

Abstract

A major purpose of exploratory metabolic profiling is for the identification of molecular species that are statistically associated with specific biological or medical outcomes; unfortunately, the structure elucidation process of unknowns is often a major bottleneck in this process. We present here new holistic strategies that combine different statistical spectroscopic and analytical techniques to improve and simplify the process of metabolite identification. We exemplify these strategies using study data collected as part of a dietary intervention to improve health and which elicits a relatively subtle suite of changes from complex molecular profiles. We identify three new dietary biomarkers related to the consumption of peas (N-methyl nicotinic acid), apples (rhamnitol), and onions (N-acetyl-S-(1Z)-propenyl-cysteine-sulfoxide) that can be used to enhance dietary assessment and assess adherence to diet. As part of the strategy, we introduce a new probabilistic statistical spectroscopy tool, RED-STORM (Resolution EnhanceD SubseT Optimization by Reference Matching), that uses 2D J-resolved 1H NMR spectra for enhanced information recovery using the Bayesian paradigm to extract a subset of spectra with similar spectral signatures to a reference. RED-STORM provided new information for subsequent experiments (e.g., 2D-NMR spectroscopy, solid-phase extraction, liquid chromatography prefaced mass spectrometry) used to ultimately identify an unknown compound. In summary, we illustrate the benefit of acquiring J-resolved experiments alongside conventional 1D 1H NMR as part of routine metabolic profiling in large data sets and show that application of complementary statistical and analytical techniques for the identification of unknown metabolites can be used to save valuable time and resources.
Dietary interventions (DIs) are a cornerstone in the management of reducing the risk of noncommunicable diseases1−3 and promoting healthy aging.4 However, understanding the response to dietary change is compromised by poor compliance to dietary recommendations and the inherently inaccurate self-reporting dietary recording tools available, with prevalence of misreporting estimated at 30–88%,5,6 lowering the value of such studies and data
It has been demonstrated that dietary biomarkers can reflect consumption of specific foods and enhance dietary intake assessment at individual and population levels.7−17 Dietary biomarkers are based on the concept that food intake is highly correlated with excretion levels of food-related compounds over a given period of time. These “biomarkers” can be compounds that are excreted unchanged10,17 or that have undergone metabolic conversion, for example, by gut bacteria.8,11,13,14 Metabolic profiling of biofluids using spectroscopic technologies18 can detect thousands of compounds simultaneously, generating a profile that can be related to specific states of health or disease. Some of the compounds in these spectral profiles are potential biomarkers of dietary intake. However, finding associations between self-reported dietary intakes and excreted metabolites19,20 in order to discover potential food biomarkers is plagued by inaccuracies of self-reporting. Thus, confirmation of a candidate biomarker is best achieved by using a controlled food challenge with subsequent validation in a larger population or study cohort.17
The complementarity of the main workhorses of metabolic profiling, one-dimensional proton nuclear magnetic resonance (1H NMR) spectroscopy and hyphenated chromatograpic-mass spectrometry (MS) techniques, has been extensively demonstrated in the past decade.21−23 However, chemical characterization of molecular species associated with an outcome still is a limiting factor for exploratory metabolic profiling. NMR spectroscopy provides an atom-centered spectroscopic tool for structure elucidation that can be enhanced by statistical spectroscopic methods24,25 or by physical hyphenation with chromatographic methods such as solid-phase extraction (SPE),26 liquid chromatography (LC)21 or LC-NMR-MS21,27 to achieve a better chemical characterization of endogenous and exogenous metabolites.
Metabolite identification in 1H NMR spectroscopy is aided by the intrinsic correlation of peaks from the same metabolite. Statistical TOtal Correlation SpectroscopY (STOCSY)28 makes use of this property by calculating the correlation between one spectral variable (driver) and all other variables to uncover structural associations. In cases where there are sufficient spectra, identification of 1H NMR peaks using statistical methods is an efficient strategy that utilizes existing spectral data without requiring additional spectroscopic experiments a priori, which has obvious advantages in usage of volume-limited samples and is cost-efficient.23 Since the STOCSY method was published, many derivations have aimed to improve specific properties such as differentiation between structural and pathway correlations by clustering, subset selection, or stoichiometric relationships.24
Statistical correlation can be undermined by overlapped signals unrelated to the metabolite of interest in a 1D-NMR spectrum, and 2D-NMR experiments are still required for unambiguous structure elucidation.29 In addition, the structural information obtained using statistical algorithms is dependent on criteria such as correlation thresholds28,30 or correlation-distance cut-offs.31 SubseT Optimization by Reference Matching (STORM)32 is a derivation of STOCSY that aims to separate out confounding spectra that do not match a supplied reference spectrum of a potential biomarker signal, thereby showing clearer spectral correlations between variables for both low and high intensity signals. The reference spectrum is a single spectrum that contains the signal of interest. The peak segment is correlated with the same region of all samples, and a high correlation indicates the samples contain the same signal and are likely “informative”, whereas samples with a low correlation do not have this signal and are uninformative. Subsets of spectra and variable correlations are found by carefully correcting for multiple testing in both phases and using statistical shrinkage. Here, we describe a holistic strategy for the identification of unknown metabolites that combines the strengths of statistical spectroscopy, NMR, multiple separation techniques, and MS, and apply the strategy to identify three novel dietary biomarkers. In addition, we demonstrate an extension of STORM to 2D-NMR spectra to uncover the identity of unknown metabolites.
Experimental Section
Food Challenges (FCs)
For the discovery of urinary biomarkers of peas, apples, and onions, three FCs were designed. A total of nine healthy participants (4 women, 5 men; nonsmokers; age 22–32 years; BMI 21.2–25.3 kg/m2) were recruited and assigned to one of the three FCs. Participants were provided with the assigned foods as part of a standardized dinner (including 125 g of chicken breast as a protein source). Incremental amounts of the designated food were consumed over three consecutive days: 60/120/180 g for (boiled) peas, 40/80/160 g for (raw) apples, and 20/40/60 g for (fried) onions. For 24 h preceding the FC, and throughout the FC, participants were asked to consume their habitual diet and avoid consumption of coffee/tea/cocoa and any additional amounts of assigned foods. Cumulative urine samples were collected into sterilized single-use urine containers (International Scientific Supplies Ltd., Bradford, U.K.) from dinner up to and including the first morning void. Urine samples were stored at −80 °C until analysis.
Controlled Clinical Trial (CCT)
There were 19 healthy participants (9 women, 10 men; nonsmokers; age 25–60 years; BMI 21.1–33.3 kg/m2) who attended the NIHR/Wellcome Trust Imperial Clinical Research Facility for four 3-day inpatient periods, separated by a period of >4 days, with food and drink intake tightly controlled (alcohol/coffee/tea/cocoa were not provided). In random order, participants followed all four DIs representing 100% (diet 1), 75% (diet 2), 50% (diet 3), and 25% (diet 4) of WHO healthy eating guidelines1 with respect to carbohydrates, fats, fiber, fruits, salt, sugar, and vegetables. Full details of the clinical trial design have been described previously.33 Foods consumed relevant to the present study are tabulated in Supporting Information.
Each participant collected cumulative urine samples (CS) on each day of each DI from after breakfast to before lunch (CS1), after lunch to before dinner (CS2), and after dinner to before breakfast the following day (CS3). The 24 h urine samples were obtained by pooling the cumulative samples. Aliquots of urine were transferred into Eppendorf tubes and stored at −80 °C until analysis. All participants provided informed, written consent prior to the CCT (Registration No. ISRCTN-43087333), which was approved by the London Brent Research Ethics Committee (13/LO/0078). All studies were carried out in accordance with the Declaration of Helsinki.
1H NMR Analysis
Aliquots of 600 μL of urine samples were centrifuged at 16 000 × g at 4 °C for 5 min. All available samples (nFC = 27, nCCT = 906, for missing CCT data see Supporting Information) were prepared for 1H NMR spectroscopy following the protocol described in ref (34) mixing 540 μL of supernatant with 60 μL of pH 7.4 phosphate buffer containing trimethylsilyl-[2,2,3,3,-2H4]-propionate as an internal reference standard (“NMR buffer”). Water-suppressed 1H NMR spectroscopy was performed at 300 K on a Bruker 600 MHz spectrometer (Bruker Biospin, Karlsruhe, Germany) using a standard 1D pulse sequence (RD–gz,1–90°–t–90°–tm–gz,2–90°–ACQ) with saturation of the water resonance.7 The following abbreviations apply: RD is the relaxation delay, t is a short delay (4 μs), 90° represents a radio frequency (RF) pulse that tips the magnetization by 90°, tm is the mixing time (10 ms), gz,1 and gz,2 are magnetic field z-gradients both applied for 1 ms, and ACQ is the data acquisition period of 2.73 s. 1H NMR spectra were acquired using 4 dummy scans and 32 scans, and 64K time domain points, with a spectral window of 20 ppm. Prior to Fourier transformation, free induction decays were multiplied by an exponential function corresponding to a line broadening of 0.3 Hz. 1H NMR spectra were normalized to the total urine volume to correct for differences in dilution.
1H–1H 2D J-resolved experiments7 were acquired using a pulse sequence to detect the J-couplings in the second dimension, with suppression of the water resonance (RD–90°–t1– 180°–t1–ACQ), where t1 is an incremented time period, RD is 2 s, 180° represents a 180° RF pulse, and ACQ is 0.41 s. J-resolved spectra were acquired using 16 dummy scans and 2 scans, 8K points with spectral window of 16.7 ppm for f2 and 40 increments with spectral window of 78 Hz for f1. Continuous wave irradiation was applied at the water resonance frequency using a 25 Hz RF during the RD. A sine-bell apodization function was applied to f2 and a squared sine-bell to f1 of the J-resolved data, followed by Fourier transformation, tilting by 45°, and symmetrization along f1 before data analysis.
A suite of 2D-NMR experiments including 1H–1H TOtal Correlation SpectroscopY (TOCSY), 1H–1H COrrelation SpectroscopY (COSY), 1H–13C Heteronuclear Single Quantum Coherence (HSQC), and 1H–13C Heteronuclear Multiple-Bond Correlation (HMBC) spectroscopy were used for identification purposes.25,29
SPE-NMR
Apple extracts were homogenized using a Kenwood KMix Blender for 5 min. The puree obtained was filtered using a stainless steel filter and centrifuged for 10 min at 16 000 × g. A 2 mL portion of each sample (urine/apple) was lyophilized overnight. Freeze-dried (FD) urine/apple samples were dissolved in 1 mL of 50 mM sodium phosphate pH 8.5 and briefly sonicated prior to being loaded onto a 100 mg/mL Bond Elut phenylboronic acid (PBA) SPE-cartridge (Agilent Technologies, Stockport, U.K.). The SPE-cartridge was conditioned with 1 mL of 70:30 v/v H2O/ACN, and 0.1 M HCl, followed by 1 mL of 50 mM sodium phosphate pH 10. After conditioning, the sample was loaded onto the SPE-cartridge. The loaded sample was washed with 2 mL of 50:50 v/v ACN/sodium phosphate (10 mM) pH 8.5. The sample was eluted using 1 mL of acidified solutions (0.1 M HCl) of 100% H2O, 90:10 v/v H2O/ACN, and 70:30 v/v H2O/ACN and subsequently lyophilized until dry. The dried eluent fractions were reconstituted in 540 μL of H2O and 60 μL of NMR buffer, vortexed, and centrifuged prior to NMR analysis.
LC-NMR-MS27
A 5 mL portion of urine collected overnight after consumption of onion was lyophilized overnight, reconstituted in 500 μL of the original urine sample, and vortexed, sonicated and centrifuged (20 min at 16 000 × g). The supernatant was repeatedly injected (7 × 2 μL) onto a reversed-phase HPLC column (Waters Atlantis-T3, 3 μm, 4.6 mm × 150 mm at 30 °C) in a Waters Acquity UPLC comprising a binary solvent manager and photodiode array detector with a Waters CTC autosampler with 100 μL sampling needle, and eluted at 0.8 mL/min using the following gradient: 0.0–60.0 min (99.9:0.1% H2O/formic acid), 60.01–65.0 min (99.9:0.1% methanol/formic acid), 65.01–127.5 min (99.9:0.1% H2O/formic acid). The chromatographic separation of the sample was fractionated using a Waters Fraction Collector III. A total of 120 fractions were collected, one every 29 s (starting at t = 5 min, finishing at t = 63 min), and dried under a stream of nitrogen. Each fraction was redissolved in 540 μL of H2O and 60 μL of NMR buffer and analyzed by 1H NMR. A volume of 50 μL of the fraction containing the unknown metabolite was analyzed by reversed-phase LC-MS, Waters Acquity Ultra Performance LC system coupled to Xevo G2 Q-TOF mass spectrometer (Waters, Milford, MA), following an established metabolic profiling method.35 The optimized capillary voltage, cone voltage, and collision energy were 3 kV/20 V/4 V for ESI+ and 1.5 kV/30 V/6 V for ESI–, using a source temperature of 120 °C and desolvation temperature of 600 °C. Desolvation was 1000L/h for both, and cone gas flows were 50 L/h (ESI+) and 150 L/h (ESI−). Leucine enkephalin was used as the reference lock mass at 556.2771 ([M + H]+) and 554.2615 ([M – H]−).
RED-STORM Algorithm
Here, STORM was extended in a probabilistic framework and modified for applicability to 2D data to provide a clearer signature of structural correlations in the data, and the extended algorithm was named Resolution EnhanceD SubseT Optimization by Reference Matching (RED-STORM).
High correlations in the data are of interest between samples and a reference spectrum of a signal of interest (for subset optimization) and between a driver and all other variables (for assessing variable importance). However, high correlations are not normally distributed because of the upper bound on the correlation ([−1, 1]); this results in their distributions being negatively skewed. Therefore, the correlation (ρ) is transformed to a Fisher z-score, which results in approximately normally distributed data that can be analyzed using parametric methods:
| 1 |
Next, the distribution of all z-scores is modeled using a Gaussian mixture model (GMM). A GMM is a weighted sum of k Gaussian clusters and is defined as
| 2 |
Here, z has n data points. μ are the k means and σ2 the k variances. π is the mixture weights for each of k Gaussians (∑ j = 1kπj = 1), and φ0,1 is a normalized Gaussian distribution with specified μ and σ. In order to obtain clusters of variables from the Fisher z-transformed correlations, a parameter-free GMM is used.36 The model automatically learns the optimal number of clusters from the data. For completeness, a description of the method in brief follows; for mathematical proofs, see ref (36).
Each cluster mean (μj) and precision (σj–2) from eq 2 are given Gaussian (p(μj|λ, r) ∼ φ0,1(λ, r–1)) and Gamma (p(σj–2|γ, β) ∼ Γ(γ, β–1)) priors, respectively. Here, λ and r are hyperparameters for the means of all clusters. Similarly, γ (shape) and β (scale) are hyperparameters for the cluster precisions. The hyperparameters themselves are initialized from the observation mean (μz) and variance (σz2) as vague priors (p(λ) ∼ φ0,1(μz, σz2); p(r) ∼ Γ(1, σz2); p(γ–1) ∼ Γ(1, 1); p(β) ∼ Γ(1, σz–2)). The posterior distributions of the cluster means are obtained from
| 3 |
where z̃j is the sum of observations from cluster j, mj the number of observations with highest probability for cluster j, ci the cluster with highest probability for observation i, and δci,j a Kronecker delta product. Similarly, the posterior distributions of the cluster precisions are obtained from
 |
4 |
The posteriors for λ ( ), r (
), r (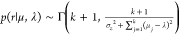 ), and β (
), and β (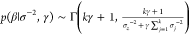 ) are obtained similarly
to the priors,
whereas posterior for γ takes a different form and makes use
of the fact that p(ln(γ)|σ–2, β) is log-concave:36
) are obtained similarly
to the priors,
whereas posterior for γ takes a different form and makes use
of the fact that p(ln(γ)|σ–2, β) is log-concave:36
 |
5 |
Each ci is directly related to π and can be written
in terms
of k and mj with concentration hyperparameter α (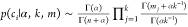 ). The prior for α is sampled
from
an inverse Gamma distribution (p(α–1) ∼ Γ(1, 1)) and the posterior from
). The prior for α is sampled
from
an inverse Gamma distribution (p(α–1) ∼ Γ(1, 1)) and the posterior from 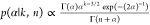 . The “doubling and shrinkage”
Markov-chain Monte Carlo (MCMC) slice sampler37 is used to generate independent samples from the nonstandard distributions
for γ and α.
. The “doubling and shrinkage”
Markov-chain Monte Carlo (MCMC) slice sampler37 is used to generate independent samples from the nonstandard distributions
for γ and α.
The posterior of observation i given class j is
 |
6 |
The same goes for a potential new cluster (k + 1), where the mean and precision are sampled from Gaussian and Gamma distributions using the posteriors from global hyperparameters λ, r, γ, and β (p(μk+1|λ, r) ∼ φ0,1(λ, r–1),p(σk+1–2|γ, β) ∼ Γ(γ, β–1)) and the posterior is as above (class j is now k + 1).
The cumulative sum
of the cluster probabilities (Cj) for ci are calculated
(Cj = ∑i = 1jp(ci = j|j ≤ k + 1)), and a new cluster is
added if and only if none of the previous k clusters
pass an arbitrary threshold of the cumulative sum of the new cluster
(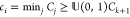 ); otherwise, ci becomes the first cluster
to pass the threshold.
At any stage when any mj = 0, cluster j is removed.
); otherwise, ci becomes the first cluster
to pass the threshold.
At any stage when any mj = 0, cluster j is removed.
This process is repeated for a set number of (burn-in) iterations (typically >100) to achieve some stability in finding a suitable k, depending on the data, before continuing with the remainder of (post burn-in) iterations of the Markov-chain. The final predictive distribution is a weighted average over the post burn-in iteration clusters. The order (for i of z) is randomized in each iteration to avoid bias.
For subset selection, the variables that make up the reference segment of interest are correlated with the same variables from all spectra (STORM); the correlations are z-transformed, and the distribution is fitted using the procedure described above. The final predictive distribution is converted to a cumulative distribution function (cdf), and for each sample the probability of it resembling the reference is calculated. The subset contains all spectra that satisfy p(zi|cdf) ≥ ts, where ts is a user-defined threshold for the samples. The reference spectrum is updated by using a weighted average of the spectra in the subset. Using only the spectra in the subset, the correlations of all driver variables (reference segment of interest) with all other spectral variables are calculated. To alleviate the computational load of the algorithm, for 2D J-resolved NMR, and other 2D-spectra, the algorithm is run on the variables that make up the peaks of the reference spectrum rather than all variables. The median ρ across all driver variables is calculated for each variable and z-transformed, and the same procedure as for subset selection is performed for the variables. MATLAB code can be obtained by contacting the authors.
Results and Discussion
Identification of a Urinary Biomarker for Pea (Pisum sativum) Consumption
On comparison of the urinary spectra obtained pre- and postpea intake, the urinary concentration of N-methylnicotinic acid (NMNA, trigonelline), although present in the baseline samples, showed a dose dependent increment after increasing the consumption of peas (Figure 1A–C) during the FC, suggesting it as a candidate biomarker. However, the presence of NMNA in the baseline samples indicated that peas were not the sole source of NMNA. The CCT data showed a similar pattern (Figure 1D–F) with low levels for diet 1 (no peas provided) and incrementally higher levels for diets 2–4 (peas provided). Interestingly, NMNA appears in highest concentrations in CS3 and 24 h samples from diet 2, whereas peas were provided only during dinner in increasing amounts (0/20/40/60 g for diets 1–4). However, chocolate was provided as an afternoon snack in diets 1–3, and baked beans were provided as part of dinner in diet 2 (see Supporting Information Table S1) which may be alternative sources of NMNA.
Figure 1.

FC and CCT spectra identify NMNA as a urinary biomarker of peas. (A–C) 1H NMR spectra of a volunteer after pea FC zoomed in on NMNA signals (A, δ 9.13(s); B, δ 8.84(t); C, δ 4.44(s)). (D–F) 1H NMR spectra of different samples of one CCT volunteer zoomed in on the same regions.
It has been long known that several plant materials (including coffee, tea, and cocoa) are rich in niacin (vitamin B3) and some of its major metabolic products,38 including NMNA. In addition, NMNA has been proposed as urinary biomarker of coffee consumption.11 Thus, although it is a poor biomarker of pea intake in the sense of specificity, NMNA could still be used to detect pea intake in urine after controlling for other sources.
With nonspecific dietary biomarkers, biomarker patterns, rather than a single biomarker, can be used to differentiate between different food sources.17 For instance, 2-furoylglycine, another marker of coffee consumption,16 can be used to cross-check for coffee consumption and as secondary marker to adjust the concentration of NMNA. Broader knowledge of dietary biomarkers will contribute to solving this specificity issue.
Identification of Urinary Biomarker of Apple (Malus domestica) Consumption
After consumption of increased amounts of apple, a spectral signal at δ 1.285 was found to be increased (Figure 2A). This same signal was found in incrementally higher levels in the urine of participants who consumed increasing amounts of apple during the CCT (Figure 2B); diets 2–4 contained 50/100/150 g of apple, respectively, as a midmorning snack. The absence of this signal in urine samples from participants on diet 1 is consistent with there being no apple intake in diet 1. STORM analysis (Figure 2C) revealed high correlations of the driver (δ 1.285) with a shoulder of the 3-hydroxyisovalerate peak at δ 1.275(s), suggesting the singlet may in fact be a doublet. This was confirmed using J-resolved spectroscopy (Figure 2D). To confirm the identity of the molecule giving rise to the doublet, we performed SPE using a PBA-cartridge on both the urine (Figure 2E) and apple puree (Figure 2F) samples to isolate the signal in one of the fractions for identification purposes, which was subsequently confirmed by NMR analysis. To assess the specificity of this doublet to apple consumption, we performed PBA-SPE on urine samples post-pear-consumption (using the same protocol as for apple) and did not find the metabolite peak in the urine or in any fraction (Figure 2G). 2D-NMR experiments were performed on fraction 1 (Supporting Information) suggesting rhamnitol as potential biomarker; a chemical spike-in experiment (Figure 2H) confirmed the identity. Rhamnitol is a component in different varieties of apples,39 and taken together, these results confirm rhamnitol as specific biomarker for apple consumption. The suitability of rhamnitol for quantification of apple intake will be investigated in a follow-up study.
Figure 2.

FC, CCT spectra, and analytical experiments identify rhamnitol as a biomarker of apple consumption. (A) 1H NMR spectrum of a volunteer after pea FC, spectral expansion of a putative singlet. (B) Same section of 1H NMR spectra of different samples from one volunteer in the CCT. (C) STORM analysis of the tentative singlet reveals potential overlap of signal with 3-hydroxyisovalerate (δ 1.275(s)). (D) Tilted J-resolved NMR experiment confirms that the putative signal is a doublet which overlaps with 3-hydroxyisovalerate. (E) 1H NMR spectrum of a freeze-dried urine sample of an apple consumer after PBA-SPE. (F) 1H NMR spectrum of apple puree after freeze-drying and PBA-SPE. (G) 1H NMR spectrum of freeze-dried urine sample of a pear consumer and PBA-SPE confirms rhamnitol is specific for apple intake. (H) Spike-in of rhamnitol confirms identity of metabolite. 2D-NMR experiments can be found in Supporting Information.
Proof-of-Concept of RED-STORM
The identification of rhamnitol as urinary biomarker of apple consumption has shown how overlap in 1D spectra cannot be resolved using STORM as the signals overlap with those of 3-hydroxyisovalerate, commonly found in urine samples, which reduces the power of the statistical correlation method. J-resolved NMR spectra are able to “untangle” overlap using the J-coupling as the second dimension. In large metabolic profiling studies, both standard one-dimensional 1H NMR and J-resolved experiments are commonly run together, since the J-resolved acquisition only adds 5 min to the total acquisition time. While standard 1D 1H NMR spectra are commonly used for data analysis, the corresponding J-resolved spectra can be used for identification purposes.
Here, we illustrate the benefit of using RED-STORM (see Experimental Section for algorithm) over STORM using a well-known dietary biomarker as example. N-Acetyl-S-methyl-cysteine-sulfoxide (NAcSMCSO) is the major urinary metabolite after consumption of cruciferous and other vegetables.14 Edmands et al. have shown that the methyl-sulfoxide signal correlates mostly with the intake of its substrate, and component of cruciferous vegetables, SMCSO, and two other metabolic products, but intramolecular correlations driven from the δ 2.78 peak of NAcSMCSO were weaker than those observed between the methyl-sulfoxide signals of other related molecules. This can also been seen in our data (Figure 3A,B). Our data comes from a diverse set of samples, of which only some contain metabolic products of broccoli consumption. Here STORM was not able to uncover the structural correlations, possibly due to overlap with more intense signals and the overall high variability of samples compared with the study by Edmands et al. (high/low consumers of cruciferous vegetables). The application of RED-STORM to two-dimensional J-resolved spectra of the same individuals, however, clearly showed some intramolecular structural correlations, which were stronger than correlations between NAcSMCSO and other SMCSO-metabolites (Figure 3C). The chemical shifts identified (δ 4.38 (m) and δ 3.10 (m), with probability >0.99) indeed come from the same metabolite;14 however, δ 3.30 (m) was not observed as its signals are heavily overlapped with other multiplets (such as methylhistidines) in the same region.
Figure 3.

Proof-of-concept of the RED-STORM algorithm: application to N-acetyl-S-methyl-cysteine-sulfoxide (NAc-SMCSO), the major biomarker of cruciferous vegetable consumption.14 (A) STORM analysis of the δ 2.78 (s) of NAc-SMCSO shows correlation to three other singlets of related compounds.14 (B) Expansion of δ 2.75–2.85 region. (C) RED-STORM on J-resolved NMR spectra reveals correlations with other nonoverlapped multiplets (δ 4.37 (dd) and δ 3.10 (ABX)) in the CCT data with the peak at δ 2.78.
Complementarity of Statistical and Analytical Platforms for Identification of a Urinary Biomarker of Onion (Allium cepa) Consumption
Previously, dimethylsulfone (δ 3.16 (s)) had been proposed as biomarker of onion consumption.9 However, it has two major disadvantages; first, it is only a singlet, and thus assignment can be ambiguous, but second, and more importantly, the chemical shift of dimethylsulfone is in a region of the NMR spectrum where there are many other di- and trimethyl signals that may confound this metabolite identification. Through an FC we have identified a tentative novel biomarker of onion consumption, which is a multiplet signal at δ 1.97 (dd) (Figure 4A). The presence of this onion-related signal was confirmed using the CCT samples (Figure 4B). STORM analysis using the peak at δ 1.97 as the driver (Figure 3C) clearly shows 3 other correlated signals (δ 2.03 (s), δ 6.50 (m), and δ 6.65 (m)). Due to the clear multiplet structure, we applied RED-STORM on the J-resolved spectra and discovered additional signals (δ 3.44 (m) and δ 4.44 (m)) with a high probability of being intramolecular (>0.97) (Figure 4E).
Figure 4.

Application of RED-STORM for identification of NMR signals tentatively associated with onion consumption. (A) Section of 600 MHz 1H NMR spectra from one volunteer after the onion FC shows a characteristic multiplet. (B) Same section of 600 MHz 1H NMR spectra of different samples of one volunteer in the CCT. (C) STORM analysis shows 3 spectral peaks related to tentative multiplet. (D) Probability of samples resembling the reference signal (x-axis) versus the percentage (y-axis) of samples from each type of urine collection in the CCT. It reveals that specific samples of certain diets (CS3 and 24 h urine samples of the two healthiest diets and a minor amount in CS2 of diet 3) contain the unknown metabolite. (E) RED-STORM identifies two more multiplets compared to STORM (C) visualized in the J-resolved pseudospectrum.
To illustrate how the process for subset selection works, we show the distribution fitting procedure of RED-STORM on the z-transformed correlations of all J-resolved spectra (n = 906) with the reference spectrum of the metabolite of interest (Supporting Information Figure S8). There appear to be two main clusters of sample–reference correlations that follow different Gaussian distributions. After completion, the samples with p(zi|cdf) ≥ 0.5 were included in the subset (n = 320). Inspection of the relation between p(zi|cdf) and the percentage of samples from each unique type of urine sample (collection time, diet) that pass a certain threshold (Figure 4D) gives a very clear indication that the metabolite is found mostly in CS3 and 24 h samples of diets 3 and 4. Small amounts of onions (20 g and 40 g for diets 3 and 4, respectively) were consumed with dinner (matching the presence of the unknown metabolite in CS3) in both of these diets. While no onion was provided in diets 1 and 2, it is interesting to see that the CS2 sample from diet 2 also appeared to contain low levels of this metabolite. On further inspection of the dietary composition, onion traces were found to be present in the sausage casserole that was provided to the CCT volunteers (in diet 2) for lunch.
Using the subset with the highest signal-to-noise of the unknown compound (n = 320), the reference is updated. The z-transformed correlations of the driver peak (δ 1.97 (dd)) with all spectral peaks were calculated, and the resulting distribution was fitted (Supporting Information Figure S9); here, most of the z-scores tend to follow a Gaussian distribution around 0, and only a few have higher z-scores. It is these z-transformed correlations that are likely structural or otherwise closely related to the multiplet of interest; the resulting cdf then gives the result as shown in Figure 4E.
STORM found 4 peaks associated with the driver; the J-coupling constants of the three multiplet signals indicated these were adjacent. RED-STORM identified an additional 2 multiplets which were in regions with overlap in the 1H NMR spectrum; however, measurement of the J-couplings (δ 4.44 (m): 9.78, 8.20, and 4.21 Hz; δ 3.44 (m), 13.38, and 4.21 Hz) indicated at least one undiscovered peak. In order to uncover the complete structure, we then employed analytical methods for further structure elucidation. First, we used a urine sample, taken after a volunteer ate 90 g (dry weight) of (fried) onions over dinner, to obtain a concentrated amount of the unknown metabolite and performed LC coupled to 1H NMR to isolate the compound in an LC-fraction (Figure 5A). The full 1H NMR spectrum of the fraction (Figure 5B) was able to uncover two more signals (δ 3.28 (dd); δ 8.30 with a weak doublet-like splitting) matching with previously measured J-couplings. Analysis of the fraction using LC-MS (ESI+) provided a likely chemical formula of the unknown metabolite of interest, C8H13NO4S (Figure 5C). Using additional 2D-NMR experiments the signals could now be properly assigned (Figure 5D) which resulted in the identification of the complete structure (Figure 5E) of the onion biomarker: N-acetyl-S-(1Z)-propenyl-cysteine-sulfoxide (NAcSPCSO).
Figure 5.

Analytical experiments confirm the identity of N-acetyl-S-(1Z)-propenyl-cysteine-sulfoxide (NAcSPCSO) as a urinary biomarker of onion intake. (A) LC-NMR is used to identify an LC-fraction that contains the metabolite for further analysis. (B) 1H NMR spectrum of the fraction with highest concentration of the unknown metabolite, with multiplets assigned and integrals calculated. (C) LC-MS (positive mode) chromatogram and corresponding total ion chromatogram of the fraction reveals C8H13NO4S as likely elemental composition. (D) Identification table of NMR signals and chemical shifts. 2D-NMR and LC-MS negative mode figures can be found in Supporting Information. (E) Structure of onion biomarker.
To the best of our knowledge, this metabolite has not been reported before. On the basis of its structure, we assume that it is a direct metabolite of S-propenyl-cysteine-sulfoxide (SPCSO). SPCSO is the major flavor precursor in Allium cepa(40) and precursor to the main lachrymatory factor (Z)-propanethial-sulfoxide.41 However, NAcSPCSO does not appear to be the product of any of the known (degradation) pathways in the genus Allium,42 and we hypothesize that it is produced by means of an N-acetyltransferase acetyl-CoA conjugation mechanism, analogous to the production of NAcSMCSO.14 However, SMCSO is not specific for cruciferous vegetables, and can also be found in certain Allium species (including onion). However, SPCSO is specific for A. ascalonicum (shallot), A. cepa, A. nutans (chives), and A. schoenoprasum (chives).40,42
The integrated analytical and statistical two-dimensional spectroscopy strategy for metabolite identification outperforms existing strategies, such as STOCSY and STORM, by utilizing the full resolution J-resolved spectra including the J-coupling constants to allow detection of extra signals attributed to intramolecular correlations. Further clarity on structural assignment is provided by the differentiation of intra- and intermolecular connectivities, not easily differentiated by the basic statistical spectroscopy methods. Thus, identification of NAcSPCSO was only possible through the use of the novel structural elucidation pathway presented here combining statistical and analytical techniques.
Conclusions
Successful structure elucidation of unknown metabolites relies on a combination of the most suitable statistical and analytical strategies and is dependent on metabolite concentration and excretion kinetics, overlap of spectral peaks, and chemical characteristics of the compound. The newly introduced statistical spectroscopy tool, RED-STORM, is able to extract information about potential biomarkers that STORM and other statistical spectroscopy methods cannot provide from 1D-NMR data. Moreover, RED-STORM does not rely on arbitrary correlation-type thresholds28,30,31 or multiple testing adjusted P-values,32 but learns probabilities from the distribution of these data and is therefore less affected by sample size,43 and the effects of normalization and scaling,44 than frequentist methods can be. RED-STORM highlights the added benefit of acquiring J-resolved experiments alongside conventional 1H NMR data as part of metabolic profiling analytical routines.34 Statistical spectroscopy tools can help narrow down the number of analytical experiments that need to be performed (saving time and money). However, for biomarker identification purposes they should not be used by themselves as we have shown that analytical experiments on selected samples can provide information that cannot be gathered using statistical means alone. These analytical experiments are ideally limited to performing a chemical spike-in experiment, but often traditional analytical tools (freeze-drying, SPE-NMR,26 LC-fractionation27) are required in order to isolate the unknown metabolite for further study and confirmation by 2D-NMR and MS. As a result of performing three FCs and combining a suite of statistical and analytical tools, we were able to identify new dietary biomarkers for pea, apple, and onion. These were subsequently validated in an in-patient randomized CCT33 where all food and drink was fully controlled. Specific dietary biomarkers, such as rhamnitol (apple) and N-acetyl-S-(1Z)-propenyl-cysteine-sulfoxide (onion), can be used to assess adherence to diet and/or to increase the accuracy of self-reported dietary records that suffer from misreporting issues.
Acknowledgments
The authors wish to thank Dr. Edward Chambers, Rachel Gibson, Kevin Walsh, and Ivan Dexeus for their assistance during the CCT. I.G.-P. is supported by a NIHR postgraduate research fellowship (ref NIHR-PDF-2012-05-456) and a Wellcome Trust Value In People award. G.F. is supported by an NIHR senior investigator award. E.H., G.F., J.C.M., and J.D. are supported by an MRC grant entitled Metabolomics for Monitoring Dietary Exposure (ref MR/J010308/1). This study was supported by the NIHR/Wellcome Trust Imperial Clinical Research Facility. The Section of Investigative Medicine is funded by grants from the MRC, BBSRC, NIHR, and an Integrative Mammalian Biology (IMB) Capacity Building Award. The MRC-NIHR National Phenome Centre is supported by MRC and NIHR (ref MC_PC_12025).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b03324.
Figures (24) of distribution fitting, 2D-NMR spectra, and LC-MS experiments, and two tables containing CCT information and pseudocode of the algorithm (PDF)
Author Present Address
# J.C.H.: Pfizer, Sandwich, U.K.
Author Present Address
∇ P.B.: Instituto de Biología Molecular y Celular de Rosario (CONICET-UNR) and Plataforma Argentina de Biología Estructural y Metabolómica (PLABEM), Ocampo y Esmeralda, Predio CCT, 2000 Rosario, Argentina.
Author Contributions
○ J.M.P. and I.G.-P. contributed equally. J.M.P., I.G.-P., G.F., E.H., and J.K.N. designed research. J.M.P. and I.G.-P. wrote the paper with input from coauthors. I.G.-P. conducted the CCT and ran NMR experiments. J.M.P. developed the algorithm and conducted statistical analyses. J.M.P. and I.G.-P. analyzed data. P.B. performed NMR experiments for apple identifications. I.G.-P. and J.C.H. performed SPE, LC, and MS experiments. J.C.L. assisted with structural assignments. G.F. supervised the CCT. E.H. and J.K.N. supervised the project. All authors have given approval to the final version of the manuscript.
This article presents independent research funded by the National Institute for Health Research (NIHR) and Medical Research Council (MRC). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Global Strategy on Diet, Physical Activity and Health; WHO: Geneva, 2004. [Google Scholar]
- Ezzati M.; Riboli E. N. Engl. J. Med. 2013, 369, 954–964. 10.1056/NEJMra1203528. [DOI] [PubMed] [Google Scholar]
- O’Keefe S. J. D.; Li J. V.; Lahti L.; Ou J.; Carbonero F.; Mohammed K.; Posma J. M.; Kinross J.; Wahl E.; Ruder E.; Vipperla K.; Naidoo V.; Mtshali L.; Tims S.; Puylaert P. G. B.; DeLany J.; Krasinskas A.; Benefiel A. C.; Kaseb H. O.; Newton K.; Nicholson J. K.; De Vos W. M.; Gaskins H. R.; Zoetendal E. G. Nat. Commun. 2015, 6, 6342. 10.1038/ncomms7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers J. C. Proc. Nutr. Soc. 2013, 72, 246–250. 10.1017/S0029665112003023. [DOI] [PubMed] [Google Scholar]
- Rennie K. L.; Coward A.; Jebb S. A. Br. J. Nutr. 2007, 97, 1169–1176. 10.1017/S0007114507433086. [DOI] [PubMed] [Google Scholar]
- Poslusna K.; Ruprich J.; de Vries J. H.; Jakubikova M.; van’t Veer P. Br. J. Nutr. 2009, 101 (S2), S73–85. 10.1017/S0007114509990602. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Foxall P. J. D.; Spraul M.; Farrant R. D.; Lindon J. C. Anal. Chem. 1995, 67, 793–811. 10.1021/ac00101a004. [DOI] [PubMed] [Google Scholar]
- Stella C.; Beckwith-Hall B.; Cloarec O.; Holmes E.; Lindon J. C.; Powell J.; van der Ouderaa F.; Bingham S.; Cross A. J.; Nicholson J. K. J. Proteome Res. 2006, 5, 2780–2788. 10.1021/pr060265y. [DOI] [PubMed] [Google Scholar]
- Winning H.; Roldan-Marin E.; Dragsted L. O.; Viereck N.; Poulsen M.; Sanchez-Moreno C.; Cano M. P.; Engelsen S. B. Analyst 2009, 134, 2344–2351. 10.1039/b918259d. [DOI] [PubMed] [Google Scholar]
- Heinzmann S. S.; Brown I. J.; Chan Q.; Bictash M.; Dumas M. E.; Kochhar S.; Stamler J.; Holmes E.; Elliott P.; Nicholson J. K. Am. J. Clin. Nutr. 2010, 92, 436–443. 10.3945/ajcn.2010.29672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R.; Wahl A.; Stark T.; Hofmann T. Mol. Nutr. Food Res. 2011, 55, 1613–1623. 10.1002/mnfr.201000656. [DOI] [PubMed] [Google Scholar]
- Lloyd A. J.; Beckmann M.; Fave G.; Mathers J. C.; Draper J. Br. J. Nutr. 2011, 106, 812–824. 10.1017/S0007114511001164. [DOI] [PubMed] [Google Scholar]
- Lloyd A. J.; Fave G.; Beckmann M.; Lin W. C.; Tailliart K.; Xie L.; Mathers J. C.; Draper J. Am. J. Clin. Nutr. 2011, 94, 981–991. 10.3945/ajcn.111.017921. [DOI] [PubMed] [Google Scholar]
- Edmands W. M. B.; Beckonert O. P.; Stella C.; Campbell A.; Lake B. G.; Lindon J. C.; Holmes E.; Gooderham N. J. J. Proteome Res. 2011, 10, 4513–4521. 10.1021/pr200326k. [DOI] [PubMed] [Google Scholar]
- Ismail N. A.; Posma J. M.; Frost G.; Holmes E.; Garcia-Perez I. Electrophoresis 2013, 34, 2776–2786. 10.1002/elps.201300066. [DOI] [PubMed] [Google Scholar]
- Heinzmann S. S.; Holmes E.; Kochhar S.; Nicholson J. K.; Schmitt-Kopplin P. J. Agric. Food Chem. 2015, 63, 8615–8621. 10.1021/acs.jafc.5b03040. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez I.; Posma J. M.; Chambers E. S.; Nicholson J. K.; Mathers J. C.; Beckmann M.; Draper J.; Holmes E.; Frost G. J. Agric. Food Chem. 2016, 64, 2423–2431. 10.1021/acs.jafc.5b05878. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Lindon J. C.; Holmes E. Xenobiotica 1999, 29, 1181–1189. 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- Guertin K. A.; Moore S. C.; Sampson J. N.; Huang W. Y.; Xiao Q.; Stolzenberg-Solomon R. Z.; Sinha R.; Cross A. J. Am. J. Clin. Nutr. 2014, 100, 208–217. 10.3945/ajcn.113.078758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan A.; Gibney M. J.; Brennan L. Am. J. Clin. Nutr. 2011, 93, 314–321. 10.3945/ajcn.110.000950. [DOI] [PubMed] [Google Scholar]
- Lindon J. C.; Nicholson J. K.; Wilson I. D. J. Chromatogr., Biomed. Appl. 2000, 748, 233–258. 10.1016/S0378-4347(00)00320-0. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Lindon J. C. Nature 2008, 455, 1054–1056. 10.1038/4551054a. [DOI] [PubMed] [Google Scholar]
- Lindon J. C.; Nicholson J. K. Annu. Rev. Anal. Chem. 2008, 1, 45–69. 10.1146/annurev.anchem.1.031207.113026. [DOI] [PubMed] [Google Scholar]
- Robinette S. L.; Lindon J. C.; Nicholson J. K. Anal. Chem. 2013, 85, 5297–5303. 10.1021/ac4007254. [DOI] [PubMed] [Google Scholar]
- Dona A. C.; Kyriakides M.; Scott F.; Shephard E. A.; Varshavi D.; Veselkov K.; Everett J. R. Comput. Struct. Biotechnol. J. 2016, 14, 135–153. 10.1016/j.csbj.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson I. D.; Nicholson J. K. Anal. Chem. 1987, 59, 2830–2832. 10.1021/ac00150a025. [DOI] [PubMed] [Google Scholar]
- Shockcor J. P.; Unger S. E.; Wilson I. D.; Foxall P. J.; Nicholson J. K.; Lindon J. C. Anal. Chem. 1996, 68, 4431–4435. 10.1021/ac9606463. [DOI] [PubMed] [Google Scholar]
- Cloarec O.; Dumas M. E.; Craig A.; Barton R. H.; Trygg J.; Hudson J.; Blancher C.; Gauguier D.; Lindon J. C.; Holmes E.; Nicholson J. Anal. Chem. 2005, 77, 1282–1289. 10.1021/ac048630x. [DOI] [PubMed] [Google Scholar]
- Beckonert O.; Keun H. C.; Ebbels T. M. D.; Bundy J. G.; Holmes E.; Lindon J. C.; Nicholson J. K. Nat. Protoc. 2007, 2, 2692–2703. 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- Blaise B. J.; Navratil V.; Domange C.; Shintu L.; Dumas M. E.; Elena-Herrmann B.; Emsley L.; Toulhoat P. J. Proteome Res. 2010, 9, 4513–4520. 10.1021/pr1002615. [DOI] [PubMed] [Google Scholar]
- Robinette S. L.; Veselkov K. A.; Bohus E.; Coen M.; Keun H. C.; Ebbels T. M. D.; Beckonert O.; Holmes E. C.; Lindon J. C.; Nicholson J. K. Anal. Chem. 2009, 81, 6581–6589. 10.1021/ac901240j. [DOI] [PubMed] [Google Scholar]
- Posma J. M.; Garcia-Perez I.; De Iorio M.; Lindon J. C.; Elliott P.; Holmes E.; Ebbels T. M.; Nicholson J. K. Anal. Chem. 2012, 84, 10694–10701. 10.1021/ac302360v. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez I.; Posma J. M.; Gibson R.; Chambers E. S.; Hansen T. H.; Vestergaard H.; Hansen T.; Beckmann M.; Pedersen O.; Elliott P.; Stamler J.; Nicholson J. K.; Draper J.; Mathers J. C.; Holmes E.; Frost G. Lancet Diabetes Endocrinol. 2017, 5, 184–195. 10.1016/S2213-8587(16)30419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dona A. C.; Jimenez B.; Schafer H.; Humpfer E.; Spraul M.; Lewis M. R.; Pearce J. T.; Holmes E.; Lindon J. C.; Nicholson J. K. Anal. Chem. 2014, 86, 9887–9894. 10.1021/ac5025039. [DOI] [PubMed] [Google Scholar]
- Want E. J.; Wilson I. D.; Gika H.; Theodoridis G.; Plumb R. S.; Shockcor J.; Holmes E.; Nicholson J. K. Nat. Protoc. 2010, 5, 1005–1018. 10.1038/nprot.2010.50. [DOI] [PubMed] [Google Scholar]
- Rasmussen C. E.Advances in Neural Information Processing Systems 12; MIT Press, 2000; Vol. 12, pp 554–560 [Google Scholar]
- Neal R. M. Ann. Stat 2003, 31, 705–741. 10.1214/aos/1056562461. [DOI] [Google Scholar]
- Melnick D.; Robinson W. D.; Field H. J. Biol. Chem. 1940, 136, 131–144. [Google Scholar]
- Tomita S.; Nemoto T.; Matsuo Y.; Shoji T.; Tanaka F.; Nakagawa H.; Ono H.; Kikuchi J.; Ohnishi-Kameyama M.; Sekiyama Y. Food Chem. 2015, 174, 163–172. 10.1016/j.foodchem.2014.11.028. [DOI] [PubMed] [Google Scholar]
- Kubec R.; Svobodova M.; Velisek J. J. Agric. Food Chem. 2000, 48, 428–433. 10.1021/jf990938f. [DOI] [PubMed] [Google Scholar]
- Block E.; Penn R. E.; Revelle L. K. J. Am. Chem. Soc. 1979, 101, 2200–2201. 10.1021/ja00502a044. [DOI] [Google Scholar]
- Rose P.; Whiteman M.; Moore P. K.; Zhu Y. Z. Nat. Prod. Rep. 2005, 22, 351–368. 10.1039/b417639c. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Hero A. O. J. Comput. Biol. 2007, 14, 1311–1326. 10.1089/cmb.2006.0151. [DOI] [PubMed] [Google Scholar]
- Saccenti E. J. Proteome Res. 2017, 16, 619–634. 10.1021/acs.jproteome.6b00704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.