

Abstract
Metagenomic approach was used to understand the structural and functional diversity present in arsenic contaminated groundwater of the Ganges Brahmaputra Delta aquifer system. A metagene dataset (coded as TTGW1) of 89,171 sequences (totaling 125,449,864 base pairs) with an average length of 1406 bps was annotated. About 74,478 sequences containing 101,948 predicted protein coding regions passed the quality control. Taxonomical classification revealed abundance of bacteria that accounted for 98.3% of the microbial population of the metagenome. Eukaryota had an abundance of 1.1% followed by archea that showed 0.4% abundance. In phylum based classification, Proteobacteria was dominant (62.6%) followed by Bacteroidetes (11.7%), Planctomycetes (7.7%), Verrucomicrobia (5.6%), Actinobacteria (3.7%) and Firmicutes (1.9%). The Clusters of Orthologous Groups (COGs) analysis indicated that the protein regulating the metabolic functions constituted a high percentage (18,199 reads; 39.3%) of the whole metagenome followed by the proteins regulating the cellular processes (22.3%). About 0.07% sequences of the whole metagenome were related to genes coding for arsenic resistant mechanisms. Nearly 50% sequences of these coded for the arsenate reductase enzyme (EC. 1.20.4.1), the dominant enzyme of ars operon. Proteins associated with iron acquisition and metabolism were coded by 2% of the metagenome as revealed through SEED analysis. Our study reveals the microbial diversity and provides an insight into the functional aspect of the genes that might play crucial role in arsenic geocycle in contaminated ground water of Assam.
Keywords: Metagenomic, Arsenic, Assam, Groundwater, Bacteria, Proteobacteria, Siderophore
1. Introduction
Arsenic toxicity in drinking water is a serious human health concern affecting millions of people around the globe. The problem is especially acute in the Ganges-Brahmaputra Delta (GBD) region of India where geogenic groundwater arsenic concentration has been reported to be more than 50 fold higher than standard WHO limit [1]. There are strong evidences to suggest that microorganisms play crucial role in mobilizing arsenic in the groundwater through cascade of reduction and oxidation reactions. The secretion of siderophore by some bacteria affects this process by releasing the primary iron bound arsenic from the sediments. A detail insight of the microbial communities controlling the bio-geochemical cycle of arsenic in the natural system is challenging due to their extreme diversity and uncultivated status (Fig. 1). Metagenomic analysis has offered an unprecedented opportunity to examine the response and adaptation strategies of the microbial communities to the environmental toxicity [2], [3], [4]. Studies on microbial communities from several environments viz., acid-mine drainage [5], marine water and sediments [6], [7] including arsenic contaminated soils [8], [9] have provided novel insights on the microbial community structure their function along with evolution pattern and have led to the discovery of novel gene.
Fig. 1.

Analysis strategy performed to analyze microbial diversity prevalent in the arsenic contaminated groundwater sample. DNA from composite groundwater sample was used for Whole Genome Shotgun (WGS) sequencing.
Arsenic contamination in groundwater of Assam, a north-eastern state of India was first reported in 2004 [10]. Since then studies indicating alarming increase in the arsenic content in the ground water of several districts in the state has been reported. Several sites (Titabor, Dhakgorah, Seleng-hat and Moriani) in the district of Jorhat of Assam have presence of very high arsenic content (194–657 g/μl) in the groundwater [1], [11]. The level of arsenic in these localities is far above the WHO and BIS approved guidelines of 10 μg/l and 50 μg/μl respectively [12], [13]. Such highly contaminated sites offer unique opportunity to investigate the role of microorganisms in arsenic geogenic cycle and its mobilization.
In this paper we report the microbial community structure and their function in a highly arsenic contaminated groundwater as revealed through shotgun sequencing method. The metagenomic library generated from our study also predicts of the roles of these microbes in arsenic geocycle.
2. Materials and methods
2.1. Ethics statement
No specific permits were required for the described field studies.
2.2. Sampling
Groundwater samples were collected from 5 different sites of Tanti Gaon, Titabor subdivision, Jorhat district (27°57″N, 94°16″E). All the samples were collected in sterile acid-washed Nalgene water bottles. Before collecting the water samples, hand-held tube-wells were pumped for 20 min to remove any unwanted residues present in the tube. Sampling was performed during November 2014. On field chemical parameters (pH and arsenic concentration) of the collected were recorded using portable pH meter (Spectronic Camspec Ltd., UK) and Arsenic Testing Kit (Merck, Germany) respectively. Samples were carried to the laboratory on ice packs and stored for further analyses using standard procedures. Concentration of arsenic was determined by atomic absorption spectrophotometer using protocol as described by Behari and Prakash [14]. Physicochemical parameters of the samples are presented in Table 1.
Table 1.
Physicochemical parameter of the contaminated groundwater sample collected for metagenomics analysis.
| Sl. no. | Parameter | Ground-water sample 1 | Ground-water sample 2 | Ground-water sample 3 | Ground-water sample 4 | Ground-water sample 5 |
|---|---|---|---|---|---|---|
| 1. | pH | 6.4 | 6.2 | 7.1 | 5.9 | 6.8 |
| 2. | Electrolytic conductivity (μS/m) | 1783 | 1532 | 1572 | 1770 | 1814 |
| 3. | Temperature (°C) | 22.0 | 24.0 | 22.0 | 21.6 | 22.0 |
| 4. | Dissolved oxygen (mg/l) | 8.4 | 7.8 | 7.6 | 8.2 | 8.7 |
| 5. | Redox (mv) | 187 | 172 | 167 | 183 | 181 |
| 6. | Arsenic concentration (μg/l) | 217 | 50 | 20 | 156 | 112 |
2.3. DNA extraction from contaminated water sample
Aliquots of 10 ml of water samples collected from 5 locations were thoroughly mixed to generate a 50 ml volume and considered as a composite sample for further analysis. The DNA was extracted from the filtrate using PowerWater® DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA) in accordance with manufacturer's instructions. Extracted DNA was quantified by DNA (dsDNA)-binding dye assay on the Qubit Fluorometer which has a detection limit of as low as dsDNA at 10–100 pg/μl [15].
2.4. Preparation of 2 × 300 MiSeq libraries
A total of 3.0 μg of environmental DNA was extracted from the sample from which, 1.0 μg was subjected to restriction digestion and library construction The paired-end sequencing library was prepared using Illumina TruSeq DNA Library Preparation Kit, initiated with the fragmentation of 1.0 μg gDNA followed by paired-end adapter ligation. The ligated product was purified using 1 × Ampure beads and elution of ~ 500–800 bp to further PCR amplify as described in the kit protocol. The amplified library was analyzed in Bioanalyzer 2100 (Agilent Technologies) using High Sensitivity (HS) DNA chip as per the manufacturer's instructions.
2.5. Cluster generation and sequencing
Based on the data obtained from the Qubit concentration for the library and the mean peak size (708 bp) from Bioanalyzer profile, 10 pM of the library was loaded onto Illumina MiSeq for cluster generation and sequencing. Paired-end sequencing allows the template fragments to be sequenced in both the forward and reverse directions on MiSeq. High-quality metagenome reads were assembled using CLC workbench (CLC bio, Denmark) with default parameter (minimum contig length: 200) for trimming and de novo assembly [16].
2.6. MG-RAST analysis
The MG-RAST portal offers automated quality control, annotation, comparative analysis and archiving services. The uploaded data is usually preprocessed through SolexaQA [17], to trim low-quality regions from FASTQ data. More than two standard deviations away from the mean read length are discarded [18]. A simple k-mer approach is used to rapidly identify all 20 character prefix identical sequences. This step is required in order to remove Artificial Duplicate Reads (ADRs) [19]. The set of ADRs is kept aside to be analyzed by DRISEE (Duplicate Read Inferred Sequencing Error Estimation) [20], in order to determine the degree of variation among prefix-identical sequences derived from the same template. The MG-RAST pipeline also provides the option of removing reads that are near-exact matches to the genomes of a handful of model organisms, including fly, mouse, cow, and human. The screening stage uses Bowtie [21] (a fast, memory-efficient, short read aligner), and only reads that do not match the model organisms pass into the next stage of the annotation pipeline.
Refined and annotated reads were distributed into different categories with rRNA reads, protein reads (both known and unknown functions), and unknown reads based on the similarity search result as compared with rRNA and protein database. Taxonomic analyses were performed using SILVA small subunit (SSU) database. It is used as an annotation source for 16S rRNA read with an e-value cut-off of 1e− 5, minimum identity cut-off of 60% and minimum alignment length cut-off of 15aa. SEED subsystem and KEGG database were used for functional analysis of the sample. Similarity search between the proteins reads and the SEED/KEGG database was done by using maximum e-value cut-off of 1e− 5, minimum identity cut-off of 60% and minimum alignment length cut-off of 15aa. The annotated reads were stored in three subsystem levels viz., Level 1–Level 3 (Level-1: highest level consisting of similarity search plot, LCA Plot; Level-2: KEGG Pathway and Level-3: functional annotation with characteristics features of proteins).
2.7. Functional annotation and domain information analysis
The putative ORFs were identified and their corresponding sequences were subjected to BLAST against the M5NR (non-redundant protein database) in the MG-RAST server to annotate their function. The M5NR is an integrated database containing the NCBI GenBank, Clusters of Orthologous Groups (COGs), Kyoto Encyclopedia of Genes and Genomes (KEGG) and SEED in a single searchable database [22].
2.8. Taxonomic classification
Taxonomic classification was conducted through BLASTN analysis against SILVA, SSUref and LSUref databases with an e-value of 1e− 5 [23] followed by annotation of BLAST output files using MEGAN [24]. This was performed by the lowest common ancestor algorithm that assigns rDNA or rRNA sequences to the lowest common ancestor in the taxonomy from a subset of best scoring matches in the BLAST result (cut-off: BLAST bit score 86, relative cut-off: 10% of the top hit) using MEGAN according to these cut-offs to select hit reads for annotation [23]. Random sequence reads exhibit very different levels of evolutionary conservation. Therefore, it is important to make use of all ranks of the NCBI taxonomy, placing more conserved sequences higher up in the taxonomy (i.e. closer to the root) and more distinct sequences onto nodes that are more specific (i.e. closer to the leaves, which represent species and strains).
2.9. Statistical analysis
Fisher's exact test [25], available from the Cenargen Bioinformatics platform, was used to compare and find out the levels and significance of contig expression between generated libraries that had passed through quality control.
3. Results
3.1. Sample description
Five randomly collected samples from Tanti Gaon, Titabor (GPS 26.58.101, 94.16.391) had arsenic concentration of 217 μg/l, 50 μg/l, 20 μg/l, 156 μg/l and 112 μg/l; and pH values of 6.4, 6.2, 7.1, 5.9 and 6.8 respectively. Electrolytic conductivities of the samples were found to be 1783, 1532, 1572, 1770 and 1814 μS/m respectively. Redox potentials of the samples were recorded to be 187, 172, 167,183 and 181 mv respectively (Table. 1).
3.2. Nucleotide sequence accession number
The extracted gDNA was sequenced with Miseq shotgun sequencing method. Refined reads were annotated by MG-RAST online server (version 3.5) and submitted to the MG-RAST Database for further references (URL: http://metagenomics.anl.gov/mgmain.html?mgpage=project&project=cb26695b0b6d67703132303338) (Date of Submission: 01–06-2015). The metagenome dataset was entitled as Arsenic_Contaminated_Groundwater (TTGW1).
3.3. Metagenome
The dataset of TTGW1 containing 89,171 sequences (totaling 125,449,864 base pairs) with an average length of 1406 bp was uploaded to the MG-RAST server. Out of these 89,171 sequences, 14,693 failed to pass through quality control. There were no de-replications as identified through Artificial Duplicate Reads (ADRs). The remaining 74,478 sequences that passed through the quality control step contained 101,948 predicted protein coding regions. Of these 101,948 predicted protein features, 66,248 (65.0% of features) were assigned an annotation using protein databases (M5NR). Rest of the 35,700 (35.0% of features) sequences had no significant similarities to the protein databases. Functional categories were assigned to 79.5% of annotated features as presented in Table 2, Fig. 2.
Table 2.
Statistical analysis of the raw and processed sequences of the metagenome.
| Raw data uploaded | |
|---|---|
| Number of base pair uploaded | 125,449,864 bp |
| Coding sequence count | 89,171 |
| Mean sequence length | 1406 ± 6901 |
| Mean GC percent | 58 ± 10% |
| Post quality control analyses | |
|---|---|
| Number of base pair which passed the QC | 66,516,956 |
| Coding sequence count after QC | 74,478 |
| Mean sequence length | 893 ± 1013 bp |
| Mean GC percent | 58 ± 10% |
| Processed sequences | |
|---|---|
| Predicted Protein features | 101,948 |
| Predicted rRNA feature | 101,948 |
| Post-alignment and BLAST tool analyses | |
|---|---|
| Identified Protein feature | 146 |
| Identified rRNA features | 40 |
| Identified functional categories | 52,689 |
Fig. 2.
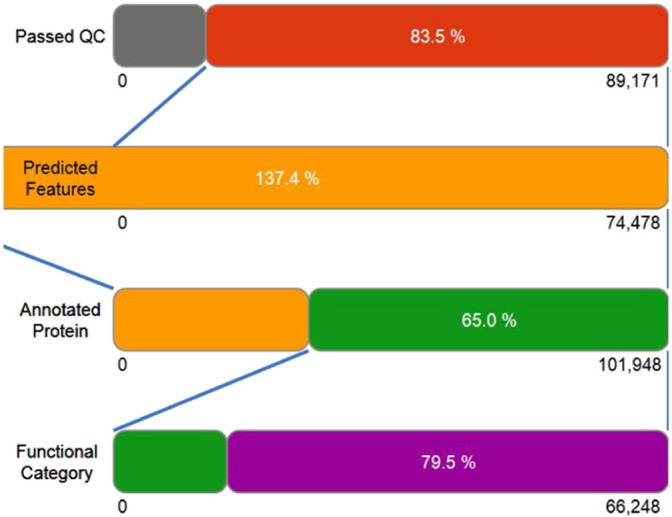
Graphical representation of sequence analysis chart.
3.4. Microbial community structure
Database search with the MG-RAST server provided an insight into microbial community structure of the arsenic contaminated groundwater sample collected from Titabor. Bacteria was the most abundant among the three domains and accounted for 98.3% of the microbial population of the metagenome. Eukaryota had an abundance of 1.1% while archea had 0.4% of abundance. Among the bacteria, proteobacteria were the most abundant (62.6%) followed by alphaproteobacteria (30.1%), betaproteobacteria (19%), deltaproteobacteria (2.8%), gammaproteobacteria (10.4%), bacteroidetes (11.7%), planctomycetes (7.7%), verrucomicrobia (5.6%), actinobacteria (3.7%) and firmicutes (1.9%). other phyla like acidobacteria, asomycota, clamydiae, chlorobi, chloroflexi and chordata constituted 0.5–3% of the whole metagenome (Fig. 3). Nitrosomonas was found to be the most predominant genus with 3298 hits followed by Pirellua (3223), Verrucomicrobium (2892), Methylobacterium (2729), Rhodopirellula (2431), Burkholderia (2171), Bradyrhizobium (2067) and Methylocystis (1822). Bacteria like Bacillus, Clostridium, Chryseobacterium, Cytophaga, Caulobacter, Flavobacterium, Granulibacter, Arthrobacter, Beijerinckia etc. were the other members as revealed from the metagenome.
Fig. 3.

Graphical representation of taxon abundance. Proteobacteria showed the highest population in the whole metagenome followed by Bacteroidetes, Planctomycetes, Verucomicrobia, Actinobacteria, and Firmcutes etc.
3.5. Rarefaction curve
Rarefaction allows the calculation of species richness for a given number of individual samples through the generation of rare faction curves. The curves represent the average number of different species annotations for subsamples of the complete dataset. Fig. 4 depicts the rarefaction curve of annotated species richness in the metagenome. This curve is a plot of the total number of distinct species annotations as a function of the number of sequences sampled. The steep slope on the left indicated that a large fraction of the species diversity existed and the curve tapering to the right meant that a reasonable number of individuals were sampled; more intensive sampling is likely to yield few additional species. Initially, the sampling curve rose very rapidly but gradually leveled off indicating fewer new species per unit of individuals collected.
Fig. 4.

Refrection curve of species richness.
3.6. Metabolic potential and resistance mechanism of the microorganisms
3.6.1. COG analysis
The Cluster of Orthologous Groups of protein (COG) analysis is a phylogenetic classification of the protein encoded by bacteria, archaea, and eukaryotes. The COGs were constructed by applying the criterion of genome-specific best hits to the results of an exhaustive comparison of all protein sequence from the metagenome. The bar diagram (Fig. 5) illustrates the distribution of functional categories at the highest level supported by the functional hierarchies. Each bar indicated the percentage of reads with predicted protein functions annotated to the category for the given source. Results below indicated that the protein regulating the metabolic functions were higher in number which constituted about 39.3% of the whole metagenome (18,199 reads) followed by the proteins regulating the cellular processes which were 22.3% of the metagenome with 10,333 hits. Proteins responsible for signaling, information storage and processing constituted about 17.1% of the total metagenome with 7928 hits; 21.3% of the metagenome were the poorly characterized proteins which needed further refinement for the analysis.
Fig. 5.

Cluster based orthologous classification of proteins.
3.6.2. Subsystem classification
SEED subsystem annotation indicated the greatest abundance (14%) of clustering based proteins involved in the different metabolic pathways responsible for complex genome structure and phenotypic expression. Biological integration with metagenome sequence of the clustering based subsystem showed the presence of functional domains like fatty acid metabolic cluster, enzymes for biosynthesis of galactoglycans and related lipopolysaccharides, RNA polymerase associates, the protein responsible for cytochrome biogenesis respectively. Enzymes responsible for carbohydrate metabolism were expressed by 12% of the metagene sequences. About 10% of the coding sequences were for different amino acid and their derivatives, 6% of it were involved with protein metabolism. Sub-categorization this 6% metabolic protein, revealed that 57% proteins were involved with biosynthesis, 17% with protein degradation, 13% with post-transcriptional processing, 8% with protein folding and 4% were the ORFs coding for selenoproteins. The ORFs for nitrogen metabolism (0.9%), phosphorus metabolism (1%), iron acquisition and metabolism (2%), sulfur metabolism (1%) and arsenic metabolism (0.07%) were also recorded (Fig. 6).
Fig. 6.

Functional prediction of annotated proteins.
3.6.3. Iron acquisition and metabolism
Acquisition of iron through scavenging from arsenopyrite ores of the sediments plays a major role in controlling the geocycle of arsenic. The metagenomics sequence search and SEED analysis revealed that the proteins involved with iron acquisition and metabolism occupied 2% of the metagenome. Sub-categorization of the proteins responsible for iron metabolism indicated that 45% of the ORFs had similarity with iron acquisition proteins of Vibrio. The tonB like receptors (31%) were predominant followed by iron transportation proteins (15%). Apart from these, 10% of the ORFs were associated with proteins for iron metabolism in Campylobacter, 9% with the hemin uptake and utilization system of Gram's reaction negative bacteria, 7% with Hemin transportation system and 5% with siderophore activity. Rest 9% sequences showed identity with iron acquisition system of Streptococcus, hemin uptake and utilization systems in Gram's reaction positive bacteria, iron scavenging clusters as found in Thermos and ABC type iron transporter system respectively (Fig.7).
Fig. 7.

Graphical representation of genes identified in the metagenome responsible for iron acquisition and siderophore activity (Krona Chart).
In domain based functional annotation, it was found that 0.08% of the total protein sequences of the whole metagenome and 5% of the total proteins responsible for iron acquisition were the proteins associated with siderophore activity. In the siderophore oriented sequences, 46% of the sequence showed similarity with the siderophore assembly subunit i.e. siderophore synthetase AsbS [26]; 27% of the sequence showed best hit classification with pyoverdine, which are generally the fluorescent siderophore produced by the members of the pseudomonaceae family like Azotobacter, Azomonas, Pseudomonas and Rhizobacter [27]; 13% were for siderophore regulating receptor system and rest 15% showed identities with the yersiniabactin (siderophore produced by the pathogenic bacteria Yersinia pestis, Yersinia pseudotuberculosis and Yersininaenterocolitica) [28]; aerobactin (siderophore produced by E. coli) [29]; bacillibactin (siderophore produced by the genus Bacillus) [30]; achromobactin (siderophore produced by Pseudomonas syringe) [31]; enterobactin and pyochelin (siderophore produced by Pseudomonas aeruginosa) [32] respectively.
3.6.4. Arsenic resistance mechanism
About 0.07% sequences of the whole metagenome were involved in arsenic resistance mechanism of which, 50% of the sequences were associated with coding for Arsenate reductase enzyme; 26% coded for arsenic efflux mechanism or arsenic pump-driving ATPase i.e. ArsB-ArsA complex (complex responsible for arsenite extrusion from the bacterial cellular system); 12% of the sequences showed best hit with arsenical resistance protein ACR3 (a homologous efflux protein like ArsB) [33]. Remaining 12% sequence showed similarity with arsenical resistance operon transacting-repressor ArsD, arsenical resistance operon represses ArsR, arsenical resistance protein ArsH and arsenical efflux protein pump respectively (Fig. 8).
Fig. 8.

Genes involved in arsenic resistance mechanisms identified from metagenome.
4. Discussion
Metagenomic analysis can provide reliable data on the phylogenetic composition and microbial metabolism along with functional genes related to the metabolism of metalloids [34]. So far, we know very little about the microflora of arsenic contaminated aquifers that controls the mobilization of arsenic in the groundwater system of Assam. In this study, comparison of the abundance of 16S rRNA gene in the metagenome with the SILVIA dataset search revealed bacteria (98.3%) to be the most abundant domain followed by eukaryota (1.1%) and archea (0.4%). Although, the presence of archea is ubiquitous and universal in natural surroundings, high arsenic content can restrict their prevelance due to their sensitivity or lack of an ‘Ars’ detoxification systems. Low abundance (0.4%) of archea was earlier reported by Layton et al. [9] in arsenic contaminated surface and well water of Bangladesh. Previous work of Luo et al., [8] have also reported absence of archea in arsenic contaminated samples of Lengshuijiang City, Hunan Province, China. Reduction in archaeal community has been reported from impacted soil. A study by Urakawa et al., [35] reported reduced representation of the archaeal sequences to 2.7% from an initial 6% in crude oil contaminated soil.
Classification at phylum level revealed that proteobacteria (62.6%) had the highest abundance with alphaproteobacteria (30.1%), betaproteobacteria (19%), deltaproteobacteria (2.8%) and gammaproteobacteria (10.4%). The presence of epsilonproteobacteria and zetaproteobacteria in the metagenome was not observed. The abundance of proteobacteria can be correlated with their ability to survive in metal contaminated stressed environments [36]. Earlier, Sheik et al., [37] reported proteobacteria as the dominant phylum in arsenic and chromium contaminated soils. Within the proteobacteria, alphaproteobacteria or gammaproteobacteria were the most abundant classes in all soils. Bacteroidetes (11.7%), planctomycetes (7.7%) and verucomicrobia (7.7%) were other groups of bacteria recorded in the metagenome. Under the phylum bacteroidetes, the class cytophagia had 37% abundance while 25% abundance to the class flavobacteria, 25% to sphingobacteria and 9% to the class bacteroidia. Classification at genus level showed that Nitrosomonas occupied the most dominant position followed by Pirellula, Verucomicrobium, Methylobacterium and Rhodopirellua. The dominance of Nitrosomonas in contaminated water system has also been reported by Ivanova et al., [38] and White et al. [39]. Pirellula is a marine planctomycetes bacterium and has a long history of relationship with aerobic and anoxic wastewater system [40]. The complete genome sequencing of Pirellula sp. strain1 had revealed the presence of arsenate reductase gene in the whole genome along with both ArsA dependent ArsB arsenite transporter system and ArsR protein [41].
Functional and hierarchical classification of the metagenome from this study revealed that out of 66,248 identified proteins, 0.07% proteins were involved in arsenic metabolism. Of the 0.07% arsenic metabolizing proteins, 50% were arsenate reductase, which is the functional protein of Ars operon responsible for reduction of arsenate to arsenite. About 26% were ArsA dependent ArsB arsenite transporter complex; ArsA being the ATPase enzyme which provides the energy required to efflux the arsenite by ArsB permease protein. In the rest of the 24% of arsenite metabolizing proteins, ACR3, a homolog to ArsB (Arsenite Permease) protein had a total of 12% abundance. The ACR3 confers resistance to both prokaryotes and eukaryotes unlike ArsB which only confers resistance to prokaryotes [42]. The ACR3 has also been reported in Saccharomyces cerevisiae [43]. Rests of the 12% were occupied by ArsD, ArsR and ArsH protein. ArsD is metallochaperone which transfers the arsenite molecule to ArsA-ArsB efflux pump to extrude [44]. ArsR is the regulatory protein, which acts as a repressor for the arsRDABC operon when there is no arsenic in the cellular system; but in the presence of arsenic ArsR, dislocates from the operon and facilitates expression of the structural genes [45]. Both ArsR and ArsD function as regulatory protein in five gene ars operonic system [46]. In addition to this, ArsH protein which was identified from the metagenome is also homologous to arsenic regulatory protein ArsR reported in Yersinia enterocolitica and in Acidothibacillus ferroxidans [47]. Genes related to arsenite oxidation were not detected indicating that this state of arsenic may not be present in the parental material. This observation is supported by the physiochemical nature of groundwater of the aquifers of Brahmaputra Delta-Plain (BDP) which is mostly contaminated by arsenate. Groundwater samples from different geomorphological units of the Brahmaputra river and its tributaries are generally of Holocene and Pleistocene in origin where reductive dissolution of (Fe-Mn)OH mechanism is dominant [48]. Groundwater's of BDP represents a characteristic nature of abundant HCO3−, low to moderate Dissolved Oxygen (DO) and lack of sufficient soluble nutrients with more or less neutral pH [49].
Microorganisms are known for their ability to produce different biogenic chelating agents like siderophore in iron limiting environment. Siderophore solubilizes the ferric iron in the iron-starved environment and transports the Fe+ 3 into the cell and helps microbial growth in an environment where iron is the limiting factor the [50], [51]. Iron and arsenic have an inter relationship in maintaining an equilibrium in arsenopyrite. The ability of siderophore-producing bacteria in solubilizing the Arsenic from minerals like FeAsO4, FeAsS was reported earlier by Ghosh et al. [52] who reasoned that siderophore serves as a major factor for mobilizing the sediment bound arsenic in the surrounding milieu. The iron acquisition is one of the major dynamics which controls the geocycle of arsenic by scavenging the iron from arsenopyrite ores of the sediments. In the metagenome, 2% of the ORF's contained genes involved in iron acquisition and metabolism. Dominant genes of iron acquisition were tonB like receptors (31%), which are bacterial outer membrane protein that transport the siderophores in an energy dependent manner in the form of proton motive force. Few genes showed similarities with the genes expressing the proteins viz., yersiniabactin, aerobactin, bacillibactin, achromobactin, enterobactin and pyochelin which are integral parts of hemin uptake and utilization systems of gram negative bacteria and gram positive bacteria.
5. Conclusion
This study was undertaken to gain an insight into the microbial diversity structure and their activity in the arsenic contaminated groundwater of the Jorhat district of Assam located within the Ganges-Brahmaputra Delta aquifer system. Metagenome analyses revealed the dominance of bacteria over other the domains in the contaminated site. The metagenomic library generated showed high abundance of genes coding for products related to arsenic resistance metabolism. A considerable amount of sequences (0.07%) were identified to be associated with the genes for arsenate reductase enzyme, arsenic efflux mechanism or arsenic pump-driving ATPase i.e. ArsB-ArsA complex (complex responsible for arsenite extrusion from the bacterial cellular system); arsenical resistance protein ACR3 (a homologous efflux protein like ArsB). Another portion of these sequences showed identity with arsenical resistance operon transacting-repressor ArsD, Arsenical resistance operon represses ArsR, arsenical resistance protein ArsH and Arsenical efflux protein pump respectively. The metagenome also contained high percentage (2%) of iron acquisition and metabolizing contigs coding for different types of siderophores that help the bacteria to acquire iron from the arsenopyrite mineral releasing the arsenic which enters the environment. High abundance of arsenic resistance and mobilization genes in the metagenome indicated active involvement of the microorganisms in mobilization of the metalloid in groundwater. The results of the analysis indicate that bacteria harboring genes related to arsenic metabolism play an active role in the arsenic geocycle and mobilize the metalloid in groundwater of the Jorhat district of Assam.
Acknowledgment
Authors are grateful to the Head, Department of Agricultural Biotechnology, Assam Agricultural University and Director, Centre for Biotechnology and Bioinformatics, Dibrugarh University for providing the facilities to carry out the research work. The first author SD, gratefully acknowledges the financial assistance received from UGC (F1-17.1/2011-12/RGNF-SC-ASS-10152) in the form of Rajiv Gandhi Fellowship.
References
- 1.Das S., Bora S.S., Lahan J.P., Barooah M., Yadav R.N.S., Chetia M. Groundwater arsenic contamination in north eastern states of India. J. Environ. Res. Develop. 2015;9:621. [Google Scholar]
- 2.Riesenfeld C.S., Schloss P.D., Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu. Rev. Genet. 2004;38:525–552. doi: 10.1146/annurev.genet.38.072902.091216. [DOI] [PubMed] [Google Scholar]
- 3.Handelsman J., Tiedje J., Alvarez-Cohen L. The new science of metagenomics: revealing the secrets of our microbial planet. Nat. Res. Counc. Rep. 2007;13:47–84. [Google Scholar]
- 4.He J.Z., Shen J.P., Zhang L.M. Quantitative analyses of the abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea of a Chinese upland red soil under long-term fertilization practices. Environ. Microbiol. 2007;9:2364–2374. doi: 10.1111/j.1462-2920.2007.01358.x. [DOI] [PubMed] [Google Scholar]
- 5.Tyson G.W., Chapman J., Hugenholtz P. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 6.DeLong E.F., Preston C.M., Mincer T. Community genomics among stratified microbial assemblages in the ocean's interior. Science. 2006;311:496–503. doi: 10.1126/science.1120250. [DOI] [PubMed] [Google Scholar]
- 7.Yooseph S., Nealson K.H., Rusch D.B. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature. 2010;468:60–66. doi: 10.1038/nature09530. [DOI] [PubMed] [Google Scholar]
- 8.Luo J., Bai Y., Liang J., Qu J. Metagenomic approach reveals variation of microbes with arsenic and antimony metabolism genes from highly contaminated soil. PLoS One. 2014;9 doi: 10.1371/journal.pone.0108185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Layton A.C., Chauhan A., Williams D.E. Metagenomes of microbial communities in arsenic-and pathogen-contaminated well and surface water from Bangladesh. Genome Announc. 2014;2 doi: 10.1128/genomeA.01170-14. (e01170-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh A.K. Arsenic contamination in groundwater of North Eastern India. In: Jain C.K., Trivedi R.C., Sharma K.D., editors. Hydrology with Focal Theme on Water Quality. Allied Publishers; New Delhi: 2004. pp. 255–262. [Google Scholar]
- 11.Thambidurai P., Chandrashekhar A.K., Chandrasekharam D. Geochemical signature of arsenic-contaminated groundwater in Barak Valley (Assam) and surrounding areas, northeastern India. Procedia Earth Planet. Sci. 2013;7:834–837. [Google Scholar]
- 12.World Health Organization (WHO) second ed. World Health Organization; Geneva: 1993. Guidelines for Drinking Water Quality Recommendations. [Google Scholar]
- 13.Bureau of Indian Standards (BIS) 1991. Indian Standard Specification for Drinking Water; pp. 2–4. (IS 10500) [Google Scholar]
- 14.Behari J.R., Prakash R. Determination of total arsenic content in water by atomic absorption spectroscopy (AAS) using vapour generation assembly (VGA) Chemosphere. 2006;63:17–21. doi: 10.1016/j.chemosphere.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 15.Jiang W., Liang P., Wang B. Optimized DNA extraction and metagenomic sequencing of airborne microbial communities. Nat. Protoc. 2015;10:768–779. doi: 10.1038/nprot.2015.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mende D.R., Waller A.S., Sunagawa S. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS One. 2012;7:1–11. doi: 10.1371/journal.pone.0031386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox M.P., Peterson D.A., Biggs P.J. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinforma. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huse S.M., Huber J.A., Morrison H.G. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Alvarez V., Teal T.K., Schmidt T.M. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3:1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- 20.Keegan K.P., Trimble W.L., Wilkening J. A platform-independent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Comput. Biol. 2012;8 doi: 10.1371/journal.pcbi.1002541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer H.K., Pyke D.A. Defoliation effects on Bromus tectorum seed production: implications for grazing. Rangel. Ecol. Manag. 2008;61:116–123. [Google Scholar]
- 23.Urich T., Lanzén A., Qi J. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One. 2008;3:1–13. doi: 10.1371/journal.pone.0002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huson D.H., Auch A.F., Qi J., Schuster S.C. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher R.A. On the interpretation of χ2 from contingency tables, and the calculation of P. J. R. Stat. Soc. 1922;85:87–94. [Google Scholar]
- 26.Nusca T.D., Kim Y., Maltseva N. Functional and structural analysis of the siderophore synthetase AsbB through reconstitution of the petrobactin biosynthetic pathway from Bacillus anthracis. J. Biol. Chem. 2012;287:16058–16072. doi: 10.1074/jbc.M112.359349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menhart N., Thariath A., Viswanatha T. Characterization of the pyoverdines of Azotobacter vinelandii ATCC 12 837 with regard to heterogeneity. Biol. Met. 1991;4:223–232. doi: 10.1007/BF01141185. [DOI] [PubMed] [Google Scholar]
- 28.Perry R.D., Balbo P.B., Jones H.A. Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology. 1999;145:1181–1190. doi: 10.1099/13500872-145-5-1181. [DOI] [PubMed] [Google Scholar]
- 29.Johnson J.R., Moseley S.L., Roberts P.L., Stamm W.E. Aerobactin and other virulence factor genes among strains of Escherichia coli causing urosepsis: association with patient characteristics. Infect. Immun. 1988;56:405–412. doi: 10.1128/iai.56.2.405-412.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotta K., Kim C.Y., Fox D.T., Koppisch A.T. Siderophore-mediated iron acquisition in Bacillus anthracis and related strains. Microbiology. 2010;156:1918–1925. doi: 10.1099/mic.0.039404-0. [DOI] [PubMed] [Google Scholar]
- 31.Berti A.D., Thomas M.G. Analysis of achromobactin biosynthesis by Pseudomonas syringae pv. syringae B728a. J. Bacteriol. 2009;191:4594–4604. doi: 10.1128/JB.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandel J., Humbert N., Elhabiri M., Schalk I.J., Mislin G.L., Albrecht-Gary A.M. Pyochelin, a siderophore of Pseudomonas aeruginosa: physicochemical characterization of the iron (III), copper (II) and zinc (II) complexes. Dalton Trans. 2012;41:2820–2834. doi: 10.1039/c1dt11804h. [DOI] [PubMed] [Google Scholar]
- 33.Wang L., Jeon B., Sahin O., Zhang Q. Identification of an arsenic resistance and arsenic-sensing system in Campylobacter jejuni. Appl. Environ. Microbiol. 2009;75:5064–5073. doi: 10.1128/AEM.00149-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas T., Gilbert J., Meyer F. Metagenomics – a guide from sampling to data analysis. Microb. Inform. Exp. 2012;2:1–12. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urakawa H., Garcia J.C., Barreto P.D. A sensitive crude oil bioassay indicates that oil spills potentially induce a change of major nitrifying prokaryotes from the Archaea to the Bacteria. Environ. Pollut. 2012;164:42–45. doi: 10.1016/j.envpol.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Luo H., Csuros M., Hughes A.L., Moran M.A. Evolution of divergent life history strategies in marine alphaproteobacteria. MBio. 2013;4:373–413. doi: 10.1128/mBio.00373-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheik C.S., Mitchell T.W., Rizvi F.Z. Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanova I.A., Stephen J.R., Chang Y.J. A survey of 16S rRNA and amoA genes related to autotrophic ammonia-oxidizing bacteria of the beta-subdivision of the class proteobacteria in contaminated groundwater. Can. J. Microbiol. 2000;46:1012–1020. doi: 10.1139/w00-099. [DOI] [PubMed] [Google Scholar]
- 39.White C.P., Debry R.W., Lytle D.A. Microbial survey of a full-scale, biologically active filter for treatment of drinking water. Appl. Environ. Microbiol. 2012;78:6390–6394. doi: 10.1128/AEM.00308-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chouari R., Le Paslier D., Daegelen P. Molecular evidence for novel planctomycete diversity in a municipal wastewater treatment plant. Appl. Environ. Microbiol. 2003;69:7354–7363. doi: 10.1128/AEM.69.12.7354-7363.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glockner F.O., Kube M., Bauer M. Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc. Natl. Acad. Sci. U. S. A. 2003;100:8298–8303. doi: 10.1073/pnas.1431443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Indriolo E., Na G., Ellis D. A vacuolar arsenite transporter necessary for arsenic tolerance in the arsenic hyper-accumulating fern Pteris vittata is missing in flowering plants. Plant Cell. 2010;22:2045–2057. doi: 10.1105/tpc.109.069773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dziubinska M.E., Migocka M., Wysocki R. Acr3p is a plasma membrane antiporter that catalyzes As(III)/H + and Sb(III)/H + exchange in Saccharomyces cerevisiae. BBA-Biomembr. 2011;1808:1855–1859. doi: 10.1016/j.bbamem.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Yang J., Rawat S., Stemmler T.L., Rosen B.P. Arsenic binding and transfer by the ArsD As(III) metallochaperone. Biochemistry. 2010;49:3658–3666. doi: 10.1021/bi100026a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenstein R., Peschel A., Wieland B., Götz F. Expression and regulation of the antimonite, arsenite, and arsenate resistance operon of Staphylococcus xylosus plasmid pSX267. J. Bacteriol. 1992;174:3676–3683. doi: 10.1128/jb.174.11.3676-3683.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Rosen B.P. Metalloregulatory properties of the ArsD repressor. J. Biol. Chem. 1997;272:14257–14262. doi: 10.1074/jbc.272.22.14257. [DOI] [PubMed] [Google Scholar]
- 47.Maury L., Florencio F.J., Reyes J.C. Arsenic sensing and resistance system in the cyanobacterium Synechocystis sp. strain PCC 6803. J. Bacteriol. 2003;185:5363–5371. doi: 10.1128/JB.185.18.5363-5371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verma S., Mukherjee A., Choudhury R., Mahanta C. Brahmaputra river basin groundwater: solute distribution, chemical evolution and arsenic occurrences in different geomorphic settings. J. Hydrol. Reg. Stud. 2015;4:131–153. [Google Scholar]
- 49.Mahanta G., Enmark D., Nordborg O. Understanding distribution, hydrogeochemistry and mobilization mechanism of arsenic in groundwater in a low-industrialized homogeneous part of Brahmaputra river flood plain India. J. Hydrol. (Amst.) 2015;4:154–171. [Google Scholar]
- 50.Banejad H., Olyaie E. Arsenic toxicity in the irrigation water-soil plant system: a significant environmental problem. J. Am. Sci. 2011;7:125–131. [Google Scholar]
- 51.Nagoba B., Vedpathak D. Medical applications of siderophores. Eur. J. Gen. Med. 2011;8:229–235. [Google Scholar]
- 52.Ghosh P., Rathinasabapathi B., Teplitski M., Ma L.Q. Bacterial ability in AsIII oxidation and AsV reduction: relation to arsenic tolerance, P uptake, and siderophore production. Chemosphere. 2015;138:995–1000. doi: 10.1016/j.chemosphere.2014.12.046. [DOI] [PubMed] [Google Scholar]