

Abstract
Melanoma is a most dangerous and deadly type of skin cancer, and considered intrinsically resistant to both radiotherapy and chemotherapy. It has become a major public health concern as the incidence of melanoma has been rising steadily over recent decades with a 5-year survival remaining less than 5%. Detection of the disease in early stage may be curable, but late stage metastatic disease that has spread to other organs has an extremely poor prognosis with a median survival of less than 10 months. Since metastatic melanoma is unresponsive to therapy that is currently available, research is now focused on different treatment strategies such as combinations of surgery, chemotherapy and radiotherapy. The molecular basis of resistance to chemotherapy seen in melanoma is multifactorial; defective drug transport system, altered apoptotic pathway, deregulation of apoptosis and/or changes in enzymatic systems that mediate cellular metabolic machinery. Understanding of alterations in molecular processes involved in drug resistance may help in developing new therapeutic approaches to treatment of malignant melanoma.
Key words: Drug-resistance, melanoma, chemotherapy, BRAF mutation, combination therapy, review
Competing interest statement
Conflict of interest: there are no conflicts of interest.
Introduction
The development of resistance to chemotherapy continues to be the major impediment in the treatment of cancer patients. Newer agents, whether chemotherapeutic or targeted, are constantly being developed. Though most anticancer therapies will alter tumor growth, in most cases the effect is not long lasting and treatment failure has an impact on the survival of advanced skin cancer patients. Consequently, there is a significant need for newer agents with low susceptibility to common drug resistance mechanisms in order to improve response rates and potentially extend survival.1
Melanoma is a rare form of skin cancer, which develops through the malignant transformation of melanocytes. It has the fastest growing incidence of any cancer among men and the second fastest growing incidence in women.2 If recognized and treated early, most cases of melanoma are curable. However, once metastasized it is difficult to treat and is responsible for 80% of deaths related to skin cancers.3,4 The efficiency of treatments for metastatic melanoma has not significantly improved over the past 50 years with the 5-year survival rate being less than 5%.5-7 The American Cancer Society estimated that 9,730 deaths will occur due to melanoma in 2017 with an incidence of 3.1/100,000 in USA, as well as 3,400 deaths from other forms of skin cancer, excluding basal cell and squamous cell carcinomas.8 The World Health Organization has estimated that more than 65,000 people die from melanoma in a year worldwide.9 Surgery, radiotherapy and chemotherapies are the commonly used therapies for non-metastasized melanoma. However, once metastasized, the treatment options for melanoma become very limited.
Chemotherapy is an extremely ineffective and unsatisfactory means of treating malignant melanoma due to drug resistance, which is characteristic of this disease. Development of drug resistance, either by intrinsic at onset (primary or intrinsic resistance) or during application of cytostatic drug (acquired resistance), is a major problem that limits the effectiveness of chemotherapies used to treat malignant melanoma. Therefore, if drug resistance could be overcome, there may be a significant impact on the survival rate. One of the various factors contributing to sensitivity of drugs is the limited amount of drug reaching the tumour and affecting the tumour microenvironment.
In this review, the relevance of certain factors, such as transport mechanisms, various enzyme systems, alterations in drug target, processing of drug-induced damage; and regulatory aspects of apoptosis for melanoma drug resistance, has been discussed.
Molecular mechanisms of drug resistance
The mechanisms conferring this intrinsic drug resistance in melanoma cells are poorly understood. Grottke et al. found eleven genes that were differentially expressed upon acquisition of etoposide resistance in malignant melanoma cells. However, most of them were of unknown function and are yet to be characterized. Multiple mechanisms identified in chemoresistance of other tumor cell types have been investigated in melanoma as well10 (Figure 1).
Figure 1.
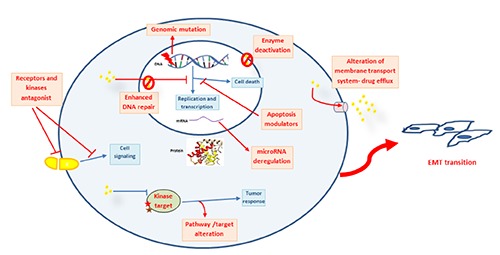
Overview of drug resistance mechanism in melanoma.
Drug transport and efflux pump mediated resistance
Drug efflux system is the most commonly observed mechanisms, responsible for reduced intracellular accumulation of cytostatic drugs in drug resistant cell lines. Drug resistance is mediated by ATP-binding cassette (ABC) transporters which actively transport drugs out of the cells.11 The two classes of ATP-dependent drug transporter proteins, P-glycoprotein (Pgp) and the multidrug resistance-associated proteins (MRPs), mediate drug efflux which reduces drug accumulation and makes tumor cells resistant to the cytotoxic effects of many anticancer agents. Among the 48 known human ABC transporters, melanoma cells express the most studied and targeted mediators of drug resistance such as ABCB1 (MDR1), ABCC1 (MRP1), ABCC2 (MRP2), ABCA9, ABCB5, ABCB8, ABCD1 and ABCG2 (MXR).12-16 Both ABCB5 and ABCB8 expression are known to facilitate doxorubicin resistance in melanoma cells13,17 and ABCC2 to mediate cisplatin resistance.18 Luo et al. isolated a subpopulation of drug effluxing cells directly from melanoma patients and named them as ‘side population’. These cells were reported to have increased efflux capacity and resistant to paclitaxel by up-regulation of ABCB1 and ABCB5.15 However, most melanoma cell lines as well as primary and metastatic tumors do not express Pgp and therefore it cannot be regarded as a major common feature mediating drug resistance in human melanoma cells.19,20 On the other hand, MRP is frequently expressed in melanoma but its expression is not up regulated significantly in response to chemotherapy. Although all drug transporters have not been studied, the present literature is suggestive that induction of drug transporter may not be the prime cause of drug-mediated resistance in skin cancer. Some studies have demonstrated association of melanoma with MRP121 and MRP222 expression to cytostatic drugs. In a phase I clinical study, only partial response was observed in a patient with melanoma who was treated with epirubicin (MRP1 inhibitor) in combination with sulindac (a non-steroidal anti-inflammatory drug).23 Therefore, the role of MRP-dependent transport mechanisms in melanoma drug resistance remains unclear.
Drug resistance mediated by altered enzyme activation
Inactivation of drugs can diminish the availability of free drug to bind to its intracellular targets. The intracellular detoxification of many anticancer drugs (mostly alkylating agents) occurs by conjugation with glutathione (GSH), catalyzed by enzyme glutathione-S-transferase (GST).24-29 GSH is a powerful antioxidant, which inhibits oxidative stress that can damage DNA and RNA. The level of GST was found to be higher in melanoma lesions compared to benign melanocytic nevi.30,31 The conjugation anticancer drugs with GSH inhibits the conversion of mono-adducts to cross-links, thereby reducing the cytotoxic potential of the adducts. The c-Myc-induced apoptosis was reported to have dependency on the intracellular GSH content in melanoma.32 However, in a panel of melanomas, no significant correlation was observed between alteration in GST/GSH-metabolism with the course of tumor progression, treatment and clinical response.26,33,34 In addition, there was neither any increase in GSH concentration in cisplatin cytotoxicity nor was the drug level affected in melanoma cells.35
Topoisomerase (Topo) is a nuclear enzyme that plays important roles for DNA transcription and recombination as well as segregation of chromatids during mitosis. It has been targeted by various inhibitory chemotherapeutic agents such as camptothecin and its derivatives, which inhibit Topo I, and doxorubicin, etoposides, mitoxantrone which inhibit Topo II.36,37 In melanoma, etoposide resistance has been associated both with mutation or deletion38 and increased activity of Topo II.39 However, Satherley et al. could not establish an association of chemosensitivity with expression of Topo II in choroidal melanoma.40 Moreover, nitric oxide (□NO) oxidation is shown to detoxify etoposide through direct nitrogen oxide radical attack.41
Drug resistance mediated by altered DNA repair
The alkylating agents induce cytotoxic O6-chloroethylguanine DNA lesions via adducts formation, followed by inter-strand DNA crosslinks and subsequent inhibition of DNA replication or RNA transcription, leading to arrest of the cell cycle in G2 phase. The DNA repair enzyme O6-alkylguanine DNA alkyltransferase helps in repair of these adducts thus impairing the cytotoxic effect and mediating a major resistance pathway for these drugs.42 Drugresistant melanoma cell lines exhibit increased (base excision) repair of DNA damage.43 In addition, decreased nuclear mismatch-repair protein expression was associated with a fotemustine-resistant phenotype in drug-resistant melanoma cell lines.39 Up-regulation associated with increased activity of O6-methylguanine-DNA methyltransferase (MGMT), an enzyme involved in repair of DNA damage caused by alkylation, was found in melanoma.44 Clinical trials proving the association of drug response and MGMT expression are inconclusive.45,46 However, in a recently performed retrospective study no significant correlation of mismatch repair (MMR) expression, alone, or in combination with MGMT levels (with clinical response to dacarbazine-based chemotherapy) could be found,47 suggesting that these molecules are not predictive of clinical response.
Drug resistance via modulation of the apoptotic pathway
Apoptosis represents a complex genetic program consisting of several pathways. Presently there are two well-characterized caspase-activating cascades that regulate caspase-mediated cell death. The extrinsic pathway is triggered by binding of ligands to their individual cell-surface death receptor (e.g., cluster of differentiation 95 (CD95), tumor necrosis factor-related apoptosis-inducing ligand receptor 1-4 (TRAIL-R1-4), tumor necrosis factor receptor 1(TNF-R1)) which, after oligomerization, recruits adapter molecules and initiator caspases (caspase-8/ caspase-10) that result in a proteolytic cascade.48,49 Interference of CD95 (Fas/APO-1) mediated apoptosis terminate immune response.50,51 The intrinsic pathway involves mitochondrial release of cytochrome c that binds to apoptotic protease activating factor-1 (Apaf-1) and thereby induces conformational changes of this apoptotic protein followed by recruitment of procaspase-9 to the complex.52 Procaspase-9 is subsequently autocatalyzed.48,49,52 These two pathways converge with the activation of effector caspases, induction of specific endonucleases resulting in DNA fragmentation and cleavage of nuclear proteins essential for nuclear and cellular structure, DNA repair and replication.53 Cell death or survival is balanced by a number of regulator molecules at multiple levels such as p53, rat sarcoma (Ras), Bcl-2-family proteins or members of the IAP family. Recently, Koetz-Ploch et al. showed that in drug-resistant melanoma cells, microRNA-125a promotes the resistance to BRAF (v-raf murine sarcoma viral oncogene homolog B) inhibitors by apoptotic suppression, as well as reactivation of signalling pathways such as mitogen-activated protein kinase (MAPK) and AKT8 virus oncogene cellular homolog (AKT).54
p53
Either overexpression of human double minute 2 (Hdm2), a negative regulator that binds and ubiquitinates p53, or loss of the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene encoding alternate reading frame protein (p14ARF), a factor that inhibits the Hdm2-dependent degradation of p53, is commonly observed in metastatic melanoma cells.49,55,56 In addition, in malignant melanomas, the anti-apoptotic protein Bcl-2 is overexpressed while Apaf-1, an important factor for apoptosome formation in the intrinsic apoptosis pathway, is downregulated.57-59 Similarly overexpression of induced myeloid leukemia cell differentiation protein (Mcl-1) was observed in drug resistant cell lines.60 Naumann et al. showed that the melanoma cell lines expressing p53 wildtype were more resistant to temozolomide (TMZ) and fotemustine than p53 mutant melanoma lines, which suggest that the role of p53 in the regulation of apoptosis upon TMZ treatment could be different in melanoma cells than in other cancer cells.61 Melanoma treated with DNA cross-linking drugs acquire resistance by p53-dependent up-regulation of DNA repair genes, xeroderma pigmentosum complementation group C (XPC) and damaged DNA-binding protein 2 (DDB2).62
The BRAF-mutated melanoma develops resistance to RAF inhibitors through several genetic changes that leads to the reactivation of the MAPK.63,64 The widely observed mechanisms are mutation of neuroblastoma-RAS (NRAS mutations-NRASQ61; NRAST58; NRASG13R),65 loss of neurofibromatosis type-1(NF1),66-68 loss of phosphatase and tensin homolog (PTEN),69,70 up-regulation of cancer Osaka thyroid kinase (COT),71 mutation of MEK,63,69 negative feedback inactivation of extracellular-related kinase (ERK) and alteration in phosphoinositide 3-kinase-AKT (PI3K-AKT) pathway.64,72
Approaches to overcoming drug resistance
The combination of targeted therapies and immunotherapy has the potential for synergism via variety of mechanisms.73 By blocking an oncogenic mutation, targeted therapies may trigger tumor cell senescence and allow tumor clearance by T-cells.74 The concurrent release of antigenic debris from tumor cell death may also contribute to the success of immunotherapy by maximizing dendritic cell activation. Lastly, the induction of tumor regression by targeted therapies and the potential decrease in tumor associated immunosuppression, may enable a more favorable environment for immunotherapy to have an effect.73 Promising agents that target programmed death (PD1) have also emerged75 and one such ligands is PD-L17.76 PD-1 protein, a T-cell coinhibitory receptor, and the ligand PD-L1 play a key role in the ability of tumor cells to evade the host immune system. Blockade of PD-1 and PD-L1 has been incontestable to mediate antitumor activity in preclinical models and early clinical evidence has emerged supporting this. In the phase I studies, anti-PD-1 (BMS-936558, nivolumab) and anti-PD-Ll (BMS-936559) antibodies demonstrated response rates of 28 and 17%, respectively in metastatic melanoma patients. A phase Ib study of the anti-PD-Ll antibody, MPDL3280A, with vemurafenib is already underway in treatment-naive patients with BRAF V600-mutant metastatic melanoma (ClinicalTrials.gov; 2012. http://www.clinicaltrials.gov (Clinical-Trials.gov Identifier NCT01656642, NCT01611675, NCT01585 415, NCT01683188, NCT01603212, NCT01659151)). Other studies using drug combination that are designed to exploit the potential benefits of selective BRAF inhibition with immune modulation, include the combinations of vemurafenib with leflunomide, white blood cell therapy, high-dose interleukin-2 (IL-2), interferon and IL-2, or lymphodepletion plus adoptive cell transfer (ClinicalTrials.gov; 2012. http://www.clinicaltrials.gov (Clinical-Trials.gov Identifier NCT01656642, NCT01611675, NCT01585 415, NCT01683188, NCT01603212, NCT01659151)). Recently deregulation of Janus kinase 1 (JAK1) was identified in melanoma resistant to BRAF inhibitors, and to overcome such resistance the use of combination therapy was suggested, which may improve the durability of the response to BRAF inhibitors.77
Conclusions
Over the past decade a variety of chemotherapeutic agents have evolved to rechallenge melanoma. However, despite progress these therapies merely gave encouraging results. Several mechanisms of drug resistance have been identified using in vivo and in vitro studies (Table 1).13,17,30-32,34,35,50,51-54,57-60,62,63-72,78-90 In light of the evidence gained till date, it appears that the lethality of malignant melanoma is mainly due to the ability of these tumor cells to resist chemotherapy. Further a deeper understanding of the chemical nature of the resistance could improve the prospects of melanoma therapy. Such studies might lead to a novel approach for overcoming chemoresistance in melanoma by modulation of apoptotic pathways. Further in vitro, in vivo and clinical controlled studies are required to establish this hypothesis.
Table 1.
Mechanisms of resistance to anticancer drugs in melanoma.
| Resistance mechanism | Examples | Reference |
|---|---|---|
| Overexpression of drug efflux proteins | Over-expression of Heat Shock Transcription Factor 1 (HSF1) Up-regulation of ABCB1 and ABCB5 |
78 13,17 |
| Alteration of enzyme activation | Increase in Glutathione (GSH) levels | 30-32,34,35 |
| Deregulation of apoptosis | Up-regulation of miR-125a expression P53-dependent up-regulation of XPC and DDB Over-expression of the MCL1 anti-apoptotic BCL-2 family member Down-regulation of Apaf-1 Down-regulation, loss and mutation of CD95/Fas-receptor |
54 62 57,59,60 58 50,51 |
| Ras mutation | NRASQ61K/R mutations (NRASQ61, NRAST58, NRASG13R) Loss of NF1 COT up-regulation MEK mutation (MEK1C121S, MEK1Q56P, MEK1K57E, MEK1E203K, MEK1V60E, MEK1G128V and MEK2F57C, MEK2C125S, MEK2V35M, MEK2L46F, MEK2N126D) CRAF up-regulation, BRAF amplifications, BRAF truncation ERK-negative feedback inactivation Loss of PTEN PI3K-AKT pathway alteration (AKT1/3 Q79K, AKT1/3 E17K, mutation in PIK3R2, PHLPP1, PIK3CA and PIK3CG) MITF amplification and feedback suppression |
65 66-68 71 63,64,69 79 80 69,70,81 64,72 82-84 |
| Epithelial to mesenchymal transition | Up-regulation of IGF-IR signaling | 85 |
| Deregulation of microRNAs expression | Overexpression miR-34a, miR-100, miR-125b miR-195, miR-638, miRNA-1246 | 86-90 |
Acknowledgments
We thank the Board of Research in Nuclear Sciences (BRNS), Department of Atomic Energy, Government of India for the financial support (Sanction no: 2013/34/8; sanctioned to DU) and Yenepoya University for Junior Research Fellowship to BSK.
References
- 1.Olszanski AJ. Current and future roles of targeted therapy and immunotherapy in advanced melanoma. J Manag Care Spec Pharm 2014;20:346-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7-30. [DOI] [PubMed] [Google Scholar]
- 3.Chang DT, Amdur RJ, Morris CG, Mendenhall WM. Adjuvant radiotherapy for cutaneous melanoma: comparing hypofractionation to conventional fractionation. Int J Radiat Oncol Biol Phys 2006;66:1051-4. [DOI] [PubMed] [Google Scholar]
- 4.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med 2006;355:51-65. [DOI] [PubMed] [Google Scholar]
- 5.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res 2006;66:6699-707. [DOI] [PubMed] [Google Scholar]
- 6.Lee CC, Faries MB, Wanek LA, Morton DL. Improved survival after lymphadenectomy for nodal metastasis from an unknown primary melanoma. J Clin Oncol 2008;26:535-41. [DOI] [PubMed] [Google Scholar]
- 7.Wu S, Singh RK. Resistance to chemotherapy and molecularly targeted therapies: rationale for combination therapy in malignant melanoma. Curr Mol Med 2011;11:553-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Society AC. Cancer facts & figures 2015. Atlanta: American Cancer Society; 2017. [Google Scholar]
- 9.Lucas R MT, Smith W, Armstrong B. Solar ultraviolet radiation: Global burden of disease from solar ultraviolet radiation. Environmental Burden of Disease Series. 2006; No. 13:World Health Organization Report. [Google Scholar]
- 10.Grottke C, Mantwill K, Dietel M, et al. Identification of differentially expressed genes in human melanoma cells with acquired resistance to various antineoplastic drugs. Int J Cancer 2000;88:535-46. [DOI] [PubMed] [Google Scholar]
- 11.Szakacs G, Paterson JK, Ludwig JA, et al. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006;5:219-34. [DOI] [PubMed] [Google Scholar]
- 12.Chen KG, Valencia JC, Gillet JP, et al. Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigment Cell Melanoma Res 2009;22:740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott AM, Al-Hajj MA. ABCB8 mediates doxorubicin resistance in melanoma cells by protecting the mitochondrial genome. Mol Cancer Res 2009;7:79-87. [DOI] [PubMed] [Google Scholar]
- 14.Fukunaga-Kalabis M, Herlyn M. Beyond ABC: another mechanism of drug resistance in melanoma side population. J Invest Dermatol 2012;132:2317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Y, Ellis LZ, Dallaglio K, et al. Side population cells from human melanoma tumors reveal diverse mechanisms for chemoresistance. J Invest Dermatol 2012;132:2440-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rockmann H, Schadendorf D. Drug resistance in human melanoma: mechanisms and therapeutic opportunities. Onkologie 2003;26:581-7. [DOI] [PubMed] [Google Scholar]
- 17.Frank NY, Margaryan A, Huang Y, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005;65:4320-33. [DOI] [PubMed] [Google Scholar]
- 18.Liedert B, Materna V, Schadendorf D, et al. Overexpression of cMOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J Invest Dermatol 2003;121:172-6. [DOI] [PubMed] [Google Scholar]
- 19.Schadendorf D, Herfordt R, Czarnetzki BM. P-glycoprotein expression in primary and metastatic malignant melanoma. Br J Dermatol 1995;132:551-5. [DOI] [PubMed] [Google Scholar]
- 20.Heimerl S, Bosserhoff AK, Langmann T, et al. Mapping ATP-binding cassette transporter gene expression profiles in melanocytes and melanoma cells. Melanoma Res 2007;17:265-73. [DOI] [PubMed] [Google Scholar]
- 21.Berger W, Hauptmann E, Elbling L, et al. Possible role of the multidrug resistance-associated protein (MRP) in chemoresistance of human melanoma cells. Int J Cancer 1997;71:108-15. [DOI] [PubMed] [Google Scholar]
- 22.Ichihashi N, Kitajima Y. Chemotherapy induces or increases expression of multidrug resistance-associated protein in malignant melanoma cells. Br J Dermatol 2001;144:745-50. [DOI] [PubMed] [Google Scholar]
- 23.O’Connor R, O’Leary M, Ballot J, et al. A phase I clinical and pharmacokinetic study of the multi-drug resistance protein-1 (MRP-1) inhibitor sulindac, in combination with epirubicin in patients with advanced cancer. Cancer Chemother Pharmacol 2007;59:79-87. [DOI] [PubMed] [Google Scholar]
- 24.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003;22:7369-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang K, Chuluyan HE, Hardie D, et al. Novel monoclonal antibodies to putative selectin carbohydrate ligands that inhibit selectin binding to myeloid cells. Hybridoma 1998;17:445-56. [DOI] [PubMed] [Google Scholar]
- 26.Schadendorf D, Jurgovsky K, Kohlmus CM, Czarnetzki BM. Glutathione and related enzymes in tumor progression and metastases of human melanoma. J Invest Dermatol 1995;105:109-12. [DOI] [PubMed] [Google Scholar]
- 27.La Porta CA. Mechanism of drug sensitivity and resistance in melanoma. Curr Cancer Drug Targets 2009;9:391-7. [DOI] [PubMed] [Google Scholar]
- 28.Grossman D, Altieri DC. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev 2001;20:3-11. [DOI] [PubMed] [Google Scholar]
- 29.Traverso N, Ricciarelli R, Nitti M, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev 2013;2013:972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannervik B, Castro VM, Danielson UH, et al. Expression of class Pi glutathione transferase in human malignant melanoma cells. Carcinogenesis 1987;8:1929-32. [DOI] [PubMed] [Google Scholar]
- 31.Moral A, Palou J, Lafuente A, et al. Immunohistochemical study of alpha, mu and pi class glutathione S transferase expression in malignant melanoma. MMM Group. Multidisciplinary Malignant Melanoma Group. Br J Dermatol 1997;136:345-50. [PubMed] [Google Scholar]
- 32.Biroccio A, Benassi B, Filomeni G, et al. Glutathione influences c-Myc-induced apoptosis in M14 human melanoma cells. J Biol Chem 2002;277:43763-70. [DOI] [PubMed] [Google Scholar]
- 33.Schadendorf D, Makki A, Stahr C, et al. Membrane transport proteins associated with drug resistance expressed in human melanoma. Am J Pathol 1995;147:1545-52. [PMC free article] [PubMed] [Google Scholar]
- 34.Drozd E, Gruber B, Marczewska J, et al. Intracellular glutathione level and efflux in human melanoma and cervical cancer cells differing in doxorubicin resistance. Postepy Hig Med Dosw (Online) 2016;70:319-28. [DOI] [PubMed] [Google Scholar]
- 35.Pendyala L, Perez R, Weinstein A, et al. Effect of glutathione depletion on the cytotoxicity of cisplatin and iproplatin in a human melanoma cell line. Cancer Chemother Pharmacol 1997;40:38-44. [DOI] [PubMed] [Google Scholar]
- 36.Rudolf K, Cervinka M, Rudolf E. Dual inhibition of topoisomerases enhances apoptosis in melanoma cells. Neoplasma 2010;57:316-24. [DOI] [PubMed] [Google Scholar]
- 37.Daud AI, Dawson J, DeConti RC, et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and phase I/II clinical trial. Clin Cancer Res 2009;15:2479-87. [DOI] [PubMed] [Google Scholar]
- 38.Campain JA, Slovak ML, Schoenlein PV, et al. Acquisition of multiple copies of a mutant topoisomerase IIalpha allele by chromosome 17 aneuploidy is associated with etoposide resistance in human melanoma cell lines. Somat Cell Mol Genet 1995;21:451-71. [DOI] [PubMed] [Google Scholar]
- 39.Lage H, Helmbach H, Dietel M, Schadendorf D. Modulation of DNA topoisomerase II activity and expression in melanoma cells with acquired drug resistance. Br J Cancer 2000;82:488-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satherley K dSL, Neale MH, Alexander RA, et al. Relationship between expression of topoisomerase II isoforms and chemosensitivity in choroidal melanoma. J Pathol 2000;192:174-81. [DOI] [PubMed] [Google Scholar]
- 41.Sinha BK, Kumar A, Bhattacharjee S, et al. Effect of nitric oxide on the anticancer activity of the topoisomerase-active drugs etoposide and adriamycin in human melanoma cells. J Pharmacol Exp Ther 2013;347:607-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012;12:104-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Runger TM, Emmert S, Schadendorf D, et al. Alterations of DNA repair in melanoma cell lines resistant to cisplatin, fotemustine, or etoposide. J Invest Dermatol 2000;114:34-9. [DOI] [PubMed] [Google Scholar]
- 44.Christmann M, Pick M, Lage H, et al. Acquired resistance of melanoma cells to the antineoplastic agent fotemustine is caused by reactivation of the DNA repair gene MGMT. Int J Cancer 2001;92:123-9. [PubMed] [Google Scholar]
- 45.Middleton MR, Lunn JM, Morris C, et al. O6-methylguanine-DNA methyltransferase in pretreatment tumour biopsies as a predictor of response to temozolomide in melanoma. Br J Cancer 1998;78:1199-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 2000;18:158-66. [DOI] [PubMed] [Google Scholar]
- 47.Ma S, Egyhazi S, Martenhed G, et al. Analysis of O(6)-methylguanine-DNA methyltransferase in melanoma tumours in patients treated with dacarbazine-based chemotherapy. Melanoma Res 2002;12:335-42. [DOI] [PubMed] [Google Scholar]
- 48.Hussein MR, Haemel AK, Wood GS. Apoptosis and melanoma: molecular mechanisms. J Pathol 2003;199:275-88. [DOI] [PubMed] [Google Scholar]
- 49.Hussein MR, Haemel AK, Wood GS. p53-related pathways and the molecular pathogenesis of melanoma. Eur J Cancer Prev 2003;12:93-100. [DOI] [PubMed] [Google Scholar]
- 50.Eberle J, Fecker LF, Hossini AM, et al. CD95/Fas signaling in human melanoma cells: conditional expression of CD95L/FasL overcomes the intrinsic apoptosis resistance of malignant melanoma and inhibits growth and progression of human melanoma xenotransplants. Oncogene 2003;22:9131-41. [DOI] [PubMed] [Google Scholar]
- 51.Aragane Y, Maeda A, Cui CY, et al. Inhibition of growth of melanoma cells by CD95 (Fas/APO-1) gene transfer in vivo. J Invest Dermatol 2000;115:1008-14. [DOI] [PubMed] [Google Scholar]
- 52.Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene 2003;22:3138-51. [DOI] [PubMed] [Google Scholar]
- 53.Helmbach H, Kern MA, Rossmann E, et al. Drug resistance towards etoposide and cisplatin in human melanoma cells is associated with drug-dependent apoptosis deficiency. J Invest Dermatol 2002;118:923-32. [DOI] [PubMed] [Google Scholar]
- 54.Koetz-Ploch L, Hanniford D, Dolgalev I, et al. MicroRNA-125a promotes resistance to BRAF inhibitors through suppression of the intrinsic apoptotic pathway. Pigment Cell Melanoma Res 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polsky D, Bastian BC, Hazan C, et al. HDM2 protein overexpression, but not gene amplification, is related to tumorigenesis of cutaneous melanoma. Cancer Res 2001;61:7642-6. [PubMed] [Google Scholar]
- 56.Lee B, Sandhu S, McArthur G. Cell cycle control as a promising target in melanoma. Curr Opin Oncol 2015;27:141-50. [DOI] [PubMed] [Google Scholar]
- 57.Selzer E, Schlagbauer-Wadl H, Okamoto I, et al. Expression of Bcl-2 family members in human melanocytes, in melanoma metastases and in melanoma cell lines. Melanoma Res 1998;8:197-203. [DOI] [PubMed] [Google Scholar]
- 58.Soengas MS, Capodieci P, Polsky D, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001;409:207-11. [DOI] [PubMed] [Google Scholar]
- 59.Borner C, Schlagbauer Wadl H, Fellay I, et al. Mutated N-ras upregulates Bcl-2 in human melanoma in vitro and in SCID mice. Melanoma Res 1999;9:347-50. [DOI] [PubMed] [Google Scholar]
- 60.Fofaria NM, Frederick DT, Sullivan RJ, et al. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget 2015;6:40535-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naumann SC, Roos WP, Jost E, et al. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br J Cancer 2009;100:322-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barckhausen C, Roos WP, Naumann SC, Kaina B. Malignant melanoma cells acquire resistance to DNA interstrand crosslinking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene 2014;33:1964-74. [DOI] [PubMed] [Google Scholar]
- 63.Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011;480:387-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krauthammer M, Kong Y, Bacchiocchi A, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet 2015;47:996-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whittaker SR, Theurillat JP, Van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 2013;3:350-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol 2013;31:1767-74. [DOI] [PubMed] [Google Scholar]
- 70.Paraiso KH, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011;71:2750-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pappalardo F, Russo G, Candido S, et al. Computational Modeling of PI3K/AKT and MAPK signaling pathways in melanoma cancer. PLoS One 2016;11:e0152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 2012;12:237-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bachireddy P, Rakhra K, Felsher DW. Immunology in the clinic review series; focus on cancer: multiple roles for the immune system in oncogene addiction. Clin Exp Immunol 2012;167:188-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim H, Frederick DT, Levesque MP, et al. Downregulation of the Ubiquitin Ligase RNF125 Underlies Resistance of Melanoma Cells to BRAF Inhibitors via JAK1 Deregulation. Cell Rep 2015;11:1458-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vydra N, Toma A, Glowala-Kosinska M, et al. Overexpression of Heat Shock Transcription Factor 1 enhances the resistance of melanoma cells to doxorubicin and paclitaxel. BMC Cancer 2013;13:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 2008;68:4853-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lito P, Saborowski A, Yue J, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 2014;25:697-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res 2013;19:4868-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pathria G, Garg B, Borgdorff V, et al. Overcoming MITF-conferred drug resistance through dual AURKA/MAPK targeting in human melanoma cells. Cell Death Dis 2016;7:e2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muller J, Krijgsman O, Tsoi J, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014;5:5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res 2015;28:390-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H, Batth IS, Qu X, et al. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer 2017;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim JH, Ahn JH, Lee M. Upregulation of microRNA-1246 is Associated with BRAF Inhibitor Resistance in Melanoma Cells with Mutant BRAF. Cancer Res Treat 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bhattacharya A, Schmitz U, Raatz Y, et al. miR-638 promotes melanoma metastasis and protects melanoma cells from apoptosis and autophagy. Oncotarget 2015;6:2966-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bhattacharya A, Schmitz U, Wolkenhauer O, et al. Regulation of cell cycle checkpoint kinase WEE1 by miR-195 in malignant melanoma. Oncogene 2013;32:3175-83. [DOI] [PubMed] [Google Scholar]
- 89.Zhou J, Xu D, Xie H, et al. miR-33a functions as a tumor suppressor in melanoma by targeting HIF-1alpha. Cancer Biol Ther 2015;16:846-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vergani E, Di Guardo L, Dugo M, et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2016;7:4428-41. [DOI] [PMC free article] [PubMed] [Google Scholar]