

Abstract
The article by Nygaard and others (2016) proposes that applying batch correction approaches to microarray data from studies with unbalanced designs may inadvertently exaggerate the differences observed. In seeking to illustrate their point, Nygaard and others (2016) utilized a dataset (GSE61901) from a study we published (Towfic and others, 2014) and showed that one analysis pipeline utilizing the traditional approach to batch correction (ComBat) yielded over 1000 differentially expressed probesets, while an alternative approach proposed by Nygaard and others (2016). (utilizing batch as a fixed effect and averaging technical replicates) recovered 11 differentially expressed probesets.
The article by Nygaard and others (2016) proposes that applying batch correction approaches to microarray data from studies with unbalanced designs may inadvertently exaggerate the differences observed. In seeking to illustrate their point, Nygaard and others (2016) utilized a dataset (GSE61901) from a study we published (Towfic and others, 2014) and showed that one analysis pipeline utilizing the traditional approach to batch correction (ComBat) yielded over 1000 differentially expressed probesets, while an alternative approach proposed by Nygaard and others (2016; utilizing batch as a fixed effect and averaging technical replicates).
recovered 11 differentially expressed probesets.
While we agree with Nygaard and others (2016) premise that the utilization of ANOVA or empirical-Bayes derived approaches to correct for batch effects may in certain cases overestimate significance, the approach they propose is surprisingly sensitive to differences in how data are preprocessed, specifically the handling of technical replicates. We therefore sought to identify alternate methods that address the points raised by Nygaard and others (2016). These efforts yielded three alternative approaches for analyzing microarray data from studies with similar designs to ours, each of which adjusts for technical variation while avoiding the risks that Nygaard and others (2016) described:
(i) Utilize the technical replicates as a random-effect variable in LIMMA.
(ii) Utilize the technical replicate as a random variable in a repeated-measures ANOVA.
(iii) Utilize a linear mixed-effects model to account for the technical replicate as a random effect while accounting for treatment as a fixed effect.
Each of the aforementioned methodologies is readily accessible through R’s base or Bioconductor packages.
Nygaard and others (2016) referenced a study (GSE61901) we conducted (Towfic and others, 2014), in which we found significant and biologically relevant differences between the gene expression profiles induced by glatiramer acetate (GA, Copaxone), and the gene expression profiles induced by a generic GA (Glatimer , Natco Pharma, Ltd., Hyderabad, India) using Illumina
, Natco Pharma, Ltd., Hyderabad, India) using Illumina Mouse-WG6 microarray chips. Each WG6 chip contains six microarrays, and each of the microarrays containing two physical strips that are to be treated as technical replicates (Shi and others, 2009). Because the distribution of the samples on each chip was heavily unbalanced (see supplementary Table 1 available at Biostatistics online), the dataset was initially analysed with ComBat including the treatments as covariates as required by the SVA package (Leek and others, 2012) and noted by Nygaard and others (2016). The dataset was further analysed using various methods including two-way ANOVA and LIMMA yielding hundreds of differentially expressed probesets between GA and generic.
Mouse-WG6 microarray chips. Each WG6 chip contains six microarrays, and each of the microarrays containing two physical strips that are to be treated as technical replicates (Shi and others, 2009). Because the distribution of the samples on each chip was heavily unbalanced (see supplementary Table 1 available at Biostatistics online), the dataset was initially analysed with ComBat including the treatments as covariates as required by the SVA package (Leek and others, 2012) and noted by Nygaard and others (2016). The dataset was further analysed using various methods including two-way ANOVA and LIMMA yielding hundreds of differentially expressed probesets between GA and generic.
In contrast, Nygaard and others (2016) performed an analysis in which all the technical replicates were averaged. After averaging all technical replicates, Nygaard and others (2016) utilized the chip ID as a blocking variable in the analysis model, finding 11 probesets that pass FDR cutoff of 0.05. Another approach is to utilize the duplicateCorrelation feature recommended by LIMMA’s authors for handling technical replicates from Illumina WG-6 BeadChips (Shi and others, 2009). While averaging technical replicates has been suggested in the literature in cases where a t-test or two-way ANOVA will be utilized for downstream analysis (Cui and Churchill, 2003), the utilization of models that can account for both technical as well as biological variation is an alternative approach to maximize power of the statistical test (Chen and others, 2004; Cui and Churchill, 2003; Smyth and others, 2005). When GSE61901 is utilized and duplicateCorrelation is applied to account for the technical replicates, we find that the approach proposed by Nygaard and others (2016) of blocking for batch effect in LIMMA identifies 1474 differentially expressed probesets. Thus, the results of Nygaard and others (2016) proposed batch correction method differ dramatically depending upon how a dataset is preprocessed.
Upon further investigation, a variety of different preprocessing and batch correction methods all yield far more differentially expressed genes than the method proposed by Nygaard and others (2016). Table 1 reports the results of various analyses we have conducted using a variety of techniques including LIMMA (Shi and others, 2009, duplicateCorrelation method: Smyth, 2013; Smyth and others, 2005) and mixed-effects ANOVA that are specifically designed to analyze unbalanced designs without reducing power (Bernal-Rusiel and others, 2013; Littell and others, 2002; Smyth, 2014). Code for these analyses is publicly available at https://github.com/immuneering/biostat_reply. Such methods take into account correlations between repeated measurements from the same subject/biological sample. These findings strongly suggest that the differences between GA and generic are robust to changes in batch correction and analysis methodologies, and support the key conclusions of Towfic and others (2014).
Table 1.
Number of differentially expressed probesets after FDR 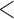 0.05 cutoff using (1) averaging technical replicates without using ComBat (2) ComBat
0.05 cutoff using (1) averaging technical replicates without using ComBat (2) ComBat  statistical analysis (common pipeline) (3) accounting for technical replicates using mixed effects models (advanced pipeline). As can be seen from the table, the common pipeline of quantile normalization
statistical analysis (common pipeline) (3) accounting for technical replicates using mixed effects models (advanced pipeline). As can be seen from the table, the common pipeline of quantile normalization  ComBat and the advanced pipeline of utilizing mixed effect models clearly detect more significant changes after FDR adjustment compared to models suffering from low power due to (1) averaging of technical arrays (2) Not utilizing a batch model that preserves power prior to analysis
ComBat and the advanced pipeline of utilizing mixed effect models clearly detect more significant changes after FDR adjustment compared to models suffering from low power due to (1) averaging of technical arrays (2) Not utilizing a batch model that preserves power prior to analysis
| Quantile normalization (averaging technical replicates), utilize CHIP as blocking variable | Quantile normalization (averaging technical replicates), apply ComBat including treatments as covariates | Quantile normalization, no averaging of technical replicates, utilize CHIP as blocking variable | |
|---|---|---|---|
| LIMMA | 11 (Nygaard and others (2016) | 974 | 1474 |
| Two-way ANOVA | 1 | 742 | — |
| Linear mixed effects model | — | — | 1968 |
| Repeated measures ANOVA | — | — | 1749 |
In summary, we present three different methods for analyzing microarray data that utilize technical replicates while correcting for batch effects. These methods yield consistent results, and therefore represent robust alternatives to traditional batch correction methods in datasets subject to the concerns articulated by Nygaard and others (2016).
Supplementary Material
ACKNOWLEDGMENTS
Conflict of Interest: All authors are employees and stockholders of Immuneering Corporation, which is majority-owned by Teva Pharmaceutical Industries, the maker of Copaxone.
SUPPLEMENTARY MATERIAL
Supplementary material is available at http://biostatistics.oxfordjournals.org.
FUNDING
This research was funded by Teva Pharmaceutical Industries.
REFERENCES
- Bernal-Rusiel J. L., Greve D. N. Reuter M. Fischl B. and Sabuncu M. R. (2013). Statistical analysis of longitudinal neuroimage data with Linear Mixed Effects models. NeuroImage 66, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. J. Delongchamp R. R. Tsai C.-A. Hsueh H., Sistare F., Thompson K. L. and Fuscoe J. C. (2004). Analysis of variance components in gene expression data. Bioinformatics 20, 1436–1446. [DOI] [PubMed] [Google Scholar]
- Cui X. and Churchill G. A. (2003). Statistical tests for differential expression in cDNA microarray experiments. Genome Biology 4, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson E. (2011 April 15). Covariates in ComBat http://groups.google.com/forum/#!msg/combat-user-forum/26FZlgU2LFQ/W6U_Lhh_64EJ(accessed December 15, 2014). [Google Scholar]
- Leek J. T. Johnson W. E. Parker H. S. Jaffe A. E. and Storey J. D. (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littell R. C. Stroup W. W. and Freund R. J. (2002). SAS for linear models. Cary, North Carolina: SAS Institute. [Google Scholar]
- Nygaard V., Rødland E. A. and Hovig E. (2016). Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics, 17, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W., Banerjee A., Ritchie M. E. Gerondakis S. and Smyth G. K. (2009). Illumina WG-6 BeadChip strips should be normalized separately. BMC Bioinformatics, 10, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. (2013). [BioC] Complex Limma design: technical replication, biological replication and repeated experiment. http://stat.ethz.ch/pipermail/bioconductor/2013-August/054615.html(accessed December 15, 2014). [Google Scholar]
- Smyth G. K. (2014). [BioC] Limma design?: What is the “best”method to infer biological replicates during differentially expression analysis. http://stat.ethz.ch/pipermail/bioconductor/2014-February/057887.html(accessed December 15, 2014). [Google Scholar]
- Smyth G. K. Michaud J. and Scott H. S. (2005). Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21, 2067–2075. [DOI] [PubMed] [Google Scholar]
- Towfic F., Funt J. M. Fowler K. D. Bakshi S., Blaugrund E., Artyomov M. N. and Zeskind B. (2014). Comparing the biological impact of glatiramer acetate with the biological impact of a generic. PLoS One 9, e83757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.