

Abstract
Objective:
To evaluate safety (primary objective) and efficacy of increasing doses (400 U up to 800 U) of incobotulinumtoxinA (Xeomin, Merz Pharmaceuticals GmbH) for patients with limb spasticity.
Methods:
In this prospective, single-arm, dose-titration study (NCT01603459), patients (18–80 years) with spasticity due to cerebral causes, who were clinically deemed to require total doses of 800 U incobotulinumtoxinA, received 3 consecutive injection cycles (ICs) with 400 U, 600 U, and 600–800 U incobotulinumtoxinA, respectively, each followed by 12–16 weeks' observation. Outcomes included adverse events (AEs), antibody testing, Resistance to Passive Movement Scale (REPAS; based on the Ashworth Scale), and Goal Attainment Scale.
Results:
In total, 155 patients were enrolled. IncobotulinumtoxinA dose escalation did not lead to an increased incidence of treatment-related AEs (IC1: 4.5%; IC2: 5.3%; IC3: 2.9%). No treatment-related serious AEs occurred. The most frequent AEs overall were falls (7.7%), nasopharyngitis, arthralgia, and diarrhea (6.5% each). Five patients (3.2%) discontinued due to AEs. No patient developed secondary nonresponse due to neutralizing antibodies. Mean (SD) REPAS score improvements from each injection to 4 weeks postinjection increased throughout the study (IC1: −4.6 [3.9]; IC2: −5.9 [4.2]; IC3: −7.1 [4.8]; p < 0.0001 for all). The proportion of patients achieving ≥3 (of 4) treatment goals also increased (IC1: 25.2%; IC2: 50.7%; IC3: 68.6%).
Conclusion:
Escalating incobotulinumtoxinA doses (400 U up to 800 U) did not compromise safety or tolerability, enabled treatment in a greater number of muscles/spasticity patterns, and was associated with increased treatment efficacy, improved muscle tone, and goal attainment.
ClinicalTrials.gov identifier:
Classification of evidence:
This study provides Class IV evidence that, for patients with limb spasticity, escalating incobotulinumtoxinA doses (400 U up to 800 U) increases treatment efficacy without compromising safety or tolerability.
Guidelines recommend botulinum toxin type A (BoNT-A) injections as a treatment option for chronic focal upper and lower limb spasticity.1–4 The efficacy and safety of different BoNT-A formulations for spasticity have been demonstrated for labeled doses.5–11 However, in multifocal disabling upper or lower limb spasticity, total doses required to fulfill goal achievement and patients' needs may exceed those currently approved.12–17 Therefore, physicians have to prioritize treating patterns whose response will have the greatest effect on overall goal achievement, but a more comprehensive treatment approach may improve outcomes and better support implemented neurorehabilitation programs. A recent survey of physicians treating spasticity with any BoNT-A formulation showed that >75% of physicians believed that using higher total doses may improve treatment outcomes and patient satisfaction.18
The safe use of higher than labeled BoNT-A doses has been reported,19–24 but not studied in large prospective clinical trials with a sufficient sample size. Furthermore, the perceived risk of increased immunogenicity and resistance associated with higher than labeled BoNT-A doses in the long term has not been addressed. In phase III trials, doses ≤400 U incobotulinumtoxinA (Xeomin, Merz Pharmaceuticals GmbH, Frankfurt am Main, Germany) were efficacious and well-tolerated by patients with upper limb spasticity.6,8–10 Due to the proven tolerability, lack of secondary nonresponse in these clinical trials, and high purity,25 incobotulinumtoxinA is a suitable BoNT-A formulation for a study investigating higher than generally used doses (400 U up to 800 U) in patients with severe upper and lower limb spasticity.
The Titration Study in Lower and Upper Limb Spasticity (TOWER) investigated the safety and efficacy of incobotulinumtoxinA for patients with spasticity due to cerebral lesions deemed to require total body doses of 800 U per injection cycle.
METHODS
Study design.
The TOWER study was a prospective, nonrandomized, single-arm, multicenter, open-label, dose-titration study. The primary objective was to investigate safety through assessments of adverse events (AEs) and investigators' global assessment of tolerability. Key efficacy data (muscle tone and resistance to passive movement scale [REPAS]; Goal Attainment Scale [GAS]; investigators' and patients' global assessment of efficacy) are also presented here. This study provides Class IV evidence that, for patients with limb spasticity, escalating incobotulinumtoxinA doses (400 U up to 800 U) increases treatment efficacy without compromising safety or tolerability because patients served as their own controls. The safety and efficacy findings from injection cycle 1, when all patients received treatment at the highest approved dose (400 U), were compared with those of cycles 2 and 3, when higher than labeled doses were administered. In addition, in the absence of a placebo control, all AEs had to be attributed to the drug, a bias against incobotulinumtoxinA. Due to word count limitations, additional efficacy data (including Disability Assessment Scale, Functional Ambulation Classification, and quality of life) will be reported separately.
The study comprised 3 injection cycles with escalating fixed total body doses of incobotulinumtoxinA (50 U/mL in normal saline) injected in the same body side (figure 1):
400 U into the upper limb only, the lower limb only, or both
600 U into the upper limb only, the lower limb only, or both
800 U into both the upper and the lower limbs (maximum dose 600 U per limb)
Figure 1. Study design.

*If a dose of 800 U was not justified for clinical or safety reasons, a lower dose of 600–800 U could be administered as an exception. TC = telephone contact; V = visit.
If a dose of 800 U incobotulinumtoxinA was clinically not indicated or in the case of safety concerns, a lower dose (≥600 U) could be administered as an exception in cycle 3. Individual doses for each clinical pattern were flexible within the range usually recommended/used/approved (table e-1 at Neurology.org). Patients were aware that they would receive 3 different doses during the study, but they did not know which dose they would receive at each visit.
Each treatment was followed by a 12- to 16-week observation period with telephone contacts at days 7 and 14, and clinic visits at weeks 4, 8, and 12–16 posttreatment to evaluate safety and efficacy. The planned regular duration of treatment was 36–48 weeks.
Standard protocol approvals, registrations, and patient consents.
This study was registered on clinicaltrials.gov (NCT01603459) and conducted in accordance with the ethical principles of the Declaration of Helsinki. The study protocol, informed consent forms, and other appropriate study-related documents were reviewed and approved by the local independent ethics committees and institutional review boards. All patients provided written informed consent.
Patients.
Men and women (aged 18–80 years) with chronic (≥12 weeks since last event leading to spasticity) upper and lower limb spasticity of the same body side due to cerebral lesions were eligible for inclusion if they were deemed by the investigator to require total body doses of 800 U incobotulinumtoxinA during the trial. Patients with bilateral symptoms were eligible if they agreed to be treated on only one side of the body.
At screening, investigators selected a target clinical pattern of spasticity (see table e-1 for patterns) to be treated in each cycle. Patients had to have a muscle tone ≥2 (Ashworth Scale [AS]) for the selected target pattern and a Disability Assessment Scale score ≥2 in the predefined principal target domain at baseline (if the upper limb was injected). Changes in antispastic/antidepressant medication, or physical/occupational therapy or other rehabilitation treatment, were not permitted from 2 weeks prior to screening. Major exclusion criteria are listed in the e-Methods.
Safety assessments.
Adverse events.
During each study visit and telephone contact (figure 1), patients were prompted to report AEs and actively questioned using a specific, extensive 5-item questionnaire (30 questions overall; questionnaire e-1) for any AEs of special interest (AESI), defined based on a prespecified list of AEs that could potentially indicate toxin spread, regardless of whether an AE was considered to be treatment-related or not.
Investigators' global assessment of tolerability.
Tolerability was assessed using a 4-point Likert scale scored at each end-of-cycle visit (1 = very good; 4 = poor).
Pulmonary function.
Forced expiratory volume in 1 second (FEV1) was assessed at screening. FEV1 and maximal inspiratory pressure (MIP) were also measured at injection visits and at 4-week control visits during cycles 2 and 3.
Anti–botulinum toxin antibody testing, laboratory assessments, and vital signs.
Blood samples were taken for antibody tests (at screening, 4 weeks after each injection, and at each end of cycle visit) and for laboratory assessments (at screening and at end of cycle visits). Details of screening assays performed are listed in the e-Methods.
Efficacy assessments.
Muscle tone and REPAS.
Muscle tone was assessed using the AS.26 All muscle groups on the treated body side were assessed to obtain the REPAS score for that side, a validated summary 26-item test (16 items for upper and 10 items for lower limbs).27 Each item is rated from 0 to 4 using the AS. Here, the 13 REPAS items for the treated body side were evaluated, resulting in a score from 0 to 52. AS and REPAS were assessed at each injection visit, 4-week control visit, and the end of study visit by the same investigator for any given patient.
Goal Attainment Scale.
At each injection visit, patients and health care teams identified 2 personal, realistic goals per limb (1 active and 1 passive allowing for up to 4 goals). Importance of and difficulty to achieve each goal were also defined. The investigators rated the GAS score for each cycle at the next injection or the end of study visit using a 5-point scale ranging from −2 (a lot less than expected) to +2 (a lot better than expected).28 A score of 0 was the expected level of achievement that should be reached if the choice of goal had been realistic.
Investigators' and patients' global assessments of efficacy.
Global assessments of efficacy for the previous cycle were performed by investigators and patients using a 4-point Likert scale (1 = very good; 4 = poor) at the next injection visit or at the end of study visit for cycle 3.
Statistical analysis.
In this exploratory trial, no distinction between primary and secondary variables was made. Safety analyses were performed on the safety evaluation set (SES; all patients who received ≥1 dose of study drug). AEs were coded according to the Medical Dictionary for Regulatory Activities version 15.0. Only treatment-emergent AEs were analyzed, i.e., AEs with onset/worsening after the first study drug administration up to and including 16 weeks after the last incobotulinumtoxinA injection or the end of study visit, whichever was later. Efficacy variables were analyzed in the full analysis set (identical to the SES in this study) using descriptive summary statistics. Continuous variables were summarized by number of nonmissing observations, mean, SD, median, quartiles, minimum, and maximum. For qualitative variables, absolute and percent frequencies were calculated. Where applicable, exploratory 95% confidence intervals (CIs) were calculated.
RESULTS
Patient disposition.
The first patient enrolled on May 24, 2012, and the last patient completed the study on September 12, 2014. Of 193 patients screened, 155 were eligible for participation and treated with incobotulinumtoxinA; 137 patients (88.4%) completed the study and 18 (11.6%) discontinued (cycle 1, n = 3; cycle 2, n = 12; cycle 3, n = 3). Reasons for discontinuation were: consent withdrawn (n = 7), AEs (n = 5), predefined discontinuation criteria met (n = 3), loss to follow-up (n = 3), noncompliance (n = 1), and administrative reasons (n = 1). For some patients, multiple discontinuation factors were entered.
Patient demographics and baseline characteristics.
Patients' mean (SD) age was 53.7 (13.1) years; approximately two-thirds were male (67.1%) and most had spasticity due to stroke (85.2%) or traumatic brain injury (7.1%) (table 1).
Table 1.
Patient demographics and baseline characteristics

Treatments.
Most patients received the scheduled doses: 91.0% (141/155) received 400 U in cycle 1; 90.8% (138/152) received 600 U in cycle 2; and 82.9% (116/140) received 800 U in cycle 3. In cycle 3, 93.6% (131/140) of patients received a dose of ≥700 U.
Safety (primary study objective).
Adverse events.
In total, 36.1% (56/155), 37.5% (57/152), and 25.7% (36/140) of patients reported AEs in cycles 1, 2, and 3, respectively. There was no increased incidence of AEs, treatment-related AEs, serious AEs, or AESIs with increasing doses or repeated injections (table 2).
Table 2.
Summary of adverse events by injection cycle
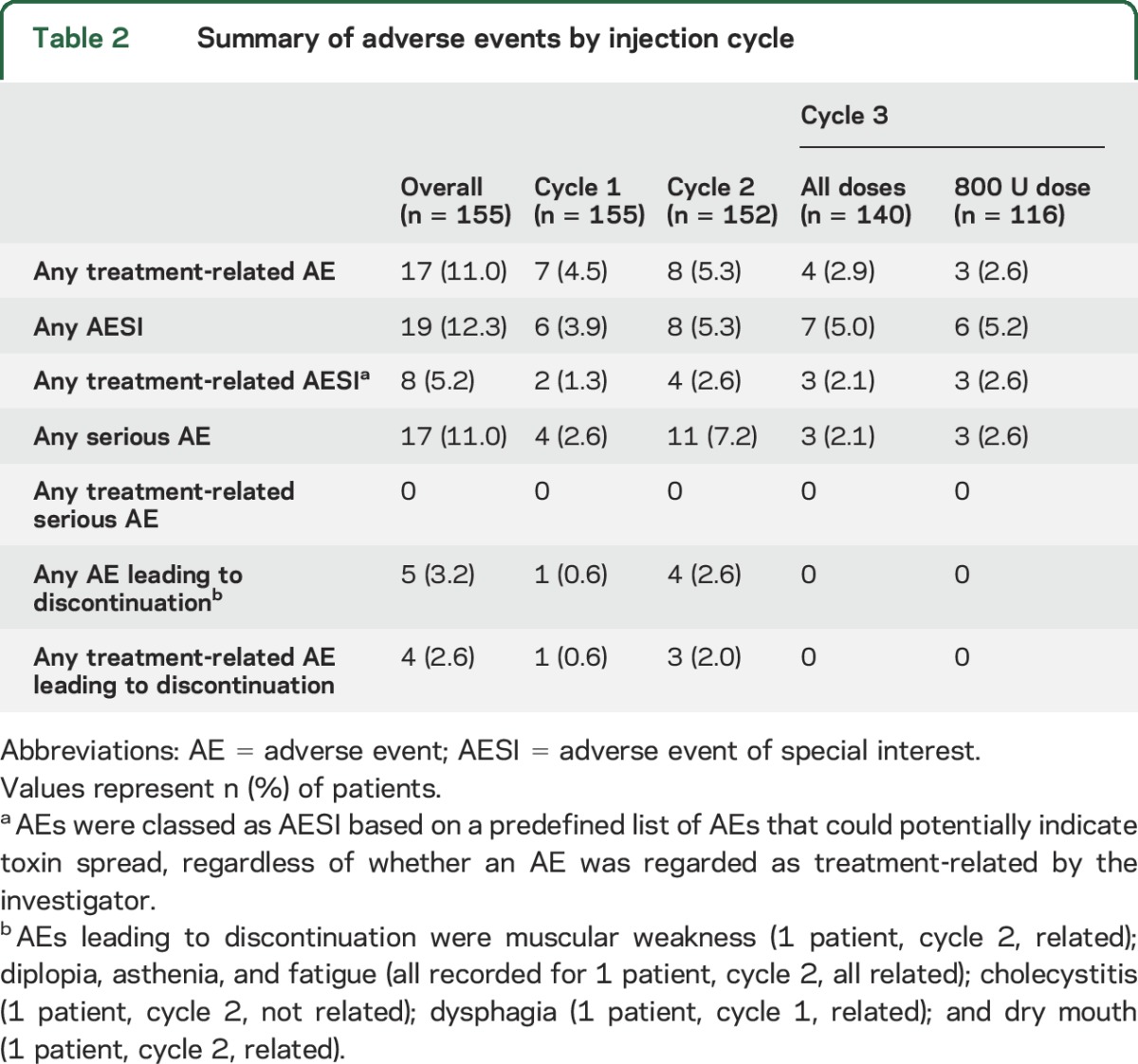
The most frequent AEs (reported by ≥5 [3.2%] patients overall) are summarized in table 3. The most common treatment-related AEs were pain in the extremity (n [patients] = 3; 1.9% [cycle 1, n = 1; cycle 2, n = 2]), dysphagia (n = 2; 1.3% [cycle 1, n = 1; cycle 3, n = 1]), and muscular weakness (n = 2; 1.3% [cycle 2, n = 1; cycle 3, n = 1]), i.e., weakness clearly exceeding the expected size of treatment effect (the investigator terms were left upper and lower limb weakness and muscle weakness of right leg and both patients had received treatment in the upper and lower limbs). These AEs resolved 4–6 weeks after the injection. All other treatment-related AEs were reported only by 1 patient. No serious AEs were related to incobotulinumtoxinA.
Table 3.
Incidence of most frequent adverse events per injection cyclea

The number of patients who reported AESIs was stable across injection cycles (table 2). The AESIs recorded were dysphagia (n [patients] = 5, 3.2%), constipation (n = 2, 1.3%), dry mouth (n = 1, 0.6%), dysphonia (n = 2, 1.3%), dyspnea (n = 2, 1.3%), pneumonia aspiration (n = 1, 0.6%), muscular weakness (n = 3, 1.9%), bradycardia (n = 2, 1.3%), diplopia (n = 1, 0.6%), blurred vision (n = 1, 0.6%), and dysarthria (n = 1, 0.6%). These AESIs were considered by investigators to be treatment-related for 2 patients with dysphagia, 1 patient with constipation, 1 patient with dry mouth, 2 patients with muscular weakness, 1 patient with bradycardia, and 1 patient with diplopia.
Investigator's global assessment of tolerability.
The tolerability of incobotulinumtoxinA treatment was rated as very good or good for 96.8% (150/155) of patients in cycle 1, 90.1% (137/152) in cycle 2, and 97.9% (137/140) in cycle 3. In contrast, tolerability was rated as poor for 0% (0/155), 1.3% (2/152), and 0% (0/140) of patients in cycles 1, 2, and 3, respectively.
Pulmonary function.
FEV1 values were >50% at all assessments, with mean and median values ranging from 82.5% to 85.1%. The mean and median values for MIP ranged from 46.0 to 57.2 cm H2O. No safety signal emerged from either the FEV1 or MIP results.
Anti–botulinum toxin antibodies.
The antibody tests showed that no patient developed secondary nonresponse due to neutralizing antibodies: no patients had positive hemidiaphragm assay (HDA) results by the end of the study, and throughout the study all patients continued to respond clinically to incobotulinumtoxinA treatment, based on changes in REPAS scores (see e-Results for further detail).
Laboratory assessments and vital signs.
At baseline and throughout the study, all mean and median laboratory values were within the respective normal ranges. Vital signs remained stable throughout the study (see e-Results for further detail).
Efficacy.
Muscle tone and REPAS.
Overall, 608 clinical patterns in 155 patients were treated in cycle 1, 743 patterns in 152 patients in cycle 2, and 811 patterns in 140 patients in cycle 3. Improvements ≥1 point on the AS scale between injection and 4-week control visits were observed in 364 (59.9%) clinical patterns treated in cycle 1, 431 (58.0%) in cycle 2, and 537 (66.2%) in cycle 3.
Mean (SD) [95% CI] improvements in REPAS scores of the treated body side from each injection to the respective 4-week control visit were as follows: cycle 1, −4.6 (3.9) [−5.2, −4.0]; cycle 2, −5.9 (4.2) [−6.6, −5.2]; cycle 3, −7.1 (4.8) [−7.9, −6.3] (p < 0.0001 for all; paired sample t test).
Goal Attainment Scale.
In cycle 1, 25.2% (39/155; 95% CI [19.0%, 32.5%]) of patients achieved ≥3 (of 4 possible) treatment goals (GAS score ≥0), compared with 50.7% (77/152; 95% CI [42.8%, 58.5%]) in cycle 2 and 68.6% (96/140; 95% CI [60.5%, 75.7%]) in cycle 3 (figure 2A). Overall, the mean (95% CI) number of goals achieved by each patient were 1.81 (1.59, 2.02) in cycle 1 (n = 155), 2.41 (2.18, 2.64) in cycle 2 (n = 152), and 3.03 (2.81, 3.24) in cycle 3 (n = 140).
Figure 2. Efficacy outcomes.

(A) Each patient and health care team identified 2 realistic treatment goals per limb (1 active and 1 passive) at each injection visit. Goal attainment for each injection cycle was rated at the next injection visit or the end of study visit. (B) The proportions of patients with a rating of very good or good are shown. Possible ratings were 1 = very good, 2 = good, 3 = moderate, 4 = poor.
Investigators' and patients' global assessments of efficacy.
The percentage of investigator assessments of very good or good increased from 55.5% (86/155; 95% CI [47.6%, 63.1%]) in cycle 1 to 72.4% (110/152; 95% CI [64.8%, 78.9%]) in cycle 2, and 89.3% (125/140; 95% CI [83.1%, 93.4%]) in cycle 3. Similarly, patient assessments of very good or good increased from 59.4% (92/155; 95% CI [51.5%, 66.8%]) in cycle 1 to 63.8% (97/152; 95% CI [55.9%, 71.0%]) in cycle 2, and 76.4% (107/140; 95% CI [68.8%, 82.7%]) in cycle 3 (figure 2B).
DISCUSSION
Patients with multifocal spasticity may benefit from BoNT-A treatment with higher total doses than currently recommended by the prescribing information of different formulations available.23–25 However, data from prospective clinical trials with a suitable sample size to evaluate higher than labeled doses are lacking. To date, our multicenter study is the largest prospective trial designed to evaluate safety and efficacy of a comprehensive treatment approach with incobotulinumtoxinA for severe and disabling multifocal spasticity. The stepwise escalation of the total dose from 400 U up to 800 U incobotulinumtoxinA allowed physicians to increase doses per muscle within the recommended ranges and the number of muscles and spasticity patterns treated according to patients' goals and needs.
With escalating total doses, a higher number of spasticity patterns was successfully treated, leading to increasing improvements in muscle tone, indicated by consistent decreases in REPAS score, which is the sum of the AS scores of different muscle groups. Moreover, higher incobotulinumtoxinA doses led to increased rates of goal attainment, with around two-thirds of patients achieving ≥3 of 4 predefined goals with the 600–800 U dose. Furthermore, improved global efficacy was reported by both investigators and patients, reinforcing the clinical relevance of the benefit of increasing incobotulinumtoxinA doses.
Treatment with up to 800 U incobotulinumtoxinA was well-tolerated, confirming previous reports.19–21,23 Importantly, no new safety concerns were identified for higher incobotulinumtoxinA doses of 600–800 U and few patients (n = 5) discontinued due to AEs. With prompted reporting for AEs and extensive active questioning for AESIs throughout the study, our findings revealed no meaningful increase in the incidence of AEs or AESIs with increasing doses or repeated injections, and no cumulative effects when injected every 12–16 weeks.
A perceived risk associated with higher than labeled BoNT doses is the development of immunogenicity and resistance to treatment. No previously BoNT treatment-naive patient had a positive HDA result for neutralizing antibodies at any point. In addition, while some pretreated patients had transient positive HDA results at various points in the study, this was not associated with nonresponsiveness to incobotulinumtoxinA in any treatment cycle (defined as a lack of response based on REPAS scores), supporting the low immunogenicity of incobotulinumtoxinA.25 Some discrepancy between the identification of neutralizing antibodies and secondary nonresponse has been described previously.29 No lasting immunogenicity was recorded with increasing incobotulinumtoxinA dose across the entire study period (up to 48 weeks) and higher than labeled doses were administered in both cycles 2 and 3. Further studies are required to investigate the effect of long-term treatment with high doses of incobotulinumtoxinA on the development of immunogenicity.
The dose escalation design of the study was chosen primarily to evaluate safety. A strength of this design was that this type of treatment regimen can be considered to be reflective of real-world clinical practice, i.e., physicians would progressively increase dosing based on patient need to optimize therapeutic outcomes. The open-label design and lack of a placebo control are the main limitations of the study design. A placebo arm was not included as BoNT-A injections are considered the standard of care for upper limb spasticity1–4 and the efficacy and tolerability of incobotulinumtoxinA for the treatment of upper limb spasticity at doses up to 400 U have been confirmed in previous clinical trials.6,8–10 Hence, ethical considerations prohibited the introduction of a placebo arm into this study. To minimize potential bias of patient-rated outcomes, patients were blinded to which dose they were receiving during which cycle.
This study addressed the previously unmet need for prospectively acquired data on the safety and efficacy of treatment with increasing incobotulinumtoxinA doses for patients with chronic upper and lower limb spasticity following brain injury. IncobotulinumtoxinA dose escalation from 400 U up to 800 U enabled treatment of a greater number of muscles and clinical spasticity patterns, resulting in increased improvements of muscle tone, goal attainment, and global efficacy, without compromising patients' safety or tolerability. Since only incobotulinumtoxinA was investigated, our findings are specific to incobotulinumtoxinA and are not interchangeable with other BoNT formulations.
IncobotulinumtoxinA up to 800 U offers the potential for comprehensive, well-tolerated, and efficacious spasticity treatment of more clinical patterns, which allows greater focus on patients' needs and goals compared with previously published studies on BoNT-A treatment with lower doses in chronic spasticity.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all investigators and patients participating in the TOWER study. Canada: Stephen McNeil, Lalith Satkunam. France: Djamel Bensmail, Isabelle Laffont, Frédéric Pellas. Germany: Manuel Dafotakis, Markus Ebke, Martin Hecht, Peter Kossmehl, David Liebetanz, Friedemann Müller, Iris Reuter, Walter Raffauf, Tobias Wächter, Jörg Wissel. Italy: Sergio Barbieri, Alessio Baricich, Mario Basciani, Giancarlo Ianieri, Franco Molteni, Maurizio Osio, Francesco Sciarrini, Nicola Smania. Norway: Tiina Ader, Tiina Rekand. Portugal: Joaquim Ferreira, Luisa Medeiros. Spain: Montserrat Abenoza Guardiola, Josefina Junyent Pares, Lourdes López de Munaín, Susana Moraleda, Marina Tirado. United States: François Bethoux, William Bockenek, Shashank Davé, John McGuire, Bruce Rubin, David M. Simpson.
GLOSSARY
- AE
adverse event
- AESI
adverse event of special interest
- AS
Ashworth Scale
- BoNT-A
botulinum toxin type A
- CI
confidence interval
- FEV1
forced expiratory volume in 1 second
- GAS
Goal Attainment Scale
- HDA
hemidiaphragm assay
- MIP
maximal inspiratory pressure
- REPAS
resistance to passive movement scale
- SES
safety evaluation set
- TOWER
Titration Study in Lower and Upper Limb Spasticity
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: TOWER study investigators, Stephen McNeil, Isabelle Laffont, Frédéric Pellas, Manuel Dafotakis, Markus Ebke, Martin Hecht, Peter Kossmehl, David Liebetanz, Friedemann Müller, Iris Reuter, Walter Raffauf, Tobias Wächter, Sergio Barbieri, Alessio Baricich, Mario Basciani, Giancarlo Ianieri, Maurizio Osio, Francesco Sciarrini, Nicola Smania, Tiina Ader, Luisa Medeiros, Montserrat Abenoza Guardiola, Josefina Junyent Pares, Lourdes López de Munaín, Marina Tirado, François Bethoux, William Bockenek, Shashank Davé, and Bruce Rubin
AUTHOR CONTRIBUTIONS
J. Wissel: study concept or design, acquisition of data, study supervision and coordination, analysis or interpretation of data, drafting the manuscript for content. D. Bensmail, J.J. Ferreira: study concept or design, acquisition of data, revising the manuscript for content. F. Molteni, L. Satkunam, S. Moraleda, T. Rekand, J. McGuire: acquisition of data, revising the manuscript for content. A. Scheschonka: study concept or design, statistical analysis, analysis or interpretation of data, revising the manuscript for content. B. Flatau-Baqué: statistical analysis, analysis or interpretation of data, revising the manuscript for content. O. Simon: study concept or design, statistical analysis, analysis or interpretation of data, revising the manuscript for content. E.T.J. Rochford: drafting the manuscript for content. D. Dressler: study concept or design, analysis or interpretation of data, revising the manuscript for content. D.M. Simpson: acquisition of data, analysis or interpretation of data, revising the manuscript for content.
STUDY FUNDING
Supported by Merz Pharmaceuticals GmbH, Frankfurt am Main, Germany. Medical writing support was provided by Complete Medical Communications and funded by Merz Pharmaceuticals GmbH. The Article Processing Charge was paid by Merz Pharmaceuticals GmbH.
DISCLOSURE
J. Wissel received research grant support from and served as a consultant for Merz, Allergan, Medtronic, and Ipsen. D. Bensmail served as a consultant for Allergan, Ipsen, Merz, Medtronic, and Almirall. J. Ferreira received research grants from GlaxoSmithKline, Grunenthal, Fundação MSD (Portugal), TEVA, MSD, Allergan, and Novartis. He served as a consultant and on advisory boards for Novartis, Lundbeck, Solvay, Abbvie, BIAL, Merck-Serono, Merz, Ipsen, and Biogen and provided expert testimony for Novartis. F. Molteni received grant support for research programs from Merz and Ipsen. L. Satkunam received grant support from and served on advisory boards for Merz and Allergan. S. Moraleda received grant support from Merz, Allergan, Ipsen, and Medtronic. T. Rekand served as a consultant for Allergan, Ipsen, Merz, Medtronic, and Almirall. J. McGuire received educational and research support from and served as a consultant for Merz, Allergan, and Medtronic. A. Scheschonka is an employee of Merz Pharmaceuticals. B. Flatau-Baqué is an employee of Merz Pharmaceuticals. Olivier Simon is an employee of Merz Pharmaceuticals. E. Rochford is an employee of Complete Medical Communications. D. Dressler received payments from Allergan, Ipsen, Merz, Syntaxin, Bayer, UCB, Abbvie, IAB-Interdisciplinary Working Group for Movement Disorders, Sintetica, and Medtronic. He holds patents on botulinum toxin and botulinum toxin therapy and he is a shareholder of Allergan. D. Simpson received research grant support from and served as a consultant for Merz, Allergan, and Ipsen. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wissel J, Ward AB, Erztgaard P, et al. European consensus table on the use of botulinum toxin type A in adult spasticity. J Rehabil Med 2009;41:13–25. [DOI] [PubMed] [Google Scholar]
- 2.Simpson DM, Hallett M, Ashman EJ, et al. Practice guideline update summary: botulinum neurotoxin for the treatment of blepharospasm, cervical dystonia, adult spasticity, and headache: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016;86:1818–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esquenazi A, Novak I, Sheean G, Singer BJ, Ward AB. International consensus statement for the use of botulinum toxin treatment in adults and children with neurological impairments: introduction. Eur J Neurol 2010;17(Suppl 2):1–8. [DOI] [PubMed] [Google Scholar]
- 4.Royal College of Physicians, British Society of Rehabilitation Medicine, Chartered Society of Physiotherapy, Association of Chartered Physiotherapists Interested in Neurology. Spasticity in adults: management using botulinum toxin: national guidelines [online]. Available at: rcplondon.ac.uk/sites/default/files/documents/spasticity-in-adults-management-botulinum-toxin.pdf. Accessed January 1, 2016.
- 5.Bakheit AM, Fedorova NV, Skoromets AA, Timerbaeva SL, Bhakta BB, Coxon L. The beneficial antispasticity effect of botulinum toxin type A is maintained after repeated treatment cycles. J Neurol Neurosurg Psychiatry 2004;75:1558–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes M, Schnitzler A, Medeiros L, Aguilar M, Lehnert-Batar A, Minnasch P. Efficacy and safety of NT 201 for upper limb spasticity of various etiologies: a randomized parallel-group study. Acta Neurol Scand 2010;122:295–302. [DOI] [PubMed] [Google Scholar]
- 7.Elovic EP, Brashear A, Kaelin D, et al. Repeated treatments with botulinum toxin type A produce sustained decreases in the limitations associated with focal upper-limb poststroke spasticity for caregivers and patients. Arch Phys Med Rehabil 2008;89:799–806. [DOI] [PubMed] [Google Scholar]
- 8.Kaňovský P, Slawek J, Denes Z, et al. Efficacy and safety of botulinum neurotoxin NT 201 in poststroke upper limb spasticity. Clin Neuropharmacol 2009;32:259–265. [DOI] [PubMed] [Google Scholar]
- 9.Kaňovský P, Slawek J, Denes Z, et al. Efficacy and safety of treatment with incobotulinum toxin A (botulinum neurotoxin type A free from complexing proteins; NT 201) in post-stroke upper limb spasticity. J Rehabil Med 2011;43:486–492. [DOI] [PubMed] [Google Scholar]
- 10.Elovic EP, Munin MC, Kaňovský P, Hanschmann A, Hiersemenzel R, Marciniak C. Randomized, placebo-controlled trial of incobotulinumtoxinA for upper-limb post-stroke spasticity. Muscle Nerve 2016;53:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaji R, Osako Y, Suyama K, Maeda T, Uechi Y, Iwasaki M. Botulinum toxin type A in post-stroke lower limb spasticity: a multicenter, double-blind, placebo-controlled trial. J Neurol 2010;257:1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allergan, Inc. Botox® US Prescribing Information [online]. Available at: allergan.com/assets/pdf/botox_pi.pdf. Accessed January 6, 2016. [Google Scholar]
- 13.Ipsen Biopharm, Ltd. Dysport® US Prescribing Information [online]. Available at: https://www.dysport.com/pdfs/Dysport_Full_Prescribing_Information.pdf. Accessed January 6, 2016. [Google Scholar]
- 14.Allergan. Botox® 100 U Summary of Product Characteristics [online]. Available at: medicines.org.uk/EMC/medicine/112/SPC/. Accessed January 6, 2016. [Google Scholar]
- 15.Ipsen. Dysport® 300 U and 500 U Summary of Product Characteristics [online]. Available at: medicines.org.uk/EMC/medicine/870/SPC/. Accessed January 6, 2016. [Google Scholar]
- 16.Merz Pharma UK Ltd. XEOMIN® 100 U Summary of Product Characteristics [online]. Available at: medicines.org.uk/emc/medicine/20666. Accessed January 6, 2016. [Google Scholar]
- 17.Merz Pharmaceuticals L. Xeomin® US Prescribing Information [online]. Available at: xeomin.com/wp-content/uploads/xeomin-full-prescribing-information.pdf. Accessed January 6, 2016. [Google Scholar]
- 18.Bensmail D, Hanschmann A, Wissel J. Satisfaction with botulinum toxin treatment in post-stroke spasticity: results from two cross-sectional surveys (patients and physicians). J Med Econ 2014;17:618–625. [DOI] [PubMed] [Google Scholar]
- 19.Dressler D. Routine use of Xeomin in patients previously treated with Botox: long term results. Eur J Neurol 2009;16(Suppl 2):2–5. [DOI] [PubMed] [Google Scholar]
- 20.Dressler D, Saberi FA, Kollewe K, Schrader C. Safety aspects of incobotulinumtoxinA high-dose therapy. J Neural Transm 2015;122:327–333. [DOI] [PubMed] [Google Scholar]
- 21.Santamato A, Panza F, Ranieri M, et al. Efficacy and safety of higher doses of botulinum toxin type A NT 201 free from complexing proteins in the upper and lower limb spasticity after stroke. J Neural Transm 2013;120:469–476. [DOI] [PubMed] [Google Scholar]
- 22.Baricich A, Grana E, Carda S, Santamato A, Cisari C, Invernizzi M. High doses of onabotulinumtoxinA in post-stroke spasticity: a retrospective analysis. J Neural Transm 2015;122:1283–1287. [DOI] [PubMed] [Google Scholar]
- 23.Intiso D, Simone V, Di Rienzo F, et al. High doses of a new botulinum toxin type A (NT-201) in adult patients with severe spasticity following brain injury and cerebral palsy. Neurorehabilitation 2014;34:515–522. [DOI] [PubMed] [Google Scholar]
- 24.Santamato A, Micello MF, Ranieri M, et al. Employment of higher doses of botulinum toxin type A to reduce spasticity after stroke. J Neurol Sci 2015;350:1–6. [DOI] [PubMed] [Google Scholar]
- 25.Dressler D. Five-year experience with incobotulinumtoxinA (Xeomin(R): the first botulinum toxin drug free of complexing proteins. Eur J Neurol 2012;19:385–389. [DOI] [PubMed] [Google Scholar]
- 26.Ashworth B. Preliminary trial of carisoprodol in multiple sclerosis. Practitioner 1964;192:540–542. [PubMed] [Google Scholar]
- 27.Platz T, Vuadens P, Eickhof C, Arnold P, Van Kaick S, Heise K. REPAS, a summary rating scale for resistance to passive movement: item selection, reliability and validity. Disabil Rehabil 2008;30:44–53. [DOI] [PubMed] [Google Scholar]
- 28.Turner-Stokes L, Baguley IJ, De Graaff S, et al. Goal attainment scaling in the evaluation of treatment of upper limb spasticity with botulinum toxin: a secondary analysis from a double-blind placebo-controlled randomized clinical trial. J Rehabil Med 2010;42:81–89. [DOI] [PubMed] [Google Scholar]
- 29.Fabbri M, Leodori G, Fernandes RM, et al. Neutralizing antibody and botulinum toxin therapy: a systematic review and meta-analysis. Neurotox Res 2016;29:105–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.