

Abstract
Transcranial low-level laser (light) therapy (LLLT) is a new non-invasive approach to treating a range of brain disorders including traumatic brain injury (TBI). We (and others) have shown that applying near-infrared light to the head of animals that have suffered TBI produces improvement in neurological functioning, lessens the size of the brain lesion, reduces neuroinflammation, and stimulates the formation of new neurons. In the present study we used a controlled cortical impact TBI in mice and treated the mice either once (4 h post-TBI, 1-laser), or three daily applications (3-laser) with 810 nm CW laser 36 J/cm2 at 50 mW/cm2. Similar to previous studies, the neurological severity score improved in laser-treated mice compared to untreated TBI mice at day 14 and continued to further improve at days 21 and 28 with 3-laser being better than 1-laser. Mice were sacrificed at days 7 and 28 and brains removed for immunofluorescence analysis. Brain-derived neurotrophic factor (BDNF) was significantly up-regulated by laser treatment in the dentate gyrus of the hippocampus (DG) and the subventricular zone (SVZ) but not in the perilesional cortex (lesion) at day 7 but not at day 28. Synapsin-1 (a marker for synaptogenesis, the formation of new connections between existing neurons) was significantly upregulated in lesion and SVZ but not DG, at 28 days but not 7 days. The data suggest that the benefit of LLLT to the brain is partly mediated by stimulation of BDNF production, which may in turn encourage synaptogenesis. Moreover the pleiotropic benefits of BDNF in the brain suggest LLLT may have wider applications to neurodegenerative and psychiatric disorders.
Keywords: traumatic brain injury, transcranial low level light therapy, synaptogenesis, neurogenesis, BDNF, Synapsin-1
Graphical Abstract
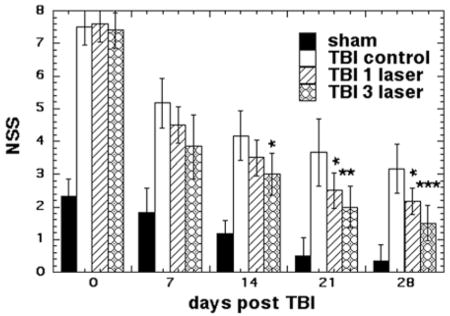
Neurological Severity Score (NSS) for TBI mice
1. Introduction
Traumatic brain injury (TBI) is caused by blunt force or penetrating external physical trauma [1]. It causes serious damage to the central nervous system (CNS) tissue and is a leading cause of disability and death after accidents [2]. If the patient survives the initial injury, TBI often causes a progressive degeneration leading to cognitive and motor deficits and is a high risk for the future development of neurodegenerative disorders such as Alzheimer’s disease [3, 4]. According to the Centers for Disease Control and Prevention approximately 5.3 million Americans are living with disabilities that resulted from TBIs [5]. Additionally, TBI is the signature injury in military personal in Iraq and Afghanistan, accounting for nearly 20% of major injuries [5]. The initial injury to the brain includes damage to neurons, glia and vascular structures, followed by a cascade of secondary injuries which can cause further deterioration to the brain tissue via multiple molecular mechanisms [6, 7]. Secondary injuries may be due to inflammation, glutamate excitotoxicity, cell necrosis, glial proliferation, dysfunction of mitochondria, apoptosis, production of oxygen free radicals and diffuse axonal injury [7, 8]. These pathological changes result in alterations in synaptogenesis, dendritic remodeling, and neurogenesis in the hippocampal, prefrontal cortical and limbic regions. As a healing response, after the primary and secondary injuries, neurons start reorganizing and repairing axonal connections by enabling synaptic growth (also called synaptogenesis) [9]. The neurotrophin, brain derived neurotrophic factor (BDNF) is a crucial regulator of synaptic plasticity, that promotes synaptic transmission and synaptogenesis [10]. At the synapse, synapsins are key presynaptic effectors for the acute action of BDNF [11]. Synapsin I is a downstream effector for the neurotrophin/Trk/MAP kinase cascade [12].
Studies suggest that BDNF acts through the mitogen-activated protein kinase (MAPK) cascade to induce phosphorylation of synapsin in both cerebro-cortical neurons and in PC12 cells. [10, 12]. In a synaptosomal preparation, BDNF increased MAPK-dependent synapsin I phosphorylation and caused a large release of glutamate [11]. PD98059, an inhibitor of MAPK activity gave a significant decrease in BDNF-induced synapsin I phosphorylation, and concurrently reduced neurotransmitter release. Moreover, in mice lacking synapsin I and/or synapsin II, the stimulation of glutamate release by BDNF was markedly attenuated. Taken togther these studies indicated a pivotal link between synapsin phosphorylation via BDNF, TrkB receptors and MAP kinase with downstream facilitation of neurotransmitter release. These pathways are proposed to contribute to the modulation of synaptic plasticity by neurotrophins. Moreover BDNF induces phosphorylation of synapsin I that also leads to increased docking of synaptic vesicles and neurotransmitter release [11].
Since multiple pathways are involved in damage to the brain, different possibilities for treatment should be explored. Unfortunately, currently there is no specific proven therapeutic strategy for TBI in humans [13]. This lack of standard treatments led us to investigate a more radical alternative to the current therapy, namely transcranial low level-laser (light) therapy (LLLT).
In recent times, there has been an increase in the use of LLLT as a mainstream treatment modality, chiefly in the areas of wound healing, physical medicine and rehabilitation medicine. Currently the experimental therapeutic applications of LLLT have expanded to include diseases such as spinal cord injury, stroke, myocardial infarction, and degenerative or traumatic brain disorders [14, 15]. LLLT involves red and/or near infra-red light absorption by mitochondrial chromophores such as cytochrome c oxidase, resulting in increased cellular respiration, including increased ATP levels and ROS generation which leads to activation of redox-sensitive genes and related transcription factors including NF-κβ [16, 18]. LLLT stimulates the expression of genes responsible for cellular proliferation, migration, and the production of cytokines and growth factors [15].
Since, LLLT (especially in the NIR) penetrates the scalp and skull and reaches the brain, it could have a role in neuroprotection and neurogenesis of traumatic and degenerative diseases. Studies have suggested that the anti-inflammatory, anti-edema and pro-angiogenic property of LLLT can act as an effective treatment modality for stroke [15, 19]. This success gives hope that LLLT might be also effective in TBI, as some features of the pathophysiology in stroke are similar to TBI. Our laboratory [20–22] and others [23–26] have shown that transcranial LLLT is beneficial in a range of different animal models of TBI [15]. The most important mechanism of transcranial LLLT for TBI might be its role in neuronal repair and neurogenesis. It would therefore be interesting to see if there is a role of LLLT not only in formation of new brain cells but also in synaptogenesis, which is the formation of new connections between existing brain cells. To answer these questions, we used a controlled cortical impact (CCI) mouse model of severe TBI treated with one or three daily transcranial NIR laser exposures, and observed the expression of genes for the neurotrophin (BDNF) and synaptogenesis (synapsin).
2. Materials and methods
2.1 Animals
Ethics statement
All animal procedures were approved by the Subcommittee on Research Animal Care (IACUC) of the Massachusetts General Hospital (protocol # 2010N000202) and met the guidelines of the National Institutes of Health.
Male BALB/c mice aged 6–8 weeks, weight 21 to 25 g; Charles River Laboratories, Wilmington, MA) were used in the study. The animals were housed as one mouse per cage and were maintained on a 12 h light & 12 h dark cycle and food and water was provided ad libitem.
2.2 Mouse model of focal controlled cortical impact (TBI)
Before the surgical procedure the mice were anesthetized with isoflurane. The hairs on the head were shaved and depilated (Nair, Carter-Wallace, NY), when the top of the skull was adequately exposed a 1 cm skin incision was made in a central and sagittal direction. Using a 4 mm trephine attached to an electric portable drill a 5 mm craniotomy was performed over the right parietal cortex taking care to avoid damaging the meninges. After the removal of bone flap the mice were subjected to a single right lateral controlled cortical impact (CCI) using a pneumatic impact device (Model AMS 201, AmScien Instruments LLC, USA) with a 3 mm flat-tip, high pressure 150 psi, low pressure 30 psi, rod speed 4.8 m/sec, rising duration 8.41 ms, and set impact depth of 2 mm with the device positioned over the right front-parietal cortex and the tip centered 3 mm anterior to lambda and 2.5 mm right of midline within the craniotomy. Immediately after generating the brain trauma the craniotomy hole was sealed with bone wax and the skin was sutured. At 1 hour post-TBI severity of injury was assessed by neurological severity score (NSS). Mice with scores of 7 or 8 were eligible for inclusion in the study, while mice with scores higher or lower that these ranges were ineligible for inclusion and were sacrifice. Sham TBI mice were subjected to all aspects of the surgical procedure including craniotomy except the actual cortical impact.
2.3 Laser treatment
Forty mice were divided into four groups of ten mice each. One group received sham TBI, and 3 groups received real TBI. One real TBI group received sham laser treatment (restraint but no light) while the two real TBI groups received 1 laser or 3 real daily laser exposures. Mice were kept in a rodent restrainer (520315, Harvard apparatus, Holliston, MA). 1 laser and 3 laser groups received a laser treatment 4 hr post TBI, using a diode laser of 810-nm wavelength and 1 W maximal power output (Photothera Inc., Carlsbad, CA), which emits continuous wave (CW) 810 nm wavelength near infrared radiation. After mice were placed on a rodent restrainer containing a 1 cm diameter hole in the plastic, the injured side of the head was exposed to a 1 cm diameter spot from the distal tip of the fiber optic at a power density of 50 mW/cm2. The laser irradiation was given for 12 min, and the total fluence was 36 J/cm2. The total energy delivered at each treatment was 28.3 J. Four mice in each group were euthanized on day 7 and four in each group were euthanized on day 28. Six mice in each group had NSS scores measured as described below.
2.4 Neurobehavioral assessment
After inducing CCI, the mice were subjected to neurological severity score (NSS) test to evaluate their neurological performance (Table 1). NSS evaluations started at 1hour post TBI (day 0) and were carried out on days 1, 4, 7, 10, 15, 19, 24, and 28. The mice were assessed by testing their motor ability, balance, and alertness. Completely normal mice score zero, each failed assignment contributed one point which could add up to a maximum score of 10. Mice with NSS score less than 5 were assigned as mild TBI; mice with NSS score of 5–6 were assigned as moderate TBI; NSS score of 7–8 indicated mice sustaining severe TBI; NSS score of 9–10 indicated mice sustaining very severe TBI. In our study, we studied mice sustaining severe TBI with NSS of 7–8.
Table 1.
Neurological Severity Score (NSS) for TBI Mice. Mice are awarded 1 point for each failure to complete a task. Mice are assessed the score for the best level they reach.
| Task | NSS |
|---|---|
| Presence of mono- or hemiparesis | 1 |
| Inability to walk on a 3-cm-wide beam | 1 |
| Inability to walk on a 2-cm-wide beam | 1 |
| Inability to walk on a 1-cm-wide beam | 1 |
| Inability to balance on a 1-cm-wide beam | 1 |
| Inability to balance on a round stick (0.5 cm wide) | 1 |
| Failure to exit a 30-cm-diameter circle (for 2 min) | 1 |
| Inability to walk straight | 1 |
| Loss of startle behavior | 1 |
| Loss of seeking behavior | 1 |
| Maximum total | 10 |
2.5 Immunofluorescence staining
Mice were anesthetized and a transcardial perfusion was given with 0.9% saline and then with 4% phosphate-buffered paraformaldehyde at days 7 and 28 days after TBI.
Brains were removed and were fixed in formaldehyde solution for 3 days after which they were embedded in paraffin. Coronal serial brain cross-sections of 10 micron-thick sections were cut from the top, middle, and bottom of the thick sliced blocks via microtome. The paraffin slides were then immersed in xylene for deparaffinization, graded ethanol for rehydration, and then passed through antigen retrieval with citrate buffer solution in microwave-oven, incubated in blocking solution consisting of 5%BSA/0.1%TritonX-100 in PBS, and immunostained with chicken anti-BDNF (Cat.# AB9042, Millipore) and rabbit anti-Synapsin I, (Cat.# AB1543, Millipore). Goat anti-chicken and goat anti-rabbit secondary antibodies (conjugated with Alexa fluor 680 and Cy3, Invitrogen) were used respectively. The slides were then mounted using mounting medium containing DAPI (Cat#H-1200, Fisher Scientific). Finally, they were cover-slipped with mounting media contained DAPI The slides were imaged with a confocal microscope (Olympus America Inc, Center Valley, PA). Red BDNF or red synapsin-1 staining and blue DAPI were quantified by the use of Image J. Ratios of red BDNF or synapsin-1 fluorescence staining to blue DAPI fluorescence staining were calculated to normalize for the number of cells present in each microscopic field.
2.6 Statistical analysis
Data are presented as mean ± SD, and statistically analyzed using one-way analysis of variance (ANOVA) followed by Tukey post-hoc test for multiple comparisons. Significance was defined as p < 0.05. SPSS statistics V17.0 software was used for statistical analyses.
3. Results
3.1 Effect of laser treatment on neurological severity score (NSS)
Figure 1 shows the NSS scores of the four groups of mice measured on 0, 7, 14, 21 and 28 days post TBI (all groups n = 6). One hour after the initial TBI injury, the NSS was similar (7–8) in all three TBI groups of mice, while the sham TBI group also showed a deficit with a NSS of 2.3 (although much lower that real TBI). There was a marked improvement in the neurobehavioral function of all groups at day 7 compared to day 0. However, the improvement was not statistically significant between the groups that received no treatment or laser. On day 14 the 3-laser group showed a significant improvement over TBI control (p < 0.05), while at day 21, both the 1-laser and 3-laser groups showed significant improvements over TBI control. At 28 days the TBI mice treated with 1-laser (p < 0.05) and 3-laser (p < 0.001) showed significant improvements in NSS test compared to TBI control. Throughout the study the mice treated with 3-laser showed a better improvement in their neurological function as compared to mice treated with only 1-laser treatment.
Figure 1.
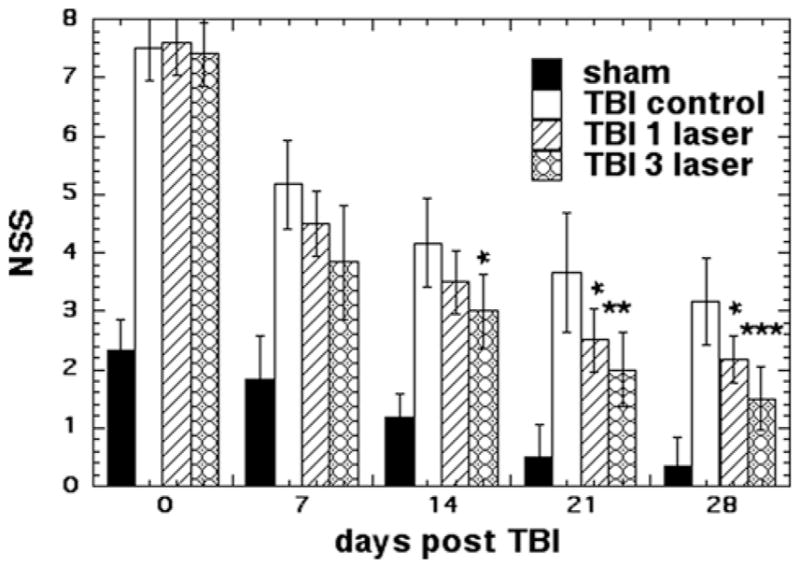
NNS scores of the mice. Mean (n = 8–14) NSS scores measured over 7 and 28 days of mice in 8 groups consisting of sham TBI mice, sham control mice, 810 nm laser treated TBI mice (18 J/cm2 delivered at 25 mW/cm2). Mice were either given 1 laser Tx or 1 sham Tx at 4 hours post TBI or 3 daily laser Tx or 3 sham Tx; *p < 0.05; **P < 0.01; ***p < 0.001.
3.2 BDNF staining
BDNF is largely expressed in the developing and adult mammalian brain, synthesized in numerous areas including the hypothalamus. Thus, BDNF mRNA and proteins are present in almost all the cortical areas as well as other tissues, including the neural soma, dendrites, fibers and amygdala [27]. To localize BDNF in brain sections, we used immunofluorescence using secondary fluorescence-labeled antibodies and DAPI counterstaining. BDNF staining was observed in: (1) the sub-granular layer of the dentate gyrus, which is a part of hippocampal formation; (2) subventricular zone (SVZ) located between the ventricular and intermediate zones in the forebrain; (3) the perilesional cortex, which is the area of actual impact.
Sample immunofluorescence images of BDNF in the dentate gyrus (DG) at 7 and 28 days are shown in Figure 2A–H and the quantification of fluorescence ratios (red/blue) from 6 slides for each group is shown in Figure 2I. A baseline level expression of BDNF was observed in sham TBI mice. Although there was a mild increase in BDNF expression in untreated TBI group at day 7 in the DG, this increase was not statistically significant. A large increase in BDNF expression was observed in the dentate gyrus with 1-laser (p < 0.01) and especially with 3-laser (p < 0.001) treatment on day 7. At day 28, the expression of BDNF was sharply decreased in all the groups, although there was still some BDNF detected in the 1-laser and 3-laser groups but this was not significant when quantifies.
Figure 2.

Protein expression of BDNF in the Dentate Gyrus area of brain sections (Mean n = 4). Immunofluorescence showing protein expression of BDNF at 400 × magnification in brain tissue of mice sacrificed at 7 or 28 days post-TBI. DAPI was used to stain the nuclei. (A) Sham- control mouse euthanized at day 7 (B) Sham-control mouse euthanized at day 28 (C) TBI-sham laser mice euthanized at day 7. (D) TBI-sham laser mice euthanized at day 28. (E) TBI mouse treated with 1 laser treatment euthanized at day 7. (F) TBI mouse treated with 1 laser treatment euthanized at day 28. (G) TBI mouse treated with 3 laser treatment euthanized at day 7 (H) TBI mouse treated with 3 laser treatment euthanized at day 28 (I) Mean fluorescent intensity (MFI) of BDNF immunostaining in brain sections measured in arbitrary units (AU) (using NIH Image J software). Data is shown as mean ± SEM from six measurements per mouse. **P < 0.01
Figure 3A–H shows analogous fluorescence images of BDNF in the SVZ at days 7 and 28 and quantification is presented in Figure 3I. Again there was a non-significant increase in untreated TBI brains compared to sham controls, and a significant increase of BDNF expression with 1-laser (p < 0.05) and 3-laser (p < 0.01) treatments compared to TBI mice at day 7, with a decrease in all groups and particularly in laser treated brains being evident at day 28.
Figure 3.

Protein expression of BDNF in the SVZ area of brain sections (Mean n = 4). Immunofluorescence showing protein expression of BDNF at 400× magnification in brain tissue of mice sacrificed at 7 or 28 days post-TBI. DAPI was used to stain the nuclei. (A) Sham- control mouse euthanized at day 7 (B) Sham- control mouse euthanized at day 28 (C) TBI-sham laser mice euthanized at day 7. (D) TBI-sham laser mice euthanized at day 28. (E) TBI mouse treated with 1 laser treatment euthanized at day 7. (F) TBI mouse treated with 1 laser treatment euthanized at day 28. (G) TBI mouse treated with 3 laser treatment euthanized at day 7 (H) TBI mouse treated with 3 laser treatment euthanized at day 28 (I) Mean fluorescent intensity (MFI) of BDNF immunostaining in brain sections measured in arbitrary units (AU) (using NIH Image J software). Data is shown as mean ± SEM from six measurements per mouse; *p < 0.05; **P < 0.01; ***p < 0.001.
There was no difference in the expression of BDNF among any of the groups in the cortex adjacent to the lesion area at either time point (data not shown).
3.3 Synapsin-1 Staining
In cell cultures, treatment with exogenous BDNF potentiates excitatory synaptic transmission [11]. To localize synapsin-1 in brain sections, we used immunofluorescence using secondary fluorescence-labeled antibodies and DAPI counterstaining. Figure 4A–H shows sample immunofluorescence images of synapsin-1 in the cortex adjacent to the lesion area at 7 and 28 days with quantification of fluorescence ratios from 6 slides for each group in Figure 4I. A baseline expression of synapsin-1 was observed in all groups but there were no differences visible between the groups at 7 days post-TBI. At 28 days there was a non-significant increase in untreated TBI mice, and a significant increase in synapsin-1 after 1-laser (p < 0.05) and especially with 3-laser (p < 0.01) treatments.
Figure 4.

Protein expression of synapsin-1 in the SVZ area of brain sections (Mean n = 4). Immunofluorescence showing protein expression of synapsin-1 at 400× magnification in brain tissue of mice sacrificed at 7 or 28 days post-TBI. DAPI was used to stain the nuclei. (A) Sham- control mouse euthanized at day 7 (B) Sham- control mouse euthanized at day 28 (C) TBI-sham laser mice euthanized at day 7. (D) TBI-sham laser mice euthanized at day 28. (E) TBI mouse treated with 1 laser treatment euthanized at day 7. (F) TBI mouse treated with 1 laser treatment euthanized at day 28. (G) TBI mouse treated with 3 laser treatment euthanized at day 7 (H) TBI mouse treated with 3 laser treatment euthanized at day 28 (I) Mean fluorescent intensity (MFI) of synapsin-1 immunostaining in brain sections measured in arbitrary units (AU) (using NIH Image J software). Data is shown as mean ± SEM from six measurements per mouse; *p < 0.05.
Figure 5I show the analogous results for synapsin-1 staining in the SVZ. The results were broadly similar to those found in the perilesional cortex with increases only seen at 28 days and not at 7 days, but the laser-induced increase was much smaller, with the only significant increase seen after 3-laser treatments at 28 days (p < 0.05).
Figure 5.

Protein expression of synapsin-1 in the lesion area of brain sections (Mean n = 4). Immunofluorescence showing protein expression of synapsin-1 at 400× magnification in brain tissue of mice sacrificed at 7 or 28 days post-TBI. DAPI was used to stain the nuclei. (A) Sham- control mouse euthanized at day 7 (B) Sham- control mouse euthanized at day 28 (C) TBI-sham laser mice euthanized at day 7. (D) TBI-sham laser mice euthanized at day 28. (E) TBI mouse treated with 1 laser treatment euthanized at day 7. (F) TBI mouse treated with 1 laser treatment euthanized at day 28. (G) TBI mouse treated with 3 laser treatment euthanized at day 7 (H) TBI mouse treated with 3 laser treatment euthanized at day 28 (I) Mean fluorescent intensity (MFI) of synapsin-1 immunostaining in brain sections measured in arbitrary units (AU) (using NIH Image J software). Data is shown as mean ± SEM from six measurements per mouse. ** P < 0.01.
Laser treatment (1 and 3 laser) failed to change the expression of synapsin-1 in the dentate gyrus either at 7 and 28 days.
4. Discussion
The present study has demonstrated an improvement in neurological functions and an increase in neurotrophin expression and a marker of neuronal repair and synaptic plasticity in the mouse brain subjected to severe CCI-TBI and treated with 1 or 3 daily transcranial NIR laser treatments. Although, the underlying mechanisms are not fully understood, LLLT might be preventing tissue damage and increasing neurogenesis and synaptogenesis.
Transcranial LLLT has shown promising results in treatment of stroke, traumatic brain injury, and neurodegenerative disease [28]. LLLT at a wavelength 808–810 nm penetrates the skull and goes into the brain tissue where it has been shown to increase ATP production in the rat cerebral cortex [29]. In an ischemic stroke model in rats, LLLT provided a significant improvement in neurological outcome in 24 hrs, with increased neurogenesis in the subventricular zone (SVZ) [30]. Oron and colleagues [24] investigated the effects of transcranial LLLT for TBI in mice. A single application of 808-nm transcranial laser was given to the brain 4 h after TBI, with two energy densities (1.2–2.4 J/cm2 calculated to be delivered to the cortex over 2 minutes irradiation with 10 and 20 mW/cm2). The study showed a significant decrease in the NSS values (26–27%), lower loss of cortical tissue at the injured site and improvement in neurobehavioral function in the laser treated groups, demonstrating that transcranial LLLT can reduce long-term neurological deficits [24]. Khuman et al. reported that a single application of 810 nm laser delivered directly to the contused brain through the craniotomy in CCI improved Morris Water Maze performance and reduced microgliosis. They suggested LLLT, could be a therapeutic option to improve cognitive recovery and limit inflammation after TBI [31].
We previously reported that transcranial LLLT at 810 nm diode laser at continuous wave (CW) and at two different pulse frequencies (PW) as a non-invasive treatment of TBI in mice. 10 Hz PW was superior to CW and 100 Hz PW in improving the neurobehavioral recovery in TBI and reducing the size of the lesion. It also had an anti-depressant effect, as revealed by significant decrease of immobility time in the forced swim test and tail suspension test, which measure depression and anxiety [20]. Xuan et al. used transcranial LLLT (continuous wave 810 m laser, 25 mW/cm2, 18 J/cm2) either once at 4 hours post-TBI, 3 daily treatments or 14 daily treatments. The 3 daily laser treatment was found to be the best in parallel with our present study and excessively repeated LLLT (14 laser Tx) had lost all its beneficial effect. The study concluded that LLLT for TBI in mice could significantly improve neural function, decrease lesion volume, augment cell proliferation, and protects the brain against neuronal damage [22].
One interesting question that should be addressed is, how does the severity of the brain lesion affect its response to transcranial LLLT? Most of the workers who have studied the beneficial effects of LLLT on the various mouse models of TBI have used severe grades of injury, rather than moderate or mild injuries. However it is presently unclear if this choice is governed more by the ease of measurement of differences in NSS and other motor and cognitive tests in severe TBI mice as compared to mild or moderate TBI mice, or by the fact that severely injured mice do in fact respond better to the LLLT than less severely injured animals. Considering the number of mild or moderate clinical TBI cases far outweighs the number of severe TBI cases both in civilian and military medicine, this is an important question that needs to be answered before widespread clinical trials are undertaken.
In the present study, the NSS performance assay in TBI mice that received the optimized LLLT repetition regimen produced improvement in neurological and cognitive function post TBI. As compared to real TBI with sham laser treatment the 1 laser treatment was effective, while the 3-laser treatment was the best and the maximum improvement was observed at 28 days. The observations from these studies suggest a role of LLLT in activating brain repair pathways. Previously, it was believed that adult brain is incapable of regenerating itself [32], but now there are studies showing neuroplasticity, neurogenesis and synaptogenesis in adult brain [33]. In the mammalian forebrain, neurogenesis occurs by the neuroprogenitor cells throughout life in subventricular zone (SVZ)-olfactory bulb pathway and hippocampal dentate gyrus [34]. In response to brain injuries suffered as a result of stroke or TBI, these neuroprogenitor cells gets stimulated and are able to migrate to the sites of brain damage to effect repairs [35].
The existence of neuroplasticity in this mouse model of TBI, is clearly shown by the fact that the neurological and cognitive performance of the mice is improving over 4 weeks, to at the same time as the size of the brain lesion is increasing over the same time period as we previously reported [22]. It should be noted that this neurological improvement happens both in untreated TBI mice, and in laser treated mice where the neurological performance improves even faster. In laser treated mice the rate of increase of the lesion size is reduced but the lesion still gets bigger over 4 weeks. The only explanation for these observations is that the uninjured part of the mouse brain is steadily taking over more of the functions of the injured part of the brain. As the injured part of the brain grows in size the normal uninjured brain takes over the functions at an even greater rate, so that the overall neurological function increases with time.
The neurotrophin BDNF plays a key role in modulating neurogenesis in the dentate gyrus and SVZ, and is also a vital mediator in the process of synaptogenesis [36]. The synapsin 1 protein is involved in vesicle clustering, neurotransmitter release, axonal elongation and maintenance of synaptic contacts. It is usually a predictor of synaptic density [37]. Studies have shown that BDNF is involved in the synthesis [38] and phosphorylation [11, 12] of synapsin-1. Some treatment approaches that enhance BDNF–related signaling via various mechanisms have been shown to restore neural connectivity that could facilitate neuroplastic changes leading to adaptive neural repair and consequently to enhance the repair of cognitive deficits in both TBI and PTSD [9]. A recent study by Meng et al. suggested that LLLT upregulates BDNF via activation of ERK/ CREB pathway and decreases neuronal loss and dendritic atrophy in mice with Alzheimer’s disease [39]. Another study used male Wistar rats that were subjected to right sciatic nerve crush injury and were irradiated on a daily basis with helium-neon laser (collimated HeNe laser, continuous emission, wavelength: 632.8 nm, power density: 0.5 mW/cm2, irradiation time: 20 sec, energy density: 10 J/cm2) during 7, 14, and 21 consecutive days, respectively. HeNe laser increased the mRNA expression of neurotrophic factors BDNF and nerve growth factor (NGF) after 14 days of LLLT, with peak expression at the 21st day. Additionally, HeNe laser reduced the inflammatory marker (inducible nitric oxide synthase iNOS) expression. The study opened a possibility of nerve regeneration using LLLT [40]. In our current study, we observed that the appropriate regimen of LLLT (1 or 3 daily laser Tx) could increase the expression of BDNF in the SVZ in 7 and 28 days.
In the dentate gyrus and the SVZ 1-laser and 3-laser Tx seemed to increase the expression of BDNF at 7 days but not at 28 days. No change in BDNF expression was observed in the lesion area. Furthermore, our results indicated that the expression of BDNF significantly decreased in SVZ at day 28 and completely diminished at day 28 in the dentate gyrus. We also observed an increase in synapsin-1 with 3-laser treatment at day 28 in the lesion area and SVZ. However, no change in synapsin-1 was observed in the dentate gyrus. 1-laser treatment failed to induce synapsin-1 expression. Our results support the hypothesis that BDNF might be involved in the production of synapsin, as the increased expression of BDNF on day 7 might have stimulated the synthesis of synapsin which was increased at day 28 and not at day 7. On the other hand, the increase in synapsin in the lesion area with no increase in BDNF in the same area might be due to some other mechanism that may be acting independently or in synergism with BDNF. It is therefore reasonable to assume that LLLT induces a cascade of processes that result in induction of neurogenesis and synaptogenesis.
Another question that needs to be addressed is how deep does the light need to penetrate into the brain to continue to have a beneficial effect. Although in humans deep TBIs are not particularly common, deep infarcts from stroke are often found in the human brain. When tLLLT was used to treat stroke in the NEST2 and NEST3 trials, it was concluded [41, 42] that deeper lesions did not respond as well the 810 nm laser therapy (delivered at a fairly high power density) as more superficial lesions. Whether the same findings will be made in TBI (In either animals or humans) remains to be discovered.
In conclusion, our data demonstrates that transcranial LLLT improves neurological function mediated (at least in part) by induction of BDNF in the DG and SVZ at 7 days that may lead to increases in synaptogenesis in the cortex at 28 days. As there is considered to be a complete lack of adverse effects after delivery of LLLT to the head, further studies on this approach are warranted as a potential therapy for traumatic brain injury and other brain disorders in humans.
Supplementary Material
Acknowledgments
This work was supported by US NIH grant R01AI050875, by Air Force Office of Scientific Research grant FA9550-13-1-0068, by US Army Medical Research Acquisition Activity grant W81XWH-09-1-0514 and by US Army Medical Research and Materiel Command grant W81XWH-13-2-0067.
Footnotes
Author biographies Please see Supporting Information online.
References
- 1.Leeds PR, Yu F, Wang Z, Chiu CT, Zhang Y, Leng Y, Linares GR, Chuang DM. ACS Chem Neurosci. 2014 doi: 10.1021/cn500040g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain KK. Drug Discov Today. 2008;13:1082–1089. doi: 10.1016/j.drudis.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Maas AI, Stocchetti N, Bullock R. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 4.Smith DH, Johnson VE, Stewart W. Nat Rev Neurol. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okie S. N Engl J Med. 2005;352:2043–2047. doi: 10.1056/NEJMp058102. [DOI] [PubMed] [Google Scholar]
- 6.Algattas H, Huang JH. Int J Mol Sci. 2014;15:309–341. doi: 10.3390/ijms15010309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blennow K, Hardy J, Zetterberg H. Neuron. 2012;76:886–899. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Kabadi SV, Faden AI. Int J Mol Sci. 2014;15:1216–1236. doi: 10.3390/ijms15011216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan GB, Vasterling JJ, Vedak PC. Behav Pharmacol. 2010;21:427–437. doi: 10.1097/FBP.0b013e32833d8bc9. [DOI] [PubMed] [Google Scholar]
- 10.Waterhouse EG, Xu B. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- 12.Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar A, Loane DJ. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Hashmi JT, Huang YY, Osmani BZ, Sharma SK, Naeser MA, Hamblin MR. PM R. 2010;2:S292–S305. doi: 10.1016/j.pmrj.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang YY, Gupta A, Vecchio D, de Arce VJ, Huang SF, Xuan W, Hamblin MR. J Biophotonics. 2012;5:827–837. doi: 10.1002/jbio.201200077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Migliario M, Pittarella P, Fanuli M, Rizzi M, Reno F. Lasers Med Sci. 2014;4:1463–1467. doi: 10.1007/s10103-014-1556-x. [DOI] [PubMed] [Google Scholar]
- 17.Avci P, Nyame TT, Gupta GK, Sadasivam M, Hamblin MR. Lasers Surg Med. 2013;45:349–357. doi: 10.1002/lsm.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avci P, Gupta GK, Clark J, Wikonkal N, Hamblin MR. Lasers Surg Med. 2014;46:144–151. doi: 10.1002/lsm.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang YY, Nagata K, Tedford CE, Hamblin MR. J Biophotonics. 2013;8:656–664. doi: 10.1002/jbio.201300125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ando T, Xuan W, Xu T, Dai T, Sharma SK, Kharkwal GB, Huang YY, Wu Q, Whalen MJ, Sato S, Obara M, Hamblin MR. PLoS One. 2011;6:e26212. doi: 10.1371/journal.pone.0026212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Q, Xuan W, Ando T, Xu T, Huang L, Huang YY, Dai T, Dhital S, Sharma SK, Whalen MJ, Hamblin MR. Lasers Surg Med. 2012;44:218–226. doi: 10.1002/lsm.22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xuan W, Vatansever F, Huang L, Wu Q, Xuan Y, Dai T, Ando T, Xu T, Huang YY, Hamblin MR. PLoS One. 2013;8:e53454. doi: 10.1371/journal.pone.0053454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khuman J, Zhang J, Park J, Carroll JD, Donahoe C, Whalen MJ. J Neurotrauma. 2011;2:408–417. doi: 10.1089/neu.2010.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oron A, Oron U, Streeter J, de Taboada L, Alexandrovich A, Trembovler V, Shohami E. J Neurotrauma. 2007;24:651–656. doi: 10.1089/neu.2006.0198. [DOI] [PubMed] [Google Scholar]
- 25.Oron A, Oron U, Streeter L, Detaboada L, Alexandrovich A, Trembovle V, Shohami E. J Neurotrauma. 2012;29:401–407. doi: 10.1089/neu.2011.2062. [DOI] [PubMed] [Google Scholar]
- 26.Quirk BJ, Torbey M, Buchmann E, Verma S, Whelan HT. Photomed Laser Surg. 2012;30:523–529. doi: 10.1089/pho.2012.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen A, Xiong LJ, Tong Y, Mao M. Biomed Rep. 2013;1:167–176. doi: 10.3892/br.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naeser MA, Saltmarche A, Krengel MH, Hamblin MR, Knight JA. Photomed Laser Surg. 2011;29:351–358. doi: 10.1089/pho.2010.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Streeter J, De Taboada L, Oron U. Mitochondrion. 2004;4:569–576. doi: 10.1016/j.mito.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 30.Oron A, Oron U, Chen J, Eilam A, Zhang C, Sadeh M, Lampl Y, Streeter J, DeTaboada L, Chopp M. Stroke. 2006;37:2620–2624. doi: 10.1161/01.STR.0000242775.14642.b8. [DOI] [PubMed] [Google Scholar]
- 31.Khuman J, Zhang J, Park J, Carroll JD, Donahue C, Whalen MJ. J Neurotrauma. 2012;29:408–417. doi: 10.1089/neu.2010.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barker JM, Boonstra R, Wojtowicz JM. Eur J Neurosci. 2011;34:963–977. doi: 10.1111/j.1460-9568.2011.07823.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhang RL, Zhang ZG, Chopp M. Neuropharmacology. 2008;55:345–352. doi: 10.1016/j.neuropharm.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parent JM. Neuroscientist. 2003;9:261–272. doi: 10.1177/1073858403252680. [DOI] [PubMed] [Google Scholar]
- 35.Kokaia Z, Thored P, Arvidsson A, Lindvall O. Cerebral cortex. 2006;16:i162–i167. doi: 10.1093/cercor/bhj174. [DOI] [PubMed] [Google Scholar]
- 36.Ambrogini P, Lattanzi D, Ciuffoli S, Betti M, Fanelli M, Cuppini R. Brain Res. 2013;1534:1–12. doi: 10.1016/j.brainres.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 37.Ferreira AF, Real CC, Rodrigues AC, Alves AS, Britto LR. Brain Res. 2011;1425:111–122. doi: 10.1016/j.brainres.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Vaynman SS, Ying Z, Yin D, Gomez-Pinilla F. Brain Res. 2006;1070:124–130. doi: 10.1016/j.brainres.2005.11.062. [DOI] [PubMed] [Google Scholar]
- 39.Meng C, He Z, Xing D. J Neurosci. 2013;33:13505–13517. doi: 10.1523/JNEUROSCI.0918-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomes LE, Dalmarco EM, Andre ES. Photo-med Laser Surg. 2012;30:642–647. doi: 10.1089/pho.2012.3242. [DOI] [PubMed] [Google Scholar]
- 41.Huisa BN, Stemer AB, Walker MG, Rapp K, Meyer BC, Zivin JA NEST-3 investigators. Nest, Int J Stroke. 2013;8:315–320. doi: 10.1111/j.1747-4949.2011.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zivin JA, Sehra R, Shoshoo A, Albers GW, Bornstein NM, Dahlof B, Kasner SE, Howard G, Shuaib A, Streeter J, Richieri SP, Hacke W NEST-3 investigators. Nest, Int J Stroke. 2012 doi: 10.1111/j.1747-4949. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.