

Abstract
The hostile environment of the microscope stage poses numerous challenges to successful imaging of morphogenesis in live tissues. This review aims to highlight some of the main practical considerations to take into account when embarking on a project to image cell behaviour in the context of cells' normal surroundings. Scrutiny of these activities is likely to be the most informative approach to understanding mechanical morphogenesis but is often confounded by the substantial technical difficulties involved in imaging samples over extended periods of time. Repeated observation of cells in live tissue requires that strategies be adopted to prioritize the stability of the sample, ensuring that it remains viable and develops normally while being held in a manner accessible to microscopic examination. Key considerations when creating reliable protocols for time-lapse imaging may be broken down into three main criteria; labelling, mounting and image acquisition. Choices and compromises made here, however, will directly influence image quality, and even small refinements can substantially improve what information may be extracted from images. Live imaging of tissue is difficult but paying close attention to the basics along with a little innovation is likely to be well rewarded.
This article is part of the themed issue ‘Systems morphodynamics: understanding the development of tissue hardware’.
Keywords: live tissue, light microscopy, time lapse, technique, imaging
1. Introduction
It has long been recognized that, from its outset at fertilization, the development of the embryo into the elaborate array of organs and tissues of the adult requires the complex interplay between the chemistry and mechanics of individual cells. Proliferation, differentiation, migration and death of cells are closely coordinated in this process to ensure the robust and reliable formation of viable and fertile offspring. Cells of the developing embryo diversify along differing pathways adopting characteristics and behaviours that drive growth and morphogenesis. Orchestration of these events culminates in the formation of the tissues and organs of the adult animal. Developmental biology aims to understand these processes and has come a long way in describing and explaining at least some aspects of this amazing story. Most often this is achieved by combining and comparing glimpses of information from many individuals at the expense of spatial and/or temporal detail. The mechanics of morphogenesis is achieved by the dynamic interplay of cells with each other as well as their environment and is hard to observe in snapshot. Rather, an understanding of these processes requires the iterative observation of the same cells in temporal and spatial context by imaging live tissue.
Much has been written about live cell imaging in two-dimensional cultures but less so about the challenges arising from attempting to image the behaviour and interactions between groups of cells and cells in tissues in three dimensions, a basic prerequisite for understanding the development of biological form or morphogenesis. While many of the essential components of successful imaging of can be gleaned from studies of cells adhered to dishes, refinement of these basic techniques is required in order to gain the most informative data. Broadly, when designing this type of experiment, equal consideration must be given to four main categories: labelling, culture conditions, optical set-up and analysis pipeline. Inevitably the nature of the task requires that some compromises must be made in all four but a careful appraisal of what these should be can maximize the usefulness of the final dataset. It is beyond the scope of this review to cover all methods that may be useful and so the aim is to propose strategies and principals that can be readily applied leading to successful imaging of morphogenic events when performing ‘tissue microscopy’.
The first basic necessity of successful live cell microscopy is the choice of contrast. With tissue this should allow three-dimensional imaging of cells in the context of what surrounds them. This can be done with general or directed labelling with fluorescent chemical dyes but their application can be hard to control and at worst have detrimental effects on the normal behaviour of cells so for this reason they need to be tested empirically. A more generally used and reliable method for many models is to employ genetically encoded fluorescent proteins. These may be introduced as a transgenic construct or targeted to a particular locus where their expression is controlled by transcriptionally regulated elements.
Green fluorescent protein (GFP), originally isolated from the jellyfish Aequorea victoria [1],was the first to be isolated and introduced into living tissue [2] and remains one of the most used and reliable versions available although a rainbow of coloured variants now exist and allow the use of combinations of colours in the same sample [3]. Careful consideration to the choice of these proteins must be given as their performance in living tissue can vary dramatically [4]. Fluorescence microscopy can be used to visualize native fluorescent proteins in the cytoplasm, and isolated staining can yield morphogenic information but where there is a group of cells together expressing the same fluorescent protein then dynamics of the individual can be hard to determine as cell–cell boundaries are not clear (figure 1). Here, further strategies can be adopted to retrieve a greater amount of useful information. Photoactivatable or photo-convertible fluorescent proteins can be powerful tools as the fluorescent properties of individual cells expressing them can be changed by exposure to UV light. In the case of Kaede or Kikume green-red, for instance, green fluorescence may be rapidly transformed into red and followed over many hours (figure 1) but longevity of this signal is subject to dilution as cells divide and so becomes weaker with time. Fusion of fluorescent protein DNA with other gene sequences can be used to direct a product to any one of a number of subcellular compartments allowing different aspects of the dynamics of living cells to be visualized. Directing fluorescent signal to the cell membrane, for example, can be a more reliable reporter of cell shape and dynamics. Individual cells are relatively easy to identify within a group if the fluorescent protein is localized to their membranes and provides a more useful resource for morphological studies (figure 1). Better still, a combination of compartments such as the nucleus and the cell membrane can be labelled with different fluorescent proteins providing layers of information and significantly aiding analysis [5].
Figure 1.

Localization of fluorescent proteins to different cellular compartments allows improved discrimination of cell dynamics in tissue. (a) Cytoplasmic localization of GFP in a mouse brain slice. Single cells are hard to distinguish when neighbours also express GFP (E.P.). (b) Restricting fluorescent signal to the nucleus with a Histone2B-Citrine fusion in zebrafish tail allows discrimination of individual cells (D.S.). (c) Putting GFP cells of interest in context by expression of a red fluorescent protein in adjacent tissue in fly eye development (E.R.). (d) Membrane restricted expression of green and tomato fluorescent proteins in ROSAmTmG Cre reporter mouse. Cell shape dynamics may be observed even in clusters of similarly expressing cells (J.T). (e) Highlighting and following complex shape of a particular neuron in a zebrafish gut is achieved by photoconverting Kaede fluorescent protein from green to red with a pulse of UV light (S.M). (f) Fluorescent ubiquitination-based cell cycle indicator system (FUCCI) allows segmentation of individual nuclei but also gives a readout of the cell cycle status by the ratio of intensity of green and red fluorescence. Red channel images are shown as magenta (L.A.B).
Imaging samples more than a few micrometres thick is limited primarily by the scattering of light as it passes through the sample. Scattering varies between tissue and is predominantly of the Mie type where light is scattered by particles (in this case cells, nuclei and organelles) of a size in the same order as the wavelength used to detect them. These structures are encased in lipid membranes and have a refractive index different from that surrounding them. It is these mismatches in refractive index that leads to scattering and is hard to avoid when imaging live tissue. Where single-photon excitation is used, there is a practical limit of around 100 µm to visualizing cells although other technologies such as lightsheet or multiphoton microscopy allow visualization to slightly greater depths (box 1). Either way, imaging morphogenesis generally requires that samples be presented to the microscope in such a manner that cells of interest are reasonably peripheral and light scattering is minimized. Practically speaking, this means that either relatively transparent, samples are adopted (like zebrafish or early stage vertebrate embryos) or that the tissue of interest is exposed somehow, either by reducing the bulk of the tissue by explant or slicing or by ‘windowing’ the sample to gain better access.
Box 1. Optical sectioning methodologies.
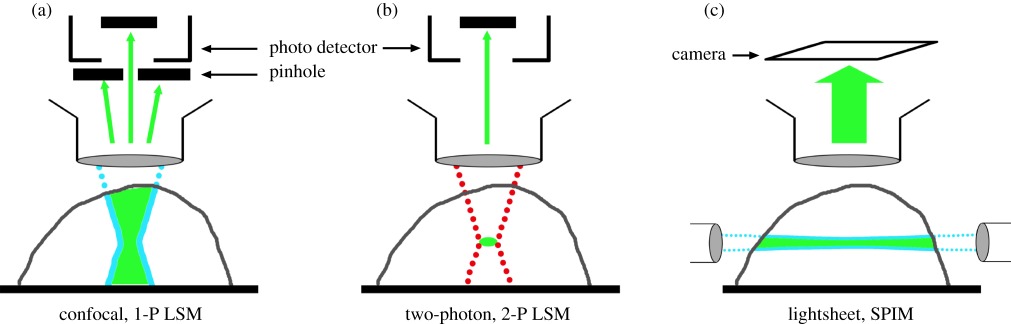
As most biological samples are highly three dimensional, techniques have been developed to obtain high contrast optical sections, removing the contribution of out of focus light that would otherwise obscure detail. Moving the sample through the focal plane and capturing a series of images creates a collection or stack of images that can be computationally combined into a three-dimensional reconstruction.
Optical sectioning of thick specimens may be achieved in a number of ways and typically single photon confocal laser-scanning microscopy (1-P LSM) is the most accessible method. Here, a point of laser light is scanned across the sample in order to excite the fluorophores within. Fluorophores, in the path of the light beam, are excited by single photons with an energy slightly higher than those subsequently emitted (a). Fluorescence emitted by the sample may then be collected and recorded but a pinhole, placed in the detection path, is required to exclude everything but emissions from the plane of focus. In this way, a high-contrast optical section may be built up point by point in a raster pattern.
Two-photon laser-scanning microscopy (2-P LSM) is executed in a similar manner as 1-P LSM but, as the name suggests, requires multiple photons (of lower energy but within a very short period of time) to excite a single fluorophore. Two-photon excitation is a rare nonlinear event and even with high intensity near-infrared lasers only occurs at a single spot, in the plane of focus, where excitation photon density is at its highest (b). Consequently, high-contrast images may be produced without a pinhole as no out of focus emission is produced. This technique has additional benefits such as considerably higher light gathering efficiency, the ability to image deeper in tissue with less fluorescence bleaching and specimen damage outside the focal plane.
Like two-photon microscopy selective plane illumination microscopy (SPIM) or lightsheet imaging has intrinsic optical sectioning as ideally no fluorescence is produced above or below the plane of focus. Single photon excitation is commonly used to illuminate the sample from the side, with a thin laminar sheet of light, and fluorescence is detected in an optical axis perpendicular to the illumination plane (c). With lightsheet there is no requirement to collect images one pixel at a time and so a camera can be used offering a significant speed and dynamic range advantages over raster scanning techniques.
Animal models able to develop normally outside of their mother are challenging but relatively straightforward to mount on the microscope. Protocols perfected here do however offer hints relevant to mammalian embryos and tissue, which sometimes can be barely possible to image. All these methods pose significant technical challenges and innovation, determination and attention to detail are required to mount samples so that they grow normally and can be imaged successfully under the microscope.
Having labelled and mounted the sample, the next key component of any imaging experiment is the microscope, and experimental choices (and compromises) made for most revolve around what is available to them. Optical sectioning is the method of choice as it allows the creation of volume data by building up a series of two-dimensional images captured from different focal planes of the sample. This can be achieved either by discarding out-of-focus light as in confocal microscopy or by capturing images of a fluorescent signal illuminated only at the focal plane as in multiphoton microscopy or lightsheet microscopy (box 1). Recently, much attention has been devoted to the promise of the emerging technology of lightsheet microscopy. Here, optical sectioning is achieved by moving fluorescent samples through a ‘sheet’ of excitation light perpendicular to the imaging objective. The method is fast, sensitive and has begun to yield high-quality data from a variety of sample types. Unfortunately at the moment, access to these systems is quite limited but hopefully in the not too distant future this technology will be more generally available. Until such a time, laser scanning microscopy is likely to offer the most accessible method of producing high-quality volume data with the most commonly available and useful system a line scanning confocal. Since its first commercial appearance, about a quarter of a century ago, this form of confocal microscopy has become routine to many laboratories. High-contrast images are built up pixel by pixel; and serial acquisition, at different axial planes, allows three-dimensional reconstructions of topologically complex objects. Repeating volume acquisitions in time gives the required fourth dimension and therefore ‘time-lapse movies'. Thankfully control of the imaging routine can be fully automated but it requires a little bit of thought when setting up the system in order to get the required information out of the experiment. Compromises must be made between signal-to-noise ratio, spatial resolution and time resolution as the project dictates.
2. General considerations
In order to collect reliable data, great care must be taken to ensure the fidelity of samples over an extended period of time in the extreme environment of the microscope stage. There are many considerations when bringing live samples to a microscope so that they can be maintained in a healthy condition for the duration of the experiment. Observation of morphogenesis requires an attention to detail on several levels. Viability of the samples is paramount but impact of contrast choice and imaging set-up all significantly contribute to the quality of the dataset obtained. A medium- to long-term experiment, lasting hours to days, may be affected by many things, and efforts must be made to limit variability wherever possible. Small improvements in any of these criteria can build to make analysis considerably easier in the long run and it is well worth the time and effort to iteratively adjust the whole protocol in response to the requirements of the analysis pipeline. Hitting the imaging sweet spot can be very rewarding but exposes a common bottleneck in the process, namely storing handling and analysing numerous large datasets. This is a serious consideration as a single overnight experiment may easily create 100 gigabytes of data, which is hard to even open on a standard desktop computer. It is beyond the scope of this review to discuss the best way to do this, but there are several useful reviews that begin to explain how this problem may be tackled [6–9].
3. Temperature
Modern microscope systems are a significant investment for any laboratory and to get the most out of them they should be situated in a stable, quiet space where vibration is minimized and room temperature is carefully controlled. Air conditioning should be maintained at a constant temperature preferably without any significant draft falling onto the microscope, and vibration should be minimized by placing the system in a quiet spot and preferably on some form of anti-vibration table.
The wider environment of the microscope significantly affects stability of the sample. The impact is not only in maintaining a stable sample temperature but also that large fluctuations can cause expansion and contraction of the microscope itself causing lateral as well as axial drift. For this, the most reliable overall maintenance of temperature is achieved by using environmental chambers, surrounding a large part of the system. Further improvements can be made with a second, smaller chamber, surrounding the sample itself, which can often be made at trivial expense. This is required in order to maintain an even temperature (without draft) but also humidity and gas concentration where required. We routinely conduct experiments in our core facility on an upright confocal microscope with an inner imaging tent constructed from Saran™ wrap, bits of sticky tape and weights. It is not pretty but it is reliable, cheap and easily fabricated. Temperature is monitored using a small thermocouple attached to a data logger measuring room temperature as well as that immediately adjacent to the sample. Environmental chambers can be heated as required using a blown-air heater or a passive heat element but care must be taken with the placement of the control thermocouple to ensure that the temperature at the sample is what is expected. We also use the blown air even where heating is not necessarily required (fish, fly, frog experiments) in order to combat temperature creep produced when microscope components are active (a galvo-z stage in particular can cause a rise in chamber temperature of 2–3°C).
4. Gas
Gas is clearly important where bicarbonate-buffered tissue culture media is used and generally is a mixture of 5% CO2 balanced with atmospheric air (N2/O2). Sufficient oxygen supply is necessary for the tissue integrity and where submerged it is wise not to flood the sample with too much media as slow oxygen diffusion can cause hypoxia of the sample. For some embryos and tissues, oxygen concentrations different from atmospheric are beneficial for tissue integrity and in this case cylinders of mixed gas or digital gas mixers can be used to attain the correct mix for the particular set-up.
When supplying gas, it is important that it is humidified (by bubbling through warm water) but particularly that the flow rate is not too high. The gas is never perfectly humidified and high flow rates run the risk of evaporating sample media or even causing a draft detrimental to imaging stability. As long as gas is maintained at a slight positive pressure within the imaging chamber then it is usually sufficient to buffer the media and equates to about 0.1–0.2 l min−1 for standard-sized chambers (slightly bigger than a well plate). Checking the flow by observing bubble rate through the humidifying bottle is usually the most reliable way to get this right. Also, a test sample set-up with tissue culture media containing pH indicator will readily demonstrate, by its colour, which minimum flow rate is providing sufficient CO2 to allow the bicarbonate buffer to perform properly. For the actual imaging experiment, it is wise to use media without pH indicator as it is itself fluorescent and can reduce the signal-to-noise ratio. While it is possible to use CO2 independent media by the addition of HEPES, there may be a significant phototoxic effect and so it is worth optimizing conditions with a complete bicarbonate buffering method if possible [10].
5. Humidity
Tissue culture is traditionally performed in a wet incubator that should be able to maintain near 100% saturation of the atmosphere (as long as the door is not being opened too much). Similar conditions can be difficult to maintain on the microscope and result in evaporation of water from the sample set-up dramatically changing the constitution of media used to maintain the sample. Tenting the sample and having wetted tissues within the tent helps significantly (especially as a much smaller volume of air needs to be humidified). A simple innovation is to cover the sample with Teflon®-FEP (fluorinated ethylene propylene) film, which is impermeable to water but will allow gas exchange. This has a number of advantages including protecting the sample from infection but primarily its use is to prevent evaporation and maintain the correct media osmolarity. For these reasons, it is used in long-term neural culture experiments [11] and tissue culture transport bags. This thin (12.5–25 µm) film is autoclavable and has a refractive index close to water so it is possible to image through it without significantly impacting image quality. Commercial products incorporating this membrane can be bought for microscopy applications such as tissue culture dishes (Lumox®–Sarstedt) or ‘foil covers' (Pecon™) that replace the lid of various imaging dish types. Alternatively, which is what we do, is to buy a lifetime supply from American Durafilm on a large reel, cutting it and using as necessary.
6. Fluorophore choice
Transgenic reporters have been produced which express fluorescent proteins in a wide range of different cells and tissues but often their expression can be unreliable and subject to genetic background and copy number effects so must be carefully appraised before investing too much time in any particular line that ultimately does not perform as expected. The whole imaging and analysis pipeline should be considered here, and pilot studies (even with fixed tissue) are well worth performing to establish which fluorophores work best for a particular experiment rather than just making do with what is immediately available.
For many animal models, creation or import and appraisal of reporter lines can be relatively quick and inexpensive, but for mammalian studies, where mouse is the model of choice, this can be a protracted and costly exercise. An efficient strategy, however, is to choose a reliable, conditionally activated reporter system (usually Cre/loxP) where the extent of fluorescently labelled cells is controlled in a spatial manner by breeding against strains with tissue-specific Cre recombinase expression. In mice, the GT(ROSA)26Sor locus is commonly targeted to harbour reporter constructs as it reliably gives high-level, ubiquitous expression throughout development with no obvious phenotypic differences from wild-type littermates [12] and a wide array of conditional reporters are already available for live imaging different aspects of cell behaviour [13]. In addition, temporal control can be achieved where the drug-inducible Cre, CreERT2 is the form expressed. This fusion of Cre and a modified oestrogen receptor will only translocate to the nucleus, and so recombine loxP sites, when it binds tamoxifen. If this drug is administered sparingly then recombination of the Cre reporter will only occur in a proportion of CreERT2 expressing cells with the resulting mosaic expression being particularly useful to observe cell dynamics in morphogenic studies [14]. More stochastic approaches to introducing fluorophore-expressing constructs into tissue may be used if less careful regulation of expression sites can be tolerated. Such approaches include electroporation or the use of viral vectors and are particularly useful in models, such as chick, which are not accessible to more permanent forms of genetic manipulation [15,16].
7. Mounting
Mounting and growing embryos or tissues for microscopic imaging poses significant challenges but is critical in order to serially acquire high-quality images. Primary aims are to ensure that the sample remains static and healthy, and develops normally. In addition, it needs to be prepared and mounted in such a way that cells of interest are accessible to high numerical aperture objectives with short working distances and that they can be viewed through the minimal amount of intervening (scattering) tissue.
One of the simplest set-ups may be used for fly pupae. A pupa can be stuck to a small strip of modelling clay on a glass slide; a window is then carefully cut in the puparium operculum above the tissue of interest, e.g. wing disc, and gas-permeable 10S oil (Voltalef, PROLABO, Paris, France) is used to join the fly ‘window’ to a coverslip suspended above the slide on sticky tape or silicon grease (figure 2) [17]. Several samples can be arranged next to each other on a single slide allowing multiple time-lapse acquisitions within one experiment thus satisfying all the criteria of effective sample mounting.
Figure 2.

Example sample mounting techniques for live tissue imaging for both upright and inverted imaging geometries. (a) High-resolution images may be acquired in windowed fly pupae mounted against a coverglass and may be imaged from above or below by inverting the slide. (b) An array of samples mounted on a gel pad braced against the edges of a 60 mm tissue culture dish. (c) Inverted culture system for imaging samples through a coverglass or gas-permeable Lumox® dish.
Earlier-stage flies, frogs or zebrafish are maintained in a simple aqueous environment and require making of some sort of mould to constrain them. This is commonly achieved by micro-machining or three-dimensional printing a mould that is then used to cast agarose mounts in culture dishes, into which samples are put and gently held in place by a coverslip or a thin layer of low melting point (LMP) agarose [18,19]. This method, easily adapted for mammalian explants as agarose, is an excellent material that allows free flow of media to the sample but should be equilibrated before use to prevent dilution of media by residual water. Samples such as tissue slices may need additional support and can be adhered to alternative supports such as filter membranes (Millipore AABP02500) and raised off the base of the dish on agarose pads or metal grids [20–23]. This works particularly well for delicate organotypic explants that require an air/liquid interface but slices should not be too thick (in the range of 150–300 µm) thus ensuring good gas and media exchange and the health of the sample.
Commonly, we use an agarose stand cast in a 60 mm dish for a wide range of sample types (figure 2). The agarose pad is cut out around the central portion where an array of tissue can be placed (securing them with a drop of LMP agarose where necessary). The ‘legs’ (figure 2) touching the edge of the dish mean that the gel support is braced and so will not shift position as the stage moves but also allows plenty of media to be used, often negating a need for a complicated perfusion set-up. Placing several explants or slices close together in the centre of the dish means that even quite wide-diameter objectives have access and that a minimal distance is travelled between samples, saving acquisition time and alleviating problems related to media sloshing around as the stage moves. Inclusion of numerous samples dramatically increases efficiency with one imaging experiment giving many individual datasets and importantly allowing for control and experimental samples to be imaged under exactly the same conditions.
The gel pad set-up is ideally suited to upright microscope systems using dry or dipping objectives. Dry objectives are used where the sample requires an air interface to grow properly and works best with objectives that have a reasonable working distance. Additional protection from osmolarity changes can be achieved by covering the dish with gas-permeable Teflon®-FEP and imaging through it thus maintaining sample integrity whether dry or dipping objectives are used. Where a similar set-up is required for an inverted microscope an arrangement using Lumox® dishes may be created but care must be taken to allow the appropriate gas mixture to enter the bottom of the dish. Dry objectives must be used to maintain the benefit of gas access to the sample but it is easier to use higher numerical aperture (NA) objectives with shorter working distances without inadvertently dipping them in media.
Some of the trickiest samples to image live are post–implantation stage mouse embryos. Stages up to mid-gestation can, however, be grown and imaged in toto either on filter membranes [24] or by suspending them in media above a coverglass-bottomed dish [25,26] but, as with many types of mammalian explants, success requires empirically establishing the most appropriate media composition and oxygen concentration first [27].
8. Imaging
As discussed above, it is likely that a line-scanning confocal will be the method of choice in morphogenic studies and most manufacturers produce excellent systems allowing automated time-lapse recording. Although software implementations are proprietary general principals apply when setting up the system, and compromises made here will impact directly on the quality of the dataset; however, as sample integrity remains paramount, every effort should be made to limit damage to the sample while observing it. This can be achieved by optimizing the optical set-up thus allowing the least exposure to damaging light.
The most important optical component of any microscope is the objective lens, and increasing resolution and contrast of images is primarily achieved by using those with the highest numerical aperture (NA) practicable. NA of a microscope objective, as originally defined by German physicist Ernst Abbe, is a measure of its ability to gather light and hence its capacity to resolve objects. NA is always written on the objective following the magnification and will generally increase (along with cost) as field of view and working distance decreases. Dry lenses have an NA limit of about 0.95 due to the refractive index difference at the glass (liquid)/air interface. Dipping or immersion objectives can achieve higher NAs, by reducing these differences, and a little imagination in sample preparation may be needed in order to accommodate them. Needless to say, the objective lens will only perform as designed when it is pristine, and it is worth confirming this before commencing each imaging session.
Generally, microscope objectives are designed to work through a coverglass. The coverglass is critical to the performance of an objective but its importance is often overlooked. While some objectives are designed to dip directly into media with no intervening glass most are designed to have a coverglass of a particular thickness between sample and lens. Ordinarily, this thickness is 0.17 mm, which equates to a #1.5 coverglass, and it is imperative that this is used with objectives of NA higher than about 0.4. Where NA of the objective is higher than this, then a coverglass that is thicker or thinner by as little as a few micrometres has a significant impact on achievable resolution and contrast [28]. Helpfully, manufacturers also write on the side of each objective which particular glass thickness it was designed for although for some high-NA objectives there is a correction collar. If present, this must be carefully adjusted for differing glass thicknesses and temperature. Set-ups described above can work with coverglass; and on upright microscopes, it is useful to use large ones (40 mm) to avoid mixing of culture media and immersion liquid.
Detection components within the microscope are key to light efficiency, and development of detectors for newer confocal systems offers a sensitivity advantage over traditional photomultiplier tubes (PMTs) and should be adopted where possible. PMTs are the standard way to convert fluorescent signal from the sample to grey values. Within the PMT, photoelectrons are produced from photons of light by interaction with a light-sensitive photocathode. These photoelectrons are then amplified by a series of dynodes to give an analogue PMT signal that can be digitized and logged in reference to its coordinate in the raster scan. Increased sensitivity is conferred by increasing ‘gain’, which equates to increasing the potential difference between amplifying dynodes but can diminish the signal-to-noise ratio if set too high. Newer detector technologies refine the principal of the PMT. A more sensitive photocathode material, gallium arsinide phosphate (GaAsP), is used in some PMTs giving an improved amplification of the initial signal and significant improval in signal-to-noise ratio. Hybrid detectors (HyDs) also take advantage of the GaAsP photocathode but instead the amplification step is achieved using an avalanche photodiode giving an overall approximately 150 000 fold amplification in signal. This allows significantly less excitation light to be used without compromising signal-to-noise ratio but it is always worth running pilot experiments specifically to appraise the effects of light dose on sample integrity even if this means using up a few good-quality samples along the way.
Setting up imaging parameters can be a black art and should be determined empirically for each biological and imaging system. Most modern microscope software has modules that allow multiple fields to be imaged with different parameters and this can be very helpful in pilot experiments aimed at establishing what these should be. In general, the aim is to achieve sufficient resolution while minimizing light dose, increasing acquisition speed and improving signal-to-noise ratio. This can be quite a difficult balancing act as generally improvement in one parameter necessitates compromise in the others. Spatial resolution should be considered first as enough detail must exist for analysis. Relative pixel size in a confocal image may be tuned to requirements by adjusting scan format and zoom parameters as pixel size equates to the length of each axis of the image divided by the number of pixels in it. The numerical aperture of the objective used provides a guide to the minimum useful pixel size but often will outperform actual requirements. The time the laser beam illuminates each point of the specimen is referred to as the dwell time. Although slower scan speeds and hence longer dwell times will improve image quality, increased exposure to light over many cycles may lead to photobleaching and damage, especially as the entire depth of the sample is illuminated in single-photon confocal microscopy. Signal-to-noise ratio may similarly be improved by averaging which preferentially diminishes random noise but suffers the same detrimental effects as increasing dwell time so must be carefully appraised. Resonant scanners offer very fast imaging speeds without diminishing field of view too much but reduction in dwell time will probably necessitate increased averages in order to provide images of sufficient quality. When deciding acquisition settings, it is important always to respond to the requirements of the analysis pipeline and establish which compromises in image quality and/or sampling frequency are most acceptable.
Although it is worth exploring the adjustable parameters of the confocal system, there are a few that always benefit imaging live tissue. Piezo z-stages or objective scanners generally move far faster than focus drives that move heavy objective turrets skimming time from stack acquisitions. Bidirectional scanning is an option that can save significant amounts of time but care must be taken in order to adjust the phase of odd and even lines so that they line up correctly. Image format, scan-field (zoom) and scan-field rotation should be adjusted so that the required lateral resolution (pixel size) is achieved and only regions of interest are imaged. Sticking doggedly to a square image format, where much of the field does not contain anything of interest, wastes time that could otherwise be used to improve signal-to-noise ratio, collect images from multiple samples or increase temporal resolution. Axial resolution too can be readily compromised, as it is rarely necessary to collect optimally spaced z-stacks. If photobleaching is a problem then, apart from adjusting the sensitivity of the detectors, the system pinhole may be opened a little wider than the optimal. Optical sections will be thicker but there will be significant gains in signal detected, allowing lower laser powers to be used. Finally, switching image depth from the default 8-bit to 12-bit or higher provides more greyscale range that, although it may not show an obvious improvement to onscreen images, is critical when automatically segmenting them as part of the analysis pipeline.
Having established the basic imaging parameters, the time-lapse protocol can be assembled. When first introduced to the microscope, samples will undergo a period of settling down that is different from natural growth or instability of the dish on the stage. Setting of the field of view and z-stack depth should account for natural movement but samples should be checked within the first couple of hours to ensure they have not settled away from the acquired volume. The period of sequential acquisitions is dictated by the speed of the process being observed but often allows for imaging at multiple positions. If this is the case, then samples should be imaged in an order that minimizes the distance travelled between samples. This ensures that samples do not shift due to violent stage movements and minimizes problems due to media sloshing about in the dish.
9. Concluding remarks
It is possible to learn a lot by looking, and this is particularly true when attempting to understand the dynamics of tissue morphogenesis. Successfully imaging these processes as they occur presents particular challenges that may appear intimidating at first but can be overcome by paying careful attention to the basics of good tissue culture and microscopy. Although the future offers substantial improvements in speed and sensitivity, the ability to present high-quality samples to the microscope will remain paramount. Perhaps, the most valuable piece of advice is not to expect such experiments to work on the first attempt but to be assured that a little tenacity and attention to detail is likely to pay dividends.
Acknowledgements
Thanks to Emilie Pacary, Daniele Soroldoni, Emily Richardson, Jacqueline Tabler Sarah McCalum and Luis Alberto Baena for samples and images used.
Competing interests
I have no competing interests.
Funding
No funding has been received for this article.
References
- 1.Shimomura O, Johnson FH, Saiga Y. 1962. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell Comp. Physiol. 59, 223–239. ( 10.1002/jcp.1030590302) [DOI] [PubMed] [Google Scholar]
- 2.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. 1994. Green fluorescent protein as a marker for gene expression. Science 263, 802–805. ( 10.1126/science.8303295) [DOI] [PubMed] [Google Scholar]
- 3.Shaner N, Steinbach P, Tsien R. 2005. A guide to choosing fluorescent proteins. Nat. Meth. 2, 905–909. ( 10.1038/nmeth819) [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Osipovich A, Ustione A, Potter L, Hipkens S, Gangula R, Yuan W, Piston D, Magnuson M. 2011. Quantification of factors influencing fluorescent protein expression using RMCE to generate an allelic series in the ROSA26 locus in mice. Dis. Models Mech. 4, 537–547. ( 10.1242/dmm.006569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shioi G, et al. 2011. A mouse reporter line to conditionally mark nuclei and cell membranes for in vivo live-imaging. Genesis 49, 570–578. ( 10.1002/dvg.20758) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindelin J, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. ( 10.1038/nmeth.2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pietzsch T, Saalfeld S, Preibisch S, Tomancak P. 2015. BigDataViewer: visualization and processing for large image data sets. Nat. Methods 12, 481–483. ( 10.1038/nmeth.3392) [DOI] [PubMed] [Google Scholar]
- 8.Heemskerk I, Streichan SJ. 2015. Tissue cartography: compressing bio-image data by dimensional reduction. Nat. Methods 12, 1139–1142. ( 10.1038/nmeth.3648) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sbalzarini IF. 2016. Seeing Is believing: quantifying is convincing: computational image analysis in biology. Adv. Anat. Embryol. Cell Biol. 219, 1–39. ( 10.1007/978-3-319-28549-8_1) [DOI] [PubMed] [Google Scholar]
- 10.Spierenburg GT, Oerlemans FT, van Laarhoven JP, de Bruyn CH. 1984. Phototoxicity of N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid-buffered culture media for human leukemic cell lines. Cancer Res. 44, 2253–2254. [PubMed] [Google Scholar]
- 11.Potter SM, DeMarse TB. 2001. A new approach to neural cell culture for long-term studies. J. Neurosci. Methods 110, 17–24. ( 10.1016/s0165-0270(01)00412-5) [DOI] [PubMed] [Google Scholar]
- 12.Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. 1997. Disruption of overlapping transcripts in the ROSA βgeo 26 gene trap strain leads to widespread expression of β-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl Acad. Sci. USA 94, 3789–3794. ( 10.1073/pnas.94.8.3789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abe T, Fujimori T. 2013. Reporter mouse lines for fluorescence imaging. Dev. Growth Differ. 55, 390–405. ( 10.1111/dgd.12062) [DOI] [PubMed] [Google Scholar]
- 14.Panousopoulou E, Green JBA, Nusse R. 2016. Invagination of Ectodermal Placodes Is Driven by Cell Intercalation-Mediated Contraction of the Suprabasal Tissue Canopy. PLoS Biol. 14, e1002405 ( 10.1371/journal.pbio.1002405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pacary E, Haas MA, Wildner H, Azzarelli R, Bell DM, Abrous DN, Guillemot F. 2012. Visualization and genetic manipulation of dendrites and spines in the mouse cerebral cortex and hippocampus using in utero electroporation. J. Vis. Exp. 65, pii: e4163 ( 10.3791/4163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Trivedi V, Truong TV, Koos DS, Lansford R, Chuong CM, Warburton D, Moats RA, Fraser SE. 2015. Dynamic imaging of the growth plate cartilage reveals multiple contributors to skeletal morphogenesis. Nat. Commun. 6, 6798 ( 10.1038/ncomms7798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Classen AK, Aigouy B, Giangrande A, Eaton S. 2008. Imaging Drosophila pupal wing morphogenesis. Methods Mol. Biol. 420, 265–275. ( 10.1007/978-1-59745-583-1_16) [DOI] [PubMed] [Google Scholar]
- 18.Megason S. 2009. In toto imaging of embryogenesis with confocal time-lapse microscopy. In Zebrafish (eds Lieschke G, Oates A, Kawakami K), pp. 317–332. New York, NY: Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kieserman EK, Lee C, Gray RS, Park T, Wallingford JB. 2010. High-magnification in vivo imaging of Xenopus embryos for cell and developmental biology. Cold Spring Harb Prot. 2010, pdb.prot5427. ( 10.1101/pdb.prot5427) [DOI] [PubMed] [Google Scholar]
- 20.Alfaqeeh SA, Tucker AS. 2013. The slice culture method for following development of tooth germs in explant culture. J Vis Exp. 81, 50824 ( 10.3791/50824) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brachmann I, Tucker KL. 2011. Organotypic slice culture of GFP-expressing mouse embryos for real-time imaging of peripheral nerve outgrowth. J Vis Exp. 49, pii: e2309 ( 10.3791/2309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gohbara A, Katagiri K, Sato T, Kubota Y, Kagechika H, Araki Y, Araki Y, Ogawa T. 2010. In vitro murine spermatogenesis in an organ culture system. Biol. Reprod. 83, 261–267. ( 10.1095/biolreprod.110.083899) [DOI] [PubMed] [Google Scholar]
- 23.Dailey ME, Marrs GS, Kurpius D. 2011. Maintaining live cells and tissue slices in the imaging setup. Cold Spring Harb protoc. 4 pdb.top105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massarwa RA, Niswander L. 2013. In toto live imaging of mouse morphogenesis and new insights into neural tube closure. Development 140, 226–236. ( 10.1242/dev.085001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamanaka Y, Tamplin OJ, Beckers A, Gossler A, Rossant J. 2007. Live imaging and genetic analysis of mouse notochord formation reveals regional morphogenetic mechanisms. Dev. Cell 13, 884–896. ( 10.1016/j.devcel.2007.10.016) [DOI] [PubMed] [Google Scholar]
- 26.Nowotschin S, Ferrer-Vaquer A, Hadjantonakis AK. 2010. Imaging mouse development with confocal time-lapse microscopy. Methods Enzymol. 476, 351–377. ( 10.1016/S0076-6879(10)76020-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy A, Gertsenstein M, Vintersten K, Behringer R. 2003. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 28.Keller HE. 2006. Objective lenses for confocal microscopy. In Handbook of biological confocal microscopy (ed. Pawley BJ.), pp. 145–161. Boston, MA: Springer. [Google Scholar]