and E, AI growth of PANC-1 cells stably expressing vector, wild-type IKKβ or constitutively active IKKβ (S177E, S181E) treated with DMSO or two different concentrations of IRAK1/4i overnight. F, relative NF-κB reporter activity and G, western blots showing changes in p-NF-κB/p65 of BxPc-3 cells stably expressing vector, wild-type IRAK4 or kinase-dead IRAK4 (K213A, K214A) treated with DMSO or two different concentrations of IRAK1/4i overnight. H, western blots showing changes of p-IKKα/β, p-NF-κB/p65 and p-IRAK4 following stable knockdown of IRAK1, IRAK4 or both in PDAC cell lines. I, effect of IRAK1 or/and IRAK4 knockdown on AI growth of the indicated PDAC lines. J, AI growth of PDAC lines stably expressing scramble shRNA or shIRAK4. K, AI growth of IRAK4-ablated PANC-1 and Pa01c lines generated by CRISPR/Cas9n technology. L, AI growth of Capan-1 and PANC-1 cells stably expressing scramble shRNA, shIRAK1 or shIRAK4 followed by rescue with empty vector, wild-type or kinase-dead IRAKs. M, western blots showing changes in p-IKKα/β and p-NF-κB/p65 of PANC-1 cells transduced with shRNA against IRAK1 followed by reexpression of vector, wild-type IRAK1 or kinase-dead IRAK1(K239A), or sgRNA against IRAK4 followed by reexpression of vector, wild-type IRAK4 or kinase-dead IRAK4 (K213A,K214A). (*p<0.05, **p<0.01, ***p<0.001 by two-tailed t test).
To further dissect the individual role of IRAK1 and IRAK4 in NF-κb activation, we performed shRNA knockdown of IRAK1, IRAK4, or both in PDAC lines. Stable knockdown of IRAK1 or IRAK4 by shRNAs, and more so with both, potently inhibits p-IKKα/β and p-NF-κB in human PDAC lines and a murine PDAC cell line (KRas-Ink4a) derived from transgenic KRasG12D; Ink4a/Arf−/− mice (Fig. 2H, Suppl. Fig. 2D). Interestingly, knockdown of IRAK1 had little effect on p-IRAK4 in two PDAC lines tested (Fig. 2H), suggesting either insufficient IRAK1 knockdown or that IRAK4 is activated independent of IRAK1 in PDAC. Consistent with our findings with IRAK1/4i, knockdown of IRAK4 or both IRAK1 and IRAK4, and less so of IRAK1, reduce AI growth of human and murine PDAC lines (Fig. 2I, 2J, Supplementary Fig. S2E). In PANC-1 cells, the suppressive effect of shIRAK1+4 on AI growth could be rescued with re-introduction of constitutively-active IKKβ mutant (Supplementary Fig. S2F), again supporting the notion that the IRAKs function predominantly through the IKK-NF-κB cascade in PDAC. Furthermore, genetic ablation of IRAK4 with CRISPR/Cas9n technique significantly reduced AI growth of PANC-1 and a PDCL line, Pa01c (Fig. 2K).
We next performed knockdown-rescue experiments to delineate the requirement of kinase function of IRAK1 and IRAK4 in PDAC cells growth. We found that the decreased soft agar growth in shIRAK4-transduced Capan-1 and PANC-1 cells could be rescued by re-introduction of wild-type but not kinase-dead IRAK4 (KK213AA); whereas the suppressive effect of shIRAK1 could be rescued with both wild-type and kinase-dead IRAK1 (K239A). Supporting this finding and consistent with published reports(29–31), the kinase activity of IRAK4, but not IRAK1, is essential in activing IKKα/β and NF-κb in PANC-1 cells (Fig. 2M). Resonating this observation, we also found that TLR7-induced NF-κB activation in HEK293T cells requires the kinase activity of IRAK4, but not IRAK1 (Suppl. Fig. S2G). Together, our data concluded that IRAK4 kinase activity is essential in driving NF-κb activity in PDAC and should be explored as a therapeutic target.
Suppression of IRAK1/4 induces apoptosis and sensitizes pancreatic cancer cells to chemotherapeutic agents
Activated NF-κB is a major mechanism that underlie chemoresistance in PDAC(5, 6, 32). Since the IRAK kinases drive NF-κB activity in PDAC cells, we next determined whether suppression of IRAK1/4 augments the cytotoxic effect of chemotherapeutics. We found that exposure of Capan-1 and PANC-1 cells to gemcitabine significantly upregulates NF-κB activity, as reported(32). However, such induction could be stifled by IRAK1/4i (Fig. 3A). Mechanistically, knockdown of IRAK1 and/or IRAK4 was sufficient to induce PARP cleavage, an indicator of apoptosis, which was further enhanced by exposure to gemcitabine (Fig. 3B). The chemo-sensitization effect of shIRAK1+4 could be partially rescued by re-introduction of constitutively active IKKβ mutant (Fig. 3C), again supporting IRAK1/4 as therapeutic targets upstream of NF-κB. Consistently, PDAC cells stably expressing shIRAK1 or shIRAK4, and to a much greater extent, both, were significantly more sensitive to the killing effect of gemcitabine and 5-fluorouracil, two commonly used chemotherapeutics in PDAC treatment (Fig. 3D), as shown by significant reductions in IC50 of both chemotherapeutic agents compared to scramble cells. (Supplementary Table 1). To translate these findings, we perform synergism analysis by treating Capan-1, MIA Paca-2 and PANC-1 with combination mixtures of IRAK1/4i plus each chemotherapeutic agent at constant ratios, as described(23). In all three PDAC lines, we observed various degrees of synergism between IRAK1/4i and each chemotherapeutic agent (Fig. 3E). Overall, our findings provide a strong rationale for testing IRAK1/4i plus chemotherapy in vivo.
Figure 3. IRAK1/4 inhibition sensitizes PDAC cells to the cytotoxic effect of chemotherapy in vitro.

A, NF-κB luciferase reporter assay showing suppressive effect of IRAK1/4i in gemcitabine-induced NF-κB activation in Capan-1 cells. B, western blots showing increased PARP cleavage following introduction of shIRAK1, shIRAK4 or both, in PDAC lines treated with DMSO or 10µM of for 48 hours. C, western blots showing partial reversal of gemcitabine-induced PARP cleavage by re-expression of activated IKKβ in shIRAK1+4-expressing PANC-1 and Pa01c cells. D, Alamar blue assays showing increased sensitivity of shIRAK1, shIRAK4 or shIRAK1+IRAK4 PDAC cell lines to gemcitabine or 5-Fluorouracil (5-FU). Each graph represents one of three independent experiments done in triplicates; means ± S.E.M. Change in IC50 is provided in Supplementary Table 1. E, Median effect analysis showing interaction between IRAK1/4i with chemotherapeutic agents in three different PDAC lines analyzed using Compusyn software. Data represents combined means ± S.E.M of three independent experiments each performed in duplicates. Horizontal dotted lines indicate the boundaries for each interaction classification(23).
IRAK1/4 drives production of inflammatory cytokines and chemokines that drive migration, invasion and proliferation of cancer-associated fibroblasts (CAFs)
Targeting the desmoplastic tumor microenvironment (TME) is emerging as another attractive therapeutic strategy in PDAC. The highly inflamed and immunosuppressive TME is widely believed to be driven by cytokines and chemokines secreted by PDAC cells(7, 11, 33–35). Because IRAK activation induces cytokine secretion in normal innate immune response, we speculate that production of inflammatory cytokines/chemokines in PDAC is also regulated by IRAK. Indeed, the abundance of IL-1α, IL1-β, IL-8, PDGF, GM-CSF, CCL2, CXCL1 and CXCL2 in the conditioned medium of Capan-1 cells was reduced following IRAK1/4i treatment (Fig. 4A). As controls, secretion of TNF-α, TNF-β and EGF were slightly increased, whereas other factors including TGFβ and VEGF were unchanged following IRAK1/4i treatment. These changes were confirmed by qRT-PCR using Capan-1 cells treated with IRAK1/4i or transduced with IRAK1 or/and IRAK4 shRNAs (Fig. 4B and 4C). Interestingly, knockdown of IRAK1 had less pronounced effect on the transcription of these factors compared to IRAK4- or double-knockdown cells, suggesting IRAK4 as the predominant driver in cytokine/chemokine production in PDAC (Fig. 4C). As confirmation, we observed very similar, but not identical, changes in production of these secreted factors in another PDAC line, PANC-1 (Fig.4B). Notably, the suppressive of shIRAK4 on cytokine/chemokine production in PANC-1 could be partially reversed by re-introduction of activated IKKβ mutant (Fig. 3D).
Figure 4. IRAK1/4 controls secretion of inflammatory chemokines and cytokines from PDAC cells to augment invasion, migration and viability of cancer-associated fibroblasts (CAFs).

A, cytokine array showing altered secretion of chemokines/cytokines in serum-free conditioned media collected from Capan-1 cells treated with either DMSO or 10µM of IRAK1/4 inhibitor overnight. Identical results were obtained from a repeat experiment. B, RT-PCR showing fold changes in mRNA levels of the indicated genes in Capan-1 and PANC-1 cells treatment with 10µM of IRAK1/4 inhibitor relative to DMSO overnight. C, RT-PCR showing fold change in mRNA levels of the indicated genes in Capan-1 cells transfected with shRNA against IRAK1 or/and IRAK4, relative to scramble transfected cells. D, qRT-PCR showing fold change of the indicated genes in PANC-1 cells transfected with shIRAK4#1 with empty vector or activated IKKβ (SSEE) mutant. All qRT-PCR were done at least twice, each in three biological replicates and three technical triplicates normalized to GAPDH. Final fold changes were compared to either vehicle- or vector-treated cells. Data represents means ± S.E.M. E, schematics showing experimental design to elucidate the effect of conditioned media collected from PDAC cells on α−SMA+ CAFs. F, G, H, transwell collagen invasion, migration and viability by Alamar blue assay of CAFs cultured in serum-free conditioned media collected from Capan-1 or PANC-1 cells stably expressing scramble shRNA, shIRAK1, shIRAK4, or both. Data presented as means ± S.E.M. Experiments were done twice in triplicates. (*p<0.05, **p<0.01, ***p<0.001 by ANOVA).
Since many of these affected chemokines/cytokines are known to induce desmoplasia, we next asked whether IRAK1/4 inhibition would impair the ability of PDAC cells to stimulate the growth and behavior of cancer-associated fibroblasts (CAFs) (Fig. 4E). Indeed, condition media collected from IRAK1, IRAK4 or IRAK1/4 knocked-down Capan-1 and PANC-1 cells were greatly defective in inducing invasion, migration and proliferation of CAFs (Fig. 4F, 4G, and 4H). Based on these results, we speculate that, in addition to curbing the survival of neoplastic PDAC cells, IRAK1/4 inhibition can attenuate the fibrotic tumor microenvironment of PDAC and is an attractive therapeutic strategy.
IRAK4 kinase is essential for pancreatic tumorigenesis in vivo
We next tested whether inhibition of IRAK1 and/or IRAK4 can impede PDAC growth in vivo. Consistent with our findings in soft agar, knockdown of IRAK1 or IRAK4 significantly thwarted the tumorigenic potential of PANC-1 cells and extended the survival of tumor-bearing mice (Fig. 5A, 5B). Strikingly, shIRAK1+shIRAK4 or IRAK4-ablated PANC-1 cells completely lost tumorigenic potential in vivo, although one mouse from each group died unexpectedly from unknown cause (Fig. 5A, 5B). Consistent with our previous findings (Fig. 2L), the suppressive effect of shIRAK4 in vivo could be rescued by re-expression of wild-type, but not kinase-dead IRAK4 (Fig. 5C). We next confirmed these findings using another PDAC line, Capan-1 which stably expresses firefly luciferase and either scramble shRNA (Group 1) or shIRAK4 (Group 2). We chose to focus on IRAK4 since it is now the target of interest. Treatment was started once tumor establishment was confirmed by palpation and positive bioluminescence signal. Group 1 mice were randomized to receive vehicle, gemcitabine, IRAK1/4 inhibitor, or both; whereas Group 2 mice were treated with vehicle or gemcitabine. In Group 1 mice, combined gemcitabine and IRAK1/4i significantly impeded tumor growth, although IRAK1/4 inhibitor alone showed modest yet statistically significant reduction in tumor growth compared to vehicle (Fig. 5E). Consistently, Capan-1 tumors treated with both gemcitabine and IRAK1/4i, and less so with IRAK1/4i alone, had significantly lower bioluminescence signals compared to vehicle- or gemcitabine-treated tumors (Fig. 5E). Notably, mice in all four cohorts showed no sign of distress or body weight loss during treatment (Supplementary Fig. 3A). Contrary to scramble tumors, shIRAK4 (Group 2) tumors could be suppressed by gemcitabine alone (Fig. 5D), resonating previous findings that shIRAK4 sensitizes PDAC cells to gemcitabine in vitro (Fig. 3D).
Figure 5. Genetic or pharmacologic suppression of IRAK4 abrogates PDAC tumorigenesis and augments the therapeutic effect of gemcitabine in vivo.

A, growth kinetics and B, Kaplan-Meier curves of PANC-1 cells stably expressing a scramble shRNA, shIRAK1, shIRAK4 or both, or PANC-1 cells with IRAK4 ablated with by CRISPR/Cas9n, following subcutaneous inoculation in nude mice. C, growth kinetics of PANC-1 cells stably expressing the indicated shRNA and IRAK4 rescue variants following subcutaneous inoculation in nude mice. D, growth kinetics of nude mice inoculated subcutaneously at bilateral flanks with Capan-1/luciferase cells stably expressing scramble shRNA (Group 1) or shIRAK4 (Group 2), which were then treated as indicated after tumors have achieved a volume of 50–100mm3. (N = 5 mice, 10 tumors/cohort). Bioluminescence imaging (BLI) was performed at 44 days after inoculation when scramble-arm reached maximum allowed volume. E, box plot showing relative fold change of bioluminescence readings (final/initial readings), of Capan-1 tumors treated as indicated at day 44. F, Kaplan-Meier survival of FVB/NJ mice orthotopically inoculated with KRas-Ink4a cells stably expressing a scramble shRNA or shIRAK1+shIRAK4. G, final average tumor weights ± S.E.M from the pancreas of immunocompetent FVB/NJ mice orthotopically inoculated with of KRas-Ink4a cells which were then treated with either DMSO, gemcitabine, IRAK1/4i or both (N=8/cohort). (* P<0.05, **P<0.01, ***P<0.001 by ANOVA).
Next, to better mimic the native tumor microenvironment and to understand the consequence of systemic IRAK inhibition in an immunocompetent setting, we resorted to the murine KRas-Ink4a line which can be grown in syngeneic immuno-competent FVB/NJ mice(18). Strikingly, mice orthotopically inoculated with shIRAK1+shIRAK4 KRas-Ink4a cells are completely healthy and showed no signs of distress for up to 6 months, whereas mice inoculated with shLacZ KRas-Ink4a cells all died by 40 days from large palpable tumors (Fig. 5F). On necropsy, none of the ten pancreas inoculated with shIRAK1+shIRAK4 KRas-Ink4a cells showed any macro- or microscopic trace of tumor growth (not shown). This striking result again supports a critical role of IRAK in PDAC tumorigenesis. Next, we treated FVB/NJ mice with either vehicle, gemcitabine, IRAK1/4i, or both, ten days after orthotopic inoculation with KRas-Ink4a cells. All mice were sacrificed when the vehicle-treated mice reached moribund status due to large palpable tumor or ascites. We found that mice treated with both gemcitabine and IRAK1/4i showed significantly reduced pancreatic tumor weight, compared to vehicle or gemcitabine-treated cohorts (Fig. 5G). Again, no mice showed any sign of toxicity or body weight loss during treatment (Supplementary Fig. 3A). Together, our data supports testing IRAK4 inhibitor in combination with chemotherapy in PDAC treatment in clinical trial.
IRAK1/4i augments the cytotoxic effect of gemcitabine and suppresses tumor fibrosis
To study the in vivo effect of IRAK1/4i on PDAC, we analyzed tumors harvested from both the Capan-1 (Fig. 5D) and KRas-Ink4a (Fig. 5G) experiments. At lower magnifications, combo-treated tumors showed larger areas of necrosis compared to the other cohorts (Fig. 6A and Supplementary Fig. S3B). By multicolor immunofluorescence, combo-treated tumors showed significantly less proliferative cells (double Ki-67+, cytokeratin+), higher apoptotic cells (cleaved-caspase-3+; cytokeratin+) (Fig. 6A and Supplementary Fig. S3B); and less stromal fibroblasts (by α-SMA area) and fibrosis by trichrome staining (Fig. 6B and Supplementary Fig. S3B). Importantly, tumors treated with IRAK1/4i showed weaker, but not loss, of p-IRAK4 and p-TAK1 (a substrate of IRAK4) staining, compared to vehicle- or gemcitabine-treated tumors (Fig. 6C), indicating on-target effect. However, this observation also explains why knockdown of IRAK1 and IRAK4, or knockout of IRAK4 were much more effective in blocking tumorigenesis, underscoring the need to develop more potent IRAK4 inhibitor for clinical testing in the future.
Figure 6. IRAK1/4i cooperates with gemcitabine in inducing apoptosis, suppressing proliferation and fibrosis of PDAC tumor in vivo.
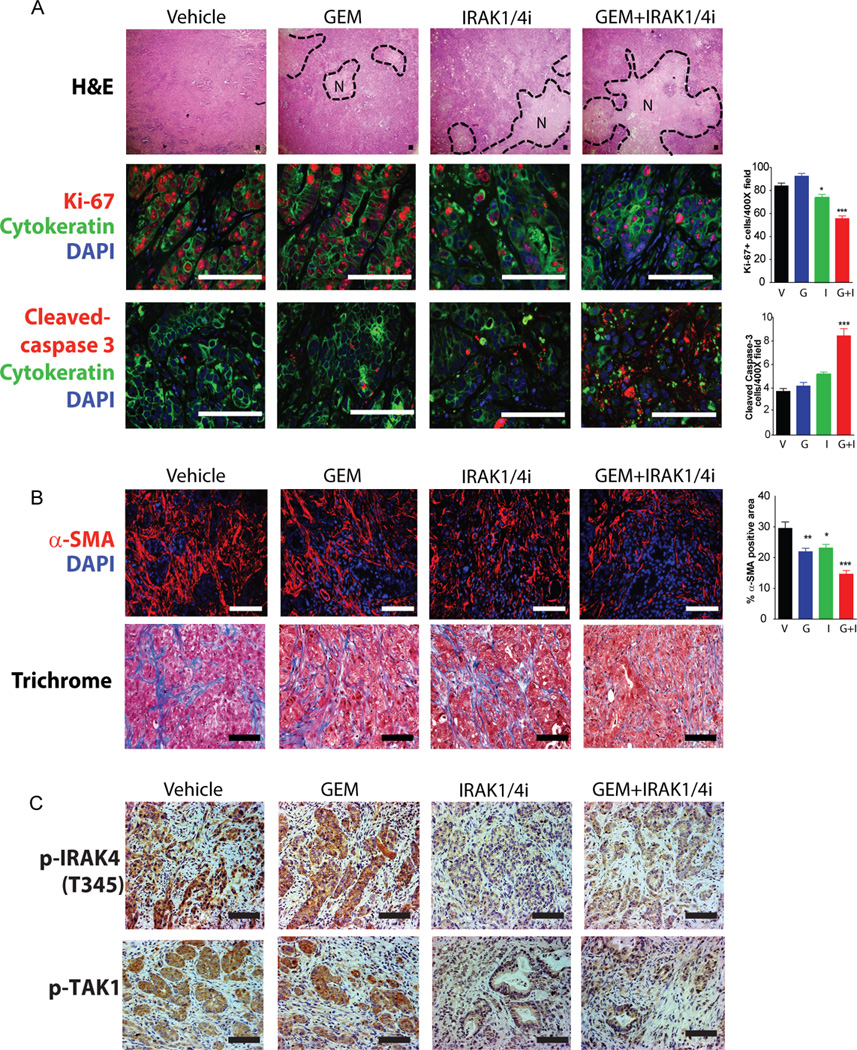
A, representative H&E and multicolor IF images of Capan-1 tumors treated as indicated. H&E staining showed large areas of necrosis (N) in tumors treated with both IRAK1/4 inhibitor and gemcitabine. Representative confocal IF images showing proliferation (Ki-67/red, cytokeratin/green, DAPI/blue) and apoptosis (cleaved-caspase-3/red, cytokeratin/green, DAPI/blue) of tumors in each treatment group. B, representative images showing changes in α-SMA+ area (CAFs/red, DAPI/blue) and fibrosis (blue) by trichrome staining. C, representative IHC images of p-IRAK4 and p-TAK1 (a substrate of IRAK4) showing on-target effect of IRAK1/4i in vivo. Quantitative analyses were performed by analyzing multiple fields (8 tumors/group and 10 random 200X or 400X fields/tumor) using NIS-Elements software, and presented as means ± SEM. (*P<0.05, **P<0.01, ***P<0.001 by ANOVA; scale bar: 50um)
DISCUSSION
Overcoming the pathogenic NF-κB signaling is an actively-pursued therapeutic strategy in PDAC(5). In this study we demonstrate that PDAC commonly activates NF-κB through the innate immune IRAK kinases. Notably, presence of p-IRAK4 is strongly associated with higher chance of relapse and poor overall prognosis. However, it should be pointed out that our analysis using surgical PDAC tumors is confined to patients with resectable disease, which represents only 10–15% of all PDAC cases(2). Patients with advanced or metastatic disease are routinely diagnosed with fine-needle aspiration, which does not allow for this kind of histologic study. Nonetheless, 80% of patients who underwent surgical resection eventually succumb to disease relapse despite adjuvant therapy(2), so identifying the mechanisms that impact patient survival is crucial for the development of effective adjuvant regimen to improve patient outcome. Herein, we provide proof-of-principle that suppression of IRAK4 can greatly cripple the growth of PDAC and augment the effect of chemotherapy in vitro and in vivo. However, in contrast to the dramatic effect of genetic ablation of IRAK4, the relatively modest antitumor effect of IRAK1/4i used in this study underscores the need to develop more potent IRAK4 inhibitors for clinical testing.
Aside from enhancing the survival of PDAC cells, IRAK4 appears to be a master regulator of a multitude of cytokines and chemokines including GM-CSF, CCL2, CXCL1, CXCL2, IL-8, and IL-β, which are all known to modulate the intensely fibrotic and immune-suppressive tumor microenvironment of PDAC. For instance, PDAC-derived GM-CSF promotes recruitment of Gr1+CD11b+ myeloid cells, which inhibits CD8+ T cells(36). The abundance of CCL2 in the PDAC microenvironment engages CCR2 receptor on tumor-associated macrophages to suppress CD8+ cells and enhances tumor-initiating stem cells(18, 37). As such, addition of CCR2 inhibitor to chemotherapy FOLFIRINOX achieved an objective response of 49% in patients with metastatic PDAC(38). Necroptosis-induced CXCL1 secretion fuels PDAC progression by promoting macrophage-induced immune suppression(39). In an elegant study by Ling et al(7), expression of oncogenic KRas activates NF-κB through enhanced secretion of IL-1α. Although we did observe increased expression of IL-1α following expression of KRasG12D in HPNE and HPDE cells, as published(7), we failed to detect upregulation of p-IRAK4 compared to vector control cells (not shown), indicating that the expression of oncogenic KRas, at least in cell culture model, is insufficient to induce IRAK4 phosphorylation. Therefore, further work using genetically-engineered mouse models is warranted to clearly establish the signaling interplay between oncogenic KRas and the IRAK-IKK-NF-κB cascades in PDAC development.
Aside from PDAC, targeting the IRAK1/4 cascade has been shown to be a promising strategy in other malignancies including DLBCL(29, 40), myelodysplastic syndrome(41), T-cell ALL(42), melanoma(27), breast cancer(43), and head and neck cancer(44), further underscoring the urgent need to develop potent IRAK inhibitors for clinical testing. Another study showed that TAK1 inhibition could augment the effect of chemotherapy in PDAC(45). Supporting this study, we found that treatment of PDAC lines with TAK1 inhibitor 5Z-7-oxozeaenol significantly reduced AI growth and p-NF-κB/p65 (not shown), and that IRAK1/4i significantly reduced p-TAK1 (Fig. 6C and not shown).
How IRAK4 is activated in PDAC is currently unknown. Since activating mutations of IRAK4 or MyD88, the adaptor protein upstream of IRAK4, are not reported in PDAC from analysis of TCGA, COSMIC database and from our own Sanger sequencing of PDAC lines (not shown); we hypothesize that IRAK4 is activated by engagement of the interleukin receptors or the TLRs by pathogen- or damage-associated molecular patterns in the tumor microenvironment. To test this, we are currently conducting loss-of-function analysis of individual TLRs, as well as IL-1R and IL-18R, in multiple PDAC cells.
In summary, our study provides a strong rationale for targeting IRAK4 as a novel therapeutic approach in PDAC. We showed that p-IRAK4 is not only a strong prognostic marker for poor outcomes in PDAC patients, but also a major driver of NF-κB activity and could be therapeutically exploited to improve chemotherapy response. Our work also suggests the need to develop and test more potent IRAK4 inhibitors in combination with chemotherapy in preclinical models. Furthermore, understanding how the IRAKs are activated in PDAC, using cell line and genetically-engineered mouse models, will add invaluable knowledge to the biology of PDAC.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Activation of the NF-κB transcription factors is a major mechanism that underlies the aggressive behavior of pancreatic ductal adenocarcinoma (PDAC). However, effective and safe inhibitor of the NF-κB is not available, underscoring a need to curb this pathway by alternative means. We now present the first evidence that activation of the Interleukin-1 Receptor Associated Kinase 4 (IRAK4) is a major driving mechanism of NF-κB activity in PDAC. Presence of phospho-IRAK4 staining in PDAC samples is associated with poor patient prognosis. Pharmacologic inhibition or silencing of IRAK4 significantly thwarted tumorigenic growth of PDAC cells and augmented the killing effect of chemotherapy. Therefore, our data provide preclinical rationale for the development of IRAK4 inhibitors as a new class of targeted agent for clinical testing in combination with chemotherapy, and indicate phospho-IRAK staining as a potential biomarker for patient selection and pharmacodynamics studies in future clinical trials involving IRAK4 inhibitor.
Acknowledgments
We wish to thank Dr. Ron Bose and Jeff Guo for critical reading of the manuscript, Maureen K. Highkin for technical advice, Julie Prior at the Molecular Imaging Center (funded under P50 CA094056 from the NCI) for assistance with animal imaging and analysis, and the Digestive Disease Research Core Center (funded by grant P30DK052574 from the NIDDK) for providing technical support. This study was supported by the NCI Cancer Center Support Grant #P30 CA091842 (K.H.L), Washington University Institute of Clinical and Translational Sciences grant UL1 TR000448 from the NCATS of the NIH (K.H.L) and Elsa Pardee Foundation Award (K.H.L and M.B.R). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
Footnotes
The authors disclose no potential conflicts of interest
REFERENCES
- 1.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371(22):2140–2141. doi: 10.1056/NEJMc1412266. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society: Cancer Facts and Figures. 2015. [Google Scholar]
- 3.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5(1):119–127. [PubMed] [Google Scholar]
- 4.Weichert W, Boehm M, Gekeler V, Bahra M, Langrehr J, Neuhaus P, et al. High expression of RelA/p65 is associated with activation of nuclear factor-kappaB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. British journal of cancer. 2007;97(4):523–530. doi: 10.1038/sj.bjc.6603878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carbone C, Melisi D. NF-kappaB as a target for pancreatic cancer therapy. Expert opinion on therapeutic targets. 2012;16(Suppl 2):S1–S10. doi: 10.1517/14728222.2011.645806. [DOI] [PubMed] [Google Scholar]
- 6.Prabhu L, Mundade R, Korc M, Loehrer PJ, Lu T. Critical role of NF-kappaB in pancreatic cancer. Oncotarget. 2014;5(22):10969–10975. doi: 10.18632/oncotarget.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D–induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(1):105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bang D, Wilson W, Ryan M, Yeh JJ, Baldwin AS. GSK-3alpha promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-kappaB. Cancer discovery. 2013;3(6):690–703. doi: 10.1158/2159-8290.CD-12-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, et al. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J. 2011;30(1):57–67. doi: 10.1038/emboj.2010.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT. 2013;2(2):e23828. doi: 10.4161/jkst.23828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5(1) doi: 10.1101/cshperspect.a011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikebe M, Kitaura Y, Nakamura M, Tanaka H, Yamasaki A, Nagai S, et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J Surg Oncol. 2009;100(8):725–731. doi: 10.1002/jso.21392. [DOI] [PubMed] [Google Scholar]
- 14.Ochi A, Graffeo CS, Zambirinis CP, Rehman A, Hackman M, Fallon N, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012;122(11):4118–4129. doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015 doi: 10.1084/jem.20142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J Exp Med. 2007;204(5):1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci U S A. 2002;99(8):5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell. 2016;29(1):75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 22.O’Hayer KM, Counter CM. A genetically defined normal somatic human cell system to study ras oncogenesis in vitro and in vivo. Methods Enzymol. 2005 doi: 10.1016/S0076-6879(05)07050-3. [DOI] [PubMed] [Google Scholar]
- 23.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 24.Baines AT, Lim KH, Shields JM, Lambert JM, Counter CM, Der CJ, et al. Use of retrovirus expression of interfering RNA to determine the contribution of activated K-Ras and ras effector expression to human tumor cell growth. Methods Enzymol. 2006;407:556–574. doi: 10.1016/S0076-6879(05)07045-X. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Frontiers in immunology. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. British journal of cancer. 2015;112(2):232–237. doi: 10.1038/bjc.2014.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava R, Geng D, Liu Y, Zheng L, Li Z, Joseph MA, et al. Augmentation of therapeutic responses in melanoma by inhibition of IRAK-1,-4. Cancer Res. 2012;72(23):6209–6216. doi: 10.1158/0008-5472.CAN-12-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Powers JP, Li S, Jaen JC, Liu J, Walker NP, Wang Z, et al. Discovery and initial SAR of inhibitors of interleukin-1 receptor-associated kinase-4. Bioorg Med Chem Lett. 2006;16(11):2842–2845. doi: 10.1016/j.bmcl.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraczek J, Kim TW, Xiao H, Yao J, Wen Q, Li Y, et al. The kinase activity of IL-1 receptor-associated kinase 4 is required for interleukin-1 receptor/toll-like receptor-induced TAK1-dependent NFkappaB activation. J Biol Chem. 2008;283(46):31697–31705. doi: 10.1074/jbc.M804779200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koziczak-Holbro M, Joyce C, Gluck A, Kinzel B, Muller M, Tschopp C, et al. IRAK-4 kinase activity is required for interleukin-1 (IL-1) receptor- and toll-like receptor 7-mediated signaling and gene expression. J Biol Chem. 2007;282(18):13552–13560. doi: 10.1074/jbc.M700548200. [DOI] [PubMed] [Google Scholar]
- 32.Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR, et al. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22(21):3243–3251. doi: 10.1038/sj.onc.1206390. [DOI] [PubMed] [Google Scholar]
- 33.Tjomsland V, Spangeus A, Valila J, Sandstrom P, Borch K, Druid H, et al. Interleukin 1alpha sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia. 2011;13(8):664–675. doi: 10.1593/neo.11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mews P, Phillips P, Fahmy R, Korsten M, Pirola R, Wilson J, et al. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. 2002;50(4):535–541. doi: 10.1136/gut.50.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vonlaufen A, Phillips PA, Xu Z, Goldstein D, Pirola RC, Wilson JS, et al. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 2008;68(19):7707–7710. doi: 10.1158/0008-5472.CAN-08-1132. [DOI] [PubMed] [Google Scholar]
- 36.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19(13):3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016 doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–249. doi: 10.1038/nature17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly PN, Romero DL, Yang Y, Shaffer AL, 3rd, Chaudhary D, Robinson S, et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. 2015;212(13):2189–2201. doi: 10.1084/jem.20151074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013;24(1):90–104. doi: 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Younger K, Gartenhaus R, Joseph AM, Hu F, Baer MR, et al. Inhibition of IRAK1/4 sensitizes T cell acute lymphoblastic leukemia to chemotherapies. J Clin Invest. 2015;125(3):1081–1097. doi: 10.1172/JCI75821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wee ZN, Yatim SM, Kohlbauer VK, Feng M, Goh JY, Bao Y, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nature communications. 2015;6:8746. doi: 10.1038/ncomms9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams AK, Bolanos LC, Dexheimer PJ, Karns RA, Aronow BJ, Komurov K, et al. IRAK1 is a novel DEK transcriptional target and is essential for head and neck cancer cell survival. Oncotarget. 2015;6(41):43395–43407. doi: 10.18632/oncotarget.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melisi D, Xia Q, Paradiso G, Ling J, Moccia T, Carbone C, et al. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J Natl Cancer Inst. 2011;103(15):1190–1204. doi: 10.1093/jnci/djr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.