

Summary
B55α is a regulatory subunit of the PP2A phosphatase. We have recently found that B55α-associated PP2A promotes partial deactivation of the HIF-prolyl-hydroxylase enzyme PHD2. Here, we show that, in turn, PHD2 triggers degradation of B55α by hydroxylating it at proline 319. In the context of glucose starvation, PHD2 reduces B55α protein levels, which correlates with MDA-MB231 and MCF7 breast cancer cell death. Under these conditions, PHD2 silencing rescues B55α degradation, overcoming apoptosis, whereas in SKBR3 breast cancer cells showing resistance to glucose starvation, B55α knockdown restores cell death and prevents neoplastic growth in vitro. Treatment of MDA-MB231-derived xenografts with the glucose competitor 2-deoxy-glucose leads to tumor regression in the presence of PHD2. Knockdown of PHD2 induces B55α accumulation and treatment resistance by preventing cell apoptosis. Overall, our data unravel B55α as a PHD2 substrate and highlight a role for PHD2-B55α in the response to nutrient deprivation.
Keywords: glucose starvation, PP2A, PHD2, breast cancer, cell death
Graphical Abstract
Highlights
-
•
PHD2 hydroxylates and degrades B55α, a regulatory subunit of PP2A phosphatase
-
•
In the absence of glucose, a high αKG-to-fumarate ratio favors PHD2 activation
-
•
Active PHD2 degrades B55α and promotes cell death under glucose starvation
-
•
PHD2-silenced xenografts showed accumulation of B55α and resistance to 2DG treatment
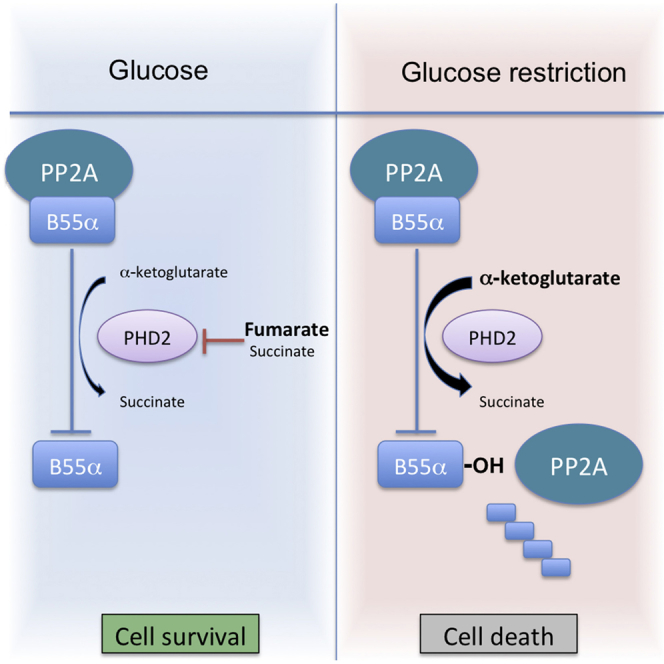
Di Conza et al. show that, following glucose starvation, a high αKG-to-fumarate ratio favors PHD2 activation that promotes apoptosis by degrading B55α. In breast cancer cells, PHD2 knockdown prevents B55α degradation and overcomes apoptosis in response to glucose starvation. PHD2-silenced MDA-MB231 xenografts show accumulation of B55α and resistance to 2DG treatment.
Introduction
B55α is a regulatory B subunit of the phosphatase complex PP2A (Sontag, 2001). PP2A participates in an immense number of pathways, and this explains the lethal phenotype associated with knockout of the catalytic C subunit in mice (Götz et al., 1998). Few B-subunit knockout mice have been generated, and none has shown lethality; instead, these mice have tissue-specific pathological features (Louis et al., 2011, Varadkar et al., 2014). Indeed, distinct from the catalytic and scaffold subunits of PP2A, the regulatory B subunits only contribute to pathways ascribed to their specific substrates so that they act uniquely under different pathological contexts (Sablina et al., 2010). We recently found that PP2A/B55α promotes the growth of colorectal cancer by dephosphorylating PHD2 and modifying its enzymatic properties (Di Conza et al., 2017). PHD2 (or EGLN1) is a member of the hypoxia-inducible factor (HIF)-prolyl hydroxylase domain proteins (PHDs) family (Epstein et al., 2001). These enzymes are crucial for HIF activation and the cellular response to hypoxia (Chan et al., 2005). Indeed, under normoxia, PHDs utilize oxygen and alpha-ketoglutarate (α-KG) to carry out a hydroxylation reaction on specific HIF prolines that lead to ubiquitination and proteasomal degradation (Keith et al., 2011). In addition to oxygen and cosubstrates, the mTOR pathway, specifically P70S6K, can further activate PHD2 through serine-125 phosphorylation. When dephosphorylated by PP2A/B55α, PHD2 shows impaired enzymatic activity, allowing full activation of HIF1 in hypoxia (Di Conza et al., 2017). Here, we investigate the molecular interaction between PHD2 and B55α in response to glucose starvation in the context of breast cancer cells.
Results
PHD2 Targets B55α to the Proteasome by Promoting Its Hydroxylation
The main function of PHD2 is the hydroxylation at specific proline residues within the alpha subunit of HIFs. Based on our previous findings showing the interaction and negative regulation of PHD2 by B55α/PP2A (Di Conza et al., 2017), we wondered whether PHD2 was able to degrade B55α in a feed-forward loop. We found that co-overexpression of PHD2 and B55α in HEK293T cells resulted in reduced B55α protein levels (Figure 1A) that was linked to its increased ubiquitination (Figure 1B) and proteasomal degradation (Figure 1C). The control of B55α protein levels was specific for PHD2 only, because either PHD1 or PHD3 was not able to induce B55α proteasomal degradation (Figure S1A). Mirroring the overexpression data, PHD2 knockdown increased the half-life of B55α as assessed by translation blockade with cycloheximide (Figures 1D and S1B). Inhibition of PHD2 enzymatic activity by IOX2 or dimethyloxaloylglycine (DMOG) impaired both B55α ubiquitination and degradation (Figures 1E and 1F). Moreover, physiological inhibition of PHD2 by exposure to hypoxia was able to induce B55α accumulation as well (Figures S1C and S1D). Overall, these data indicate that PHD2 hydroxylase function is required for targeting B55α to the proteasome.
Figure 1.

PHD2 Induces Ubiquitination and Degradation of B55α
(A) HEK293T cells were transfected with plasmids encoding for nothing (EV [empty vector]), B55α, PHD2, or both. After 24 hr, whole-cell extracts (WCEs) were lysed and analyzed by western blot (WB).
(B) HEK293T cells were transfected with FLAG-B55α alone or in combination with plasmids carrying Ubiquitin-HA and with PHD2 or a control vector. After 16 hr, cells were treated with MG132 (10 μM) for 4 hr, lysed, and immunoprecipitated using anti-FLAG M2 beads to detect Ubiquitin-HA. WB on WCEs is shown on the right.
(C) WB analysis of HEK293T cells transfected with B55α alone or in combination with PHD2 and treated with MG132 (10 μM) or vehicle for 8 hr.
(D) HEK293T cells stably expressing a shRNA specifically targeting PHD2 (shPHD2) or control (shCTR) were transfected with B55α. Then, cells were treated with 100 μg/mL cycloheximide for the indicated time points; WCEs were collected and analyzed by WB. The graph represents the quantification of three independent experiments.
(E) HEK293T cells were transfected with Ubiquitin-HA alone or in combination with FLAG-B55α or control vector. After 8 hr, cells were treated with the PHD2 inhibitor IOX2 (50 μM) for 16 hr and immunoprecipitated using anti-FLAG M2 beads to detect Ubiquitin-HA. WB on WCEs is shown on the right.
(F) HEK293T cells were transfected with B55α alone or in combination with PHD2. After 24 hr, cells were incubated for 8 hr in the presence or absence of the PHD2 inhibitor DMOG (1 or 2 mM). Protein levels were analyzed by WB.
(G) WB analysis of HEK293T cells stably transfected with plasmids carrying wild-type B55α (WT) or B55α mutants (P159A, P236A, P319A) in the presence or absence of PHD2.
(H) In vitro hydroxylation assays were performed with increasing concentrations of a B55α peptide (spanning from W311 to L329). Km was determined by using Michaelis-Menten curve (left panel) or L-B plot (right panel).
(I) In vitro hydroxylation assays were performed on three different peptides: HIF1α (556–575), B55α (311–329), and B55α P-OH (where P319 is hydroxylated). ∗p < 0.05 versus the other conditions. The graph shows the mean ± SEM.
(J) HEK293T cells were transfected with either empty vector (EV), FLAG-B55αWT, or FLAG-B55αP319A. After 16 hr, cells were lysed and immunoprecipitated using anti-FLAG M2 beads to detect the A and C subunits of the complex PP2A. WB on whole-cell extracts (WCEs) is shown on the left.
(K) HEK293T cells were transfected with either empty vector (EV), FLAG-B55αWT, or FLAG-B55αP319A. Cell lysates expressing FLAG-B55αWT or FLAG-B55αP319A were incubated in vitro for 2 hr with recombinant PHD2. Then an anti-FLAG immunoprecipitation was performed in order to detect by WB the levels of the Ab subunit as readout of PP2A complex information.
See also Figure S1.
PHD2 Hydroxylates B55α on Proline 319
Mass spectrometry analysis of B55α isolated from HEK293T cells, upon co-expression of PHD2, revealed three hydroxylation sites in P159, P236, and P319 of B55α that we singularly mutated into an alanine. Of the three B55α point mutants, only B55αP319A exhibited increased resistance to PHD2-mediated degradation (Figure 1G). To confirm that this residue is hydroxylated by PHD2, we performed an in vitro hydroxylation assay where recombinant PHD2 was incubated with increasing doses of a B55α peptide and the Km was determined by using a Michaelis-Menten curve or a Lineweaver-Burk (L-B) plot (Figure 1H). The same assay was then performed by using a B55α peptide spanning from amino acid 311 to 329, therefore containing P319, or B55α P-OH (where P319 is synthetically hydroxylated) and HIF1α (556–575) peptides as negative and positive controls, respectively. This assay confirmed that PHD2 was able to directly hydroxylate B55α in P319, although with a lower efficiency than for HIF1α (Figure 1I). However, when we tested the ubiquitination levels of this mutant, we could not detect any difference (Figure S1E). Because proline 319 lies in the region of B55α that allows its binding to the scaffold subunit of the PP2A complex (Li and Virshup, 2002), we hypothesized that the hydroxylation of this residue is important to detach B55α from the complex, therefore making it more available for degradation. When measuring the binding of B55αWT and B55αP319A to the scaffold Aβ and the catalytic Cα subunits of the PP2A complex, we found that B55αP319A had higher affinity for the complex than B55αWT (Figure 1J). Moreover, recombinant PHD2 was able to reduce the binding of B55αWT (but not of B55αP319A) to the A subunit of PP2A complex (Figure 1K). Altogether, these data suggest that B55α hydroxylation is important to disassemble the PP2A complex, rendering B55α available for degradation.
PHD2 Degrades B55α and Induces Cell Death in Response to Glucose Deprivation
To analyze the degradation of endogenous B55α, we silenced PHD2 in three breast cancer cell lines, MDA-MB231, MCF7, and SKBR3. In all cell lines, we observed an increase of B55α protein levels upon silencing of PHD2 (Figure 2A) that was not linked to an augmentation in the mRNA levels (Figures 2B and 2C). Contrariwise, overexpression of PHD2 could reduce endogenous B55α protein levels (Figure S1F). Because a previous report has shown that B55α plays an important role in response to nutrient deprivation (Reid et al., 2013), in order to investigate the biological readout of our findings, we cultured breast cancer cells in glucose-deprived medium at several time points. We observed that B55α was progressively degraded (Figures 2D and 2E) and that, in the same conditions, silencing of PHD2 was able to rescue the degradation of B55α (Figures 2F and 2G). Based on the observation that glucose starvation did not affect the overall levels of PHD2, we hypothesized that the deprivation of glucose might rather affect PHD2 activity. Accordingly, lack of glucose, by slowing down the tricarboxylic acid (TCA) cycle, would lead to a change in the amount of α-KG, a co-substrate needed for PHD2 activity, as well as fumarate and succinate, two metabolites that inhibit PHD2 and derive from the oxidation of α-KG in the TCA cycle (Isaacs et al., 2005, King et al., 2006). To test this hypothesis, we measured by mass spectrometry the levels of α-KG, succinate, and fumarate upon glucose starvation. At all the time points tested, in both MDA-MB231 and MCF7 cells, the levels of fumarate rapidly declined, whereas succinate was not affected (Figures 2H and 2I). As a consequence, the α-KG-to-fumarate ratio gradually and significantly increased, possibly playing in favor of PHD2 function (Figures 2J and 2K). To further explain why fumarate dropped more rapidly than α-KG under glucose starvation, we analyzed the levels of malate (that generates from fumarate in the TCA cycle) and we measured the amount of α-KG deriving from glutamine. In both cell lines, malate levels were tremendously reduced to a similar extent as observed for fumarate (Figures 2H and 2I). This, together with the fact that succinate levels were unaltered, suggests a block of succinate dehydrogenase (SDH) activity upon glucose starvation (Andreev et al., 2015). At least in MCF7, the amount of α-KG coming from glutamine was increasing under glucose deprivation versus fed conditions, as evidenced by U-13C-glutamine fractional labeling (Figure S2A). Reductive carboxylation (RC), a pathway that utilizes glutamine-derived α-KG to generate citrate and eventually acetyl-CoA (Metallo et al., 2011), was slightly but significantly decreased under glucose starvation as proven by a drop of the m + 1 acetyl-CoA isotopologue in the presence of 5-13C-glutamine (Figure S2B). Levels of glutamine-derived α-KG were close to the detection limit in MDA-MB231 cells (not shown). Overall, our data indicate that, in the absence of glucose, at least in breast cancer cells, a block of SDH activity, per se or together with increased glutamine anaplerosis, results in a high α-KG-to-fumarate ratio. These results prompted us to test how these metabolic changes affect PHD2 activity. We transfected fed and glucose-deprived MCF7 cells with a luciferase construct fused to the oxygen degradation domain of HIF1α (ODDD-Luc). When active, PHD2 leads to the degradation of this ODDD-Luc, resulting in overall reduction of luciferase signal. During glucose starvation, the activity of both endogenous and overexpressed PHD2 was strongly increased (Figure 2L), and addition of fumarate to the medium was able to rescue this induction (Figure S2C), thus supporting that increased PHD2 activation in glucose starvation leads to B55α degradation.
Figure 2.

PHD2 Depletion Prevents Degradation of B55α in Response to Glucose Starvation
(A) WB analysis of MCF7, MDA-MB231, and SKBR3 cells transiently transfected with small interfering RNA (siRNA) specifically targeting PHD2 (siPHD2) or control (siCTR) for 48 hr.
(B and C) qPCR of RNA extracted from MDA-MB231 cells (B) and MCF7 cells (C) transfected with siRNA specifically targeting PHD2 (siPHD2) or control (siCTR).
(D and E) MDA-MB231 cells (D) and MCF7 cells (E) were cultured in complete (Mock) or glucose-deprived (GS) medium at the indicated time points. Then WCEs were collected and analyzed by WB.
(F and G) MDA-MB231 cells (F) and MCF7 cells (G) were transfected with siRNA targeting PHD2 (siPHD2) or control (siCTR). Cells were cultured in complete or glucose-deprived medium (GS) for 16 hr. WCEs were then collected and analyzed by WB.
(H–K) MDA-MB231 cells (H and J) and MCF7 cells (I and K) were cultured in complete (Mock) or glucose-deprived (GS) medium for the indicated time points. LC-MS was then performed for metabolite quantification on biological triplicates as described in Experimental Procedures. The α-KG-to-fumarate ratio in MDA-MB231 or MCF7 cells is shown in (J) and (K), respectively.
(L) MCF7 were transfected with plasmids expressing ODDD (oxygen degradation domain) alone (EV) or together with PHD2 (PHD2). After 8 hr, medium was replaced with complete or glucose-deprived medium (GS) for 16 hr. Then cells were lysed and luciferase activity was measured and normalized for protein concentration. #p < 0.05 versus other conditions; ∗p < 0.05 versus Mock. All graphs show mean ± SEM.
See also Figure S2.
Silencing of PHD2 Rescues B55α Degradation and Overcomes Apoptosis in Response to Glucose Starvation
We then wondered whether the degradation of B55α was necessary for the activation of survival/apoptotic pathways in response to nutrient deprivation (Sun et al., 2015). Cell-cycle analysis showed that deprivation of glucose induced a high percentage of cell death in both MDA-MB231 and MCF7 cell lines (Figure 3A). However, cells silenced for PHD2 showed 30% of reduction in cell death (Figures 3A–3C), whereas the cell-cycle phases were unchanged (Figures S2D and S2E). Furthermore, this protection against cell death was B55α dependent because combined knockdown of B55α and PHD2 was able to restore apoptosis in cells silenced for PHD2 only (Figures 3D, 3E, S2F, and S2G). Similarly to PHD2 silencing, overexpression of B55αWT or B55αP319A was able to induce protection against cell death as well (Figure 3F). Complementing these data, physiological expression of B55αWT in MCF7 silenced for PHD2 was able to reduce glucose starvation-induced cell death, whereas un-degradable B55αP319A was ineffective (Figure 3G). These data demonstrate that PHD2-mediated B55α degradation is responsible, at least in part, for cell apoptosis induced by glucose deprivation.
Figure 3.

PHD2 Depletion Promotes Resistance to Cell Death in Response to Glucose Starvation
(A–C) MCF7 and MDA-MB231 cells were transfected with siRNA targeting PHD2 (siPHD2) or control (siCTR). After 24 hr, cells were cultured in complete (Mock) or glucose-deprived (GS) medium for 24 hr. Cell death was assessed by propidium iodide (A and B) or Annexin V staining (C) and analyzed by FACS.
(D and E) MDA-MB231 cells (D) and MCF7 cells (E) were transfected with siRNA targeting control (siCTR), PHD2 (siPHD2), or the combination of PHD2 and B55α (siPHD2/siB55α). After 24 hr, cells were cultured in complete (Mock) or glucose-deprived (GS) medium. Cell death was assessed by propidium iodide staining at 24 hr.
(F) MCF7 were transfected with plasmids encoding empty vector (EV), B55αWT, or B55αP319A. After 10 hr, cells were cultured in complete (Mock) or glucose-deprived (GS) medium. Cell death was assessed by propidium iodide staining at 16 hr.
(G) MCF7 were transfected with siRNA targeting PHD2 (siPHD2) or control (siCTR). After 16 hr, cells were transfected with plasmids encoding empty vector (EV), B55α-WT, or B55α-P319A. After 10 hr, cells were cultured in complete (Mock) or glucose-deprived (GS) medium for 16 hr. Cell death was assessed by propidium iodide staining.
(H) SKBR3 cells were transfected with siRNA targeting PHD2 (siPHD2) or control (siCTR). After 16 hr, cells were cultured in complete (Mock) or glucose-deprived (GS) medium for the indicated time points. WCEs were collected and analyzed by WB.
(I and J) SKBR3 cells were transfected with siRNA targeting PHD2 (siPHD2) and control (siCTR) in (I) or with siRNA targeting B55α (siB55α) and control (siCTR) in (J). Cell death was assessed by propidium iodide staining and FACS analysis after 24 hr in complete (Mock) or glucose-deprived (GS) medium. ∗p < 0.05 versus siCTR in (A–C); versus all other conditions in (D), (E), and (J); versus EV in (F); versus siCTR/GS in (G). All graphs show mean ± SEM.
See also Figure S2.
Unlike MDA-MB231 and MCF7, SKBR3 cells were partially resistant to starvation-induced cell death. When SKBR3 cells were cultured in glucose-deprived medium, the protein levels of B55α were unchanged and silencing of PHD2 did not significantly affect cell death (Figures 3H and 3I). Depletion of B55α rendered these cells responsive to glucose deprivation and ultimately able to activate the apoptotic pathway (Figure 3J). In a focus-forming assay, glucose starvation partly decreased the number of colonies formed by control SKBR3 cells, but combined B55α silencing resulted in a further and stronger reduction (Figures 4A and 4B), confirming the relevance of B55α degradation in response to glucose starvation.
Figure 4.

Degradation of B55α by PHD2 Blocks Neoplastic Growth in Response to Glucose Deprivation
(A and B) Colony-forming assay from SKBR3 cells stably silenced for B55α (shB55α) or control (shCTRL). Representative images from one of three independent experiments are shown (B).
(C) In vivo growth curve of xenograft tumors derived from subcutaneous injection of CD1 mice of MDA-MB231 cells stably silenced for control (shCTRL) or PHD2 (shPHD2).
(D) Graph shows tumor weight of MDA-MB231 cells 38 days after cancer cell injection.
(E) Graph shows quantification of B55α protein levels upon WB analysis of tumor samples derived from the experiment in (C).
(F) Morphometric quantification and representative images of TUNEL+ apoptotic cells in MDA-MB231 tumor sections.
Representative images and morphometric quantification of MDA-MB231 tumor sections stained with TUNEL assay, showing cell death. ∗p < 0.05 versus all other conditions in (A) and (C–F). All graphs show mean ± SEM.
In order to test whether the molecular interaction between PHD2 and B55α had relevance in an in vivo context, we injected subcutaneously MDA-MB231 cells stably silenced for CTRL (shCTRL) or PHD2 (shPHD2) in nude mice. When the tumors reached 50–100 mm3, we treated the mice with glycolysis inhibitor 2-DG (2-deoxy-glucose) (in order to mimic glucose starvation) or a vehicle control. In the presence of PHD2 (shCTRL), treatment with 2-DG blocks tumor growth with strong efficiency. However, when PHD2 is silenced (shPHD2), the tumors showed complete resistance to the treatment (Figures 4C and 4D). Consistent with our in vitro findings, the levels of B55α were reduced in 2-DG-treated tumors, whereas this degradation was rescued in tumors lacking PHD2 (Figure 4E). Histological analysis of tumor sections revealed a strong increase of TUNEL positivity in 2-DG-treated cells that was completely canceled by silencing of PHD2, confirming the relevance of its activity in the response to glucose starvation (Figure 4F). Altogether, these data prove that PHD2-mediated B55α degradation allows breast cancer cell death in response to chronic glucose deprivation.
Discussion
In this study, we uncover two molecular players in response to nutrient restriction: the phosphatase B55α/PP2A and the prolyl-hydroxylase PHD2. The pathways involved in the response to nutrient deprivation have been widely investigated. In the absence of nutrients, the stress sensor AMPK is activated, leading to the inhibition of mTOR and the induction of a metabolic response able to initiate adaptation to nutrient restriction in order to allow the cell to survive in this harsh situation (Laplante and Sabatini, 2012). Although all of the kinases of this pathway have been studied for decades (Brown et al., 1994), the relevance of phosphatases in this matter has only recently received more attention (Reid et al., 2013, Wong et al., 2015).
Upon nutrient starvation, PP2A activity is liberated as a consequence of mTOR repression, and this event leads to a PP2A-mediated block of c-myc that thus causes inhibition of proliferation (Cianfanelli et al., 2015, Di Conza et al., 2017). Moreover, the inactivation of mTOR and the simultaneous activation of PP2A/B55α are necessary to switch on the autophagic pathway in a very early response to amino acid deprivation (Wong et al., 2015). In this study, we have further investigated the link between PP2A/B55α and nutrient shortage, unravelling how a late response to glucose restriction is accomplished by degradation of PP2A/B55α. Induced prevention of its degradation (by silencing of PHD2) or impaired control of its levels, cause a resistance to cell death. It is thus possible that PP2A/B55α participates in a first-wave reaction in response to nutrient stress by controlling the phosphorylation cascade responsible for the growth arrest/survival of the cells. However, prolonged cell stress leads to the activation of apoptosis, signaled by a reduction in B55α levels that is mediated by PHD2. The role of PP2A in response to nutrient restriction has been highlighted by several studies (Cianfanelli et al., 2015, Wong et al., 2015), and what is mostly emerging from all of them is the tight and reverse relationship between PP2A and mTOR signaling under starved conditions. Similarly, we have shown that hypoxic blockade of mTOR (via the upregulation of REDD1) releases PP2A/B55α activity, allowing dephosphorylation of PHD2 and full HIF1α stabilization (Di Conza et al., 2017). In the current report, we demonstrate that, in a feed-forward loop, PHD2 is able to harness its own inhibitor PP2A/B55α by inducing its hydroxylation, ubiquitination, and degradation through the proteasome. These data indicate that B55α is a substrate of the oxygen-sensing enzyme PHD2. However, different from what occurs for HIF1α, the B55α hydroxylation is not a signal for ubiquitin ligase, but it rather facilitates the detachment of B55α from the main complex PP2A. Indeed, it has been demonstrated that the domain where the proline 319 lies, is involved in the binding with the scaffold A subunit (Li and Virshup, 2002). Therefore, the hydroxylation in proline 319 by inhibiting this binding favors B55α ubiquitination and degradation. Several reports have highlighted the pro-tumoral activity of B55α (Gilan et al., 2015, Hein et al., 2016, Reid et al., 2013). On the other hand, the role of PHD2 in cancer is more controversial, having shown even opposite effects in different tumor contexts (Chan et al., 2009, Klotzsche-von Ameln et al., 2011). Our data help to define the dual role of PHD2 in cancer. Together with hypoxia, nutrient restriction is a major feature of the tumor microenvironment. If, on one hand, we show that oxygen shortage enables B55α accumulation, on the other hand, we speculate that in breast cancer cells glucose deprivation reduces the production of fumarate mainly because of a block of SDH (Andreev et al., 2015), tilting the balance toward an excess of α-KG at the expense of the PHD2 inhibitor fumarate, overall resulting in enhanced PHD2 activity and increased B55α degradation. In parallel, prolonged glucose starvation results in P70S6K reactivation (G.D.C. and M.M., unpublished data) that further boosts PHD2 function and therefore B55α degradation (Di Conza et al., 2017). Yet, it remains to be defined how glucose starvation causes this SDH block in this specific cell context. Altogether, this study shows that PHD2 takes part in the network of nutrient-sensing proteins by regulating PP2A in breast cancer cells.
Experimental Procedures
More detailed methods can be found in Supplemental Experimental Procedures.
Cell Culture and Transfection
HEK293T, MCF7, MDA-MB231, and SKBR3 cell lines were cultured in DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco), 2 mM glutamine (Life Technologies), and 100 units/mL penicillin/100 μg/mL streptomycin (Life Technologies). Cells were maintained in a humidified incubator at 37°C and 5% CO2. For glucose starvation experiments, DMEM glucose-free (Life Technologies) has been supplemented with heat-inactivated FBS previously dialyzed with slide A lyzer cassette (Thermo Scientific). Transfections were performed with Lipofectamine 2000 Transfection Reagent or Lipofectamine RNAiMax (Life Technologies), according to the manufacturer’s instructions.
Mass Spectrometry of Polar Metabolites
Polar metabolites were extracted using a methanol-water extraction (Fendt et al., 2013). Following extraction, the extract was centrifuged for 10 min at 4°C at 20,000 × g, and the supernatant was transferred to MS vials. Measurements by gas chromatography (GC) or liquid chromatography (LC) were performed as described in Supplemental Experimental Procedures.
In Vitro Decarboxylation Assay
An in vitro decarboxylation assay was performed as previously published (Zheng et al., 2014) to assess the hydroxylation of a specific peptide by recombinant PHD2. The sequences of the peptides used are as follows: B55α peptide, WDLNMENRPVETYQVHEYL; B55α P-OH peptide, WDLNMENRP#VETYQVHEYL (# denotes hydroxylation); HIF1α (556–575), DLDLEMLAPYIPMDDDFQLR.
Xenograft Tumors
MDA-MB231 cells were harvested and single-cell suspensions of 10 × 106 cells in 100 μL of PBS and Matrigel solution (1:1) were injected subcutaneously into the flank of CD1 mice. Tumor volumes were measured three times a week with a caliper and calculated using the formula: V = π × [d2 × D]/6, where d is the minor tumor axis and D is the major tumor axis. This study was approved by the institutional ethical commission at KU Leuven.
Statistics
All statistical analyses were performed using GraphPad Prism software. Statistical significance was calculated by two-tailed unpaired t test with p < 0.05 considered statistically significant. Western blots and FACS analysis are representative of three independent experiments. All graphs show mean values ± SEM.
Author Contributions
G.D.C. performed experimental design, experiments, acquisition of data, analysis and interpretation of data and wrote the manuscript. S.T.C. performed western blot experiments. X.Z. and Q.Z. performed in vitro decarboxylation assay. M.M. performed experimental design, conducted scientific direction, and wrote the manuscript.
Acknowledgments
The authors thank B. Meeusen, G. Manzella, J. Serneels, and A. Acosta Sanchez for technical assistance; A. Sablina for sharing plasmids and reagents; and C. Frezza, B. Ghesquière, J. Aragonès, and K. De Bock for constructive discussion and advice. This work was supported by grants from FWO (1505611N00) and Stichting tegen Kanker (2010-169). G.D.C. is supported by a Pegasus FWO-Marie Curie Fellowship (1211413N); M.M. received an ERC Starting Grant (OxyMO, 308459).
Published: March 21, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and two figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.02.081.
Contributor Information
Giusy Di Conza, Email: giusy.diconza@unil.ch.
Massimiliano Mazzone, Email: massimiliano.mazzone@vib-kuleuven.be.
Supplemental Information
References
- Andreev D.E., O’Connor P.B., Zhdanov A.V., Dmitriev R.I., Shatsky I.N., Papkovsky D.B., Baranov P.V. Oxygen and glucose deprivation induces widespread alterations in mRNA translation within 20 minutes. Genome Biol. 2015;16:90. doi: 10.1186/s13059-015-0651-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E.J., Albers M.W., Shin T.B., Ichikawa K., Keith C.T., Lane W.S., Schreiber S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Chan D.A., Sutphin P.D., Yen S.E., Giaccia A.J. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol. Cell. Biol. 2005;25:6415–6426. doi: 10.1128/MCB.25.15.6415-6426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D.A., Kawahara T.L., Sutphin P.D., Chang H.Y., Chi J.T., Giaccia A.J. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell. 2009;15:527–538. doi: 10.1016/j.ccr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfanelli V., Fuoco C., Lorente M., Salazar M., Quondamatteo F., Gherardini P.F., De Zio D., Nazio F., Antonioli M., D’Orazio M. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015;17:20–30. doi: 10.1038/ncb3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Conza G., Trusso Cafarello S., Loroch S., Mennerich D., Deschoemaeker S., Di Matteo M., Ehling M., Gevaert K., Prenen H., Zahedi R.P. The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine-tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Rep. 2017;18:1699–1712. doi: 10.1016/j.celrep.2017.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein A.C., Gleadle J.M., McNeill L.A., Hewitson K.S., O’Rourke J., Mole D.R., Mukherji M., Metzen E., Wilson M.I., Dhanda A. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Fendt S.M., Bell E.L., Keibler M.A., Davidson S.M., Wirth G.J., Fiske B., Mayers J.R., Schwab M., Bellinger G., Csibi A. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73:4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilan O., Diesch J., Amalia M., Jastrzebski K., Chueh A.C., Verrills N.M., Pearson R.B., Mariadason J.M., Tulchinsky E., Hannan R.D., Dhillon A.S. PR55α-containing protein phosphatase 2A complexes promote cancer cell migration and invasion through regulation of AP-1 transcriptional activity. Oncogene. 2015;34:1333–1339. doi: 10.1038/onc.2014.26. [DOI] [PubMed] [Google Scholar]
- Götz J., Probst A., Ehler E., Hemmings B., Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proc. Natl. Acad. Sci. USA. 1998;95:12370–12375. doi: 10.1073/pnas.95.21.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein A.L., Seshacharyulu P., Rachagani S., Sheinin Y.M., Ouellette M.M., Ponnusamy M.P., Mumby M.C., Batra S.K., Yan Y. PR55α subunit of protein phosphatase 2A supports the tumorigenic and metastatic potential of pancreatic cancer cells by sustaining hyperactive oncogenic signaling. Cancer Res. 2016;76:2243–2253. doi: 10.1158/0008-5472.CAN-15-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs J.S., Jung Y.J., Mole D.R., Lee S., Torres-Cabala C., Chung Y.L., Merino M., Trepel J., Zbar B., Toro J. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Keith B., Johnson R.S., Simon M.C. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer. 2011;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A., Selak M.A., Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- Klotzsche-von Ameln A., Muschter A., Mamlouk S., Kalucka J., Prade I., Franke K., Rezaei M., Poitz D.M., Breier G., Wielockx B. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the antiproliferative activity of TGFβ. Cancer Res. 2011;71:3306–3316. doi: 10.1158/0008-5472.CAN-10-3838. [DOI] [PubMed] [Google Scholar]
- Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Virshup D.M. Two conserved domains in regulatory B subunits mediate binding to the A subunit of protein phosphatase 2A. Eur. J. Biochem. 2002;269:546–552. doi: 10.1046/j.0014-2956.2001.02680.x. [DOI] [PubMed] [Google Scholar]
- Louis J.V., Martens E., Borghgraef P., Lambrecht C., Sents W., Longin S., Zwaenepoel K., Pijnenborg R., Landrieu I., Lippens G. Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc. Natl. Acad. Sci. USA. 2011;108:6957–6962. doi: 10.1073/pnas.1018777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo C.M., Gameiro P.A., Bell E.L., Mattaini K.R., Yang J., Hiller K., Jewell C.M., Johnson Z.R., Irvine D.J., Guarente L. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid M.A., Wang W.I., Rosales K.R., Welliver M.X., Pan M., Kong M. The B55α subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell. 2013;50:200–211. doi: 10.1016/j.molcel.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Sablina A.A., Hector M., Colpaert N., Hahn W.C. Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res. 2010;70:10474–10484. doi: 10.1158/0008-5472.CAN-10-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E. Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell. Signal. 2001;13:7–16. doi: 10.1016/s0898-6568(00)00123-6. [DOI] [PubMed] [Google Scholar]
- Sun L., Song L., Wan Q., Wu G., Li X., Wang Y., Wang J., Liu Z., Zhong X., He X. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015;25:429–444. doi: 10.1038/cr.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadkar P., Despres D., Kraman M., Lozier J., Phadke A., Nagaraju K., Mccright B. The protein phosphatase 2A B56γ regulatory subunit is required for heart development. Dev. Dyn. 2014;243:778–790. doi: 10.1002/dvdy.24111. [DOI] [PubMed] [Google Scholar]
- Wong P.M., Feng Y., Wang J., Shi R., Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015;6:8048. doi: 10.1038/ncomms9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Zhai B., Koivunen P., Shin S.J., Lu G., Liu J., Geisen C., Chakraborty A.A., Moslehi J.J., Smalley D.M. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014;28:1429–1444. doi: 10.1101/gad.242131.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.