

Abstract
1-Deoxy-D-xylulose 5-phosphate synthase (DXS) is a thiamin diphosphate (TDP) dependent enzyme that marks the beginning of the methylerythritol 4-phosphate isoprenoid biosynthesis pathway. The mechanism of action for DXS is still poorly understood and begins with the formation of a thiazolium ylide. This TDP activation step is thought to proceed through an intramolecular deprotonation by the 4′-amino-pyrimidine ring of TDP; however, this step would occur only after an initial deprotonation of its own 4′-amino group. The mechanism of the initial deprotonation has been hypothesized, by analogy to transketolases, to occur via a histidine or an active site water molecule. Results from hybrid quantum mechanical/molecular mechanical (QM/MM) reaction path calculations reveal an ~10 kcal/mol difference in transition state energies, favoring a water mediated mechanism over direct deprotonation by histidine. This difference was determined to be largely governed by electrostatic changes induced by conformational variations in the active site. Additionally, mutagenesis studies reveal DXS to be an evolutionarily resilient enzyme. Particularly, we hypothesize that residues H82 and H304 may act in a compensatory fashion if the other is lost due to mutation. Further, nucleus-independent chemical shifts (NICSs) and aromatic stabilization energy (ASE) calculations suggest that reduction in TDP aromaticity also serves as a factor for regulating ylide formation and controlling reactivity.
Graphical abstract

INTRODUCTION
Isoprenoids are one of the largest and most diverse families of biomolecules with a number of them essential for life.1–3 An example would be vitamin A, which plays a role in human growth and development as well as immune system maintenance. Two isoprene molecules are variably employed in the construction of all isoprenoids. Isopentenyl diphosphate (IDP) and dimethylallyl diphosphate (DMADP) are produced via two distinct biosynthetic pathways (Figure 1): mevalonate pathway (MVA) and methylerythritol 4-phosphate pathway (MEP pathway).4–6 The MVA pathway was discovered in the 1950s and was considered the sole pathway until the 1990s when discrepancies in isotopic labeling studies led researchers to hypothesize an alternative, MEP pathway.7,8 Subsequent genetic studies have revealed a large variety of life (e.g., algae, bacteria, etc.) to have varying degrees of dependence upon MEP pathway for isoprenoid production, in addition to a number of human pathogens (e.g., Plasmodium spp. and M. tuberculosis).9–12 Interestingly, the MEP pathway is absent in all mammalian genomes, meaning that the enzymes of this pathway are ideal targets for novel antibiotics and antimalarials.13 For example, fosmidomycin is known to be an effective agent against several of the Plasmodium spp. (malarial pathogens) and targets MEP pathway’s second step, 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR).13–15
Figure 1.

Schematic of isoprene production via MVA or MEP pathway. MVA pathway produces DMADP via a secondary enzyme, IDP isomerase.16,17 MEP pathway directly synthesizes both isoprene molecules.
MEP pathway comprises eight reactions catalyzed by seven enzymes beginning with 1-deoxy-D-xylulose 5-phosphate synthase (DXS).18–25 DXS catalyzes the condensation of glyceraldehyde 3-phosphate (G3P) and pyruvate to produce 1-deoxy-D-xylulose 5-phosphate (DXP). Aside from isoprenoid production, DXP is utilized in the production of vitamin B1 (thiamin) and vitamin B6 (pyridoxine) biosynthetic pathways,22,26,27 suggesting increased significance to understanding the mechanism of DXS. Further, DXS is believed to be a rate-limiting step due to the observed correlation between isoprenoid product levels and DXS levels.22 DXS is a member of the thiamin diphosphate (TDP) dependent family of proteins, specifically a member of the transketolase (TK) enzyme subclass, and also possesses pyruvate decarboxylase activity.18,24,28 TKs are a class of TDP dependent enzymes responsible for the transfer of a ketol donor group to an aldehyde or ketone acceptor molecule.29–31 In 2007, Xiang et al. published crystal structures and mutagenesis results of DXS from E. coli and D. radiodurans and compared them to the E1 subunit of pyruvate dehydrogenase (PDH) and yeast TK (members of the same class of enzymes).32 The comparison revealed significant similarities between these four enzymes: (1) each enzyme is composed of three domains (I, II, and III); (2) all possess a TDP cofactor; (3) all contain a GDGX25–30N motif that plays a role in producing the twisted “V” shape of the TDP cofactor in the active site.32 The strained cofactor conformation has been shown to play a role in lowering the pKa of a hydrogen on the thiazolium ring’s C2 atom (Figure 2).31,33–36 The active site of DXS contains a number of strictly/highly conserved residues (e.g., Glu370/372, Asp152/154 in E. coli/D. radiodurans, respectively) that are common among TDP dependent enzymes, particularly TKs. These similarities have led researchers to propose a DXS reaction mechanism based, primarily, on mechanistic data of other TK enzymes (Figure 2).29,30,37
Figure 2.

Proposed general mechanism for DXP biosynthesis. Pieces of each step are labeled with different colors to indicate where they originate from. Red represents pyruvate, and blue represents the pieces affiliated with G3P.
Although DXS contains many of the strictly/highly conserved residues of the TDP-dependent superfamily of enzymes (vide supra), it displays distinct structural features:32 specifically, the domain arrangement of DXS, homodimeric with a deep pocket between two domains of the same monomer rather than at the dimer interface as is the case with other TDP enzymes. These structural differences logically lead to questions of mechanistic similarity. Until the discovery of DXS, it was believed that all TDP enzymes functioned via a classical “ping-pong” mechanism (i.e., pyruvate binding → CO2 release → G3P binding).31 However, Eubanks and Poulter in 2003 concluded that DXS operates via an ordered mechanism (i.e., irreversible pyruvate binding → G3P binding → CO2 release) and hypothesized a side reaction for producing CO2 via binding of a second pyruvate molecule.38 This hypothesized side reaction was subsquently confirmed by Brammer and Meyers in 2009.39 However, the following year a steady-state kinetics study examining a herbicide metabolite (ketoclomazone, a derivative of clomazone) provided evidence of a traditional ping-pong mechanism for DXS.40 Nearly simultaneously (in 2010), a single-molecule force spectroscopy nanosensor was developed and used to observe an approximate 2-fold binding enhancement of G3P in the presence of pyruvate, suggesting an ordered DXS mechanism.41 As part of this work, the authors cast doubt on the reliability of previous results based upon assays that measure bulk phenomena rather than single-molecule behavior. To further confound the situation, Meyers and co-workers (in 2011) proposed an unprecedented TDP-based mechanism; G3P and pyruvate were found to bind independently and reversibly. Thus, they concluded DXS functions via a rapid equilibrium, random sequential mechanism.42 In the following year, Meyers and co-workers revealed a 600-fold acceleration in the decarboxylation of the lactyl-TDP intermediate upon binding of G3P. This result further distinguishes DXS from other TDP-dependent enzymes.
Two recent studies have called into question our understanding of the active sites of the large class of TDP dependent enzymes. For instance, the benzaldehyde lyase (BAL) enzyme is devoid of all but two acid–base residues around the TDP active site: a histidine and highly conserved glutamate.43,44 Most interesting is the lack of any apparent acid–base residues in glyoxylate carboligase (GCL)45. These recent discoveries represent glaring gaps in our understanding of TDP-dependent enzymes and bolster the importance of investigating distinct related enzymes (i.e., DXS).43–46 Here, computation is an ideal partner to experiment.
Of particular interest in this work is the “true first step” of this process: TDP activation, of which significant mechanistic details are still largely uncertain. For example, a proposed mechanism for ylide formation begins with an initial deprotonation of the 4′-aminopyrimidine (AP) state that produces the 1′,4′-iminopyrimidine (IP) state.18,24 A general base (GB) is required for this deprotonation; however, the identity of this group remains unknown. One hypothesized GB is a highly conserved histidine (His434 in D. radiodurans DXS) found proximal to TDP’s 4′-amino group.31,47 The aforementioned mutation studies (i.e., H434A) showed approximately 95% activity retention, which suggests an alternative mechanism. A more recent 2014 study by Querol et al. suggests H431 (E. coli equivalent of D. radiodurans H434) plays a role in transition state stabilization but not required for catalysis.48 Additionally, numerous structural differences between DXS and TK enzymes (vide supra) support the possibility of an alternative mechanism.32 One possible alternative mirrors that of human TKs, where a water molecule would replace H434 as the GB with a Gln residue acting to stabilize this via coordination.37,49,50 This results in two possible TDP activation mechanisms: a water-mediated mechanism (WMM) and an direct histidine mechanism (DHM). Even though the WMM utilizes a water molecule as the initial general base, it is possible that H434 plays a role in this mechanism either as a coordination site for the water molecule or as the final location of the proton.
TDP has been shown to exist in four different tautomeric/ionization states (Figure 3); however, the exact mechanism for producing the final ylide form remains unclear.51–54 Figure 3 illustrates two possible mechanisms: (1) a concerted AP to IP conversion followed by ylide formation; (2) a stepwise mechanism where the AP is first ionized to a 4′-aminopyrimidinium ion (APH+) and subsequently converted to the IP and ylide, respectively. Although direct spectroscopic evidence of the APH+ state remains elusive, its existence has been inferred from alternative experiments (e.g., pH rate profiles, solid-state NMR) and hypothesized to assist in promoting IP formation via stabilization of the tautomerization reaction.52,53,55 An elevation in the pKa of TDP’s N1 atom is proposed to account for the APH+ state’s existence, which is justified by its proximity to a strictly conserved glutamate residue. This idea, however, does not consider the possibility of an accompanying elevation in the pKa of the glutamate residue. Recent studies have determined the pKa for the N1 atom in DXS to be 7.5,54 and a PROPKA56–59 calculation has estimated the E373 residue to have a pKa of 8.4.a These pKa values suggest an equilibrium between the AP, and APH+ states, which is consistent with the enzyme stabilizing the IP formation via pKa modulation. Additionally, Jordan et al. supports the equilibria presented in Figure 3, particularly for apo (TDP-bound enzyme lacking substrate) enzymes.55 Therefore, it is not necessary to select between the stepwise or concerted mechanism for the purposes of this study.
Figure 3.

Structure and relationship of the four possible tautomeric/ionization states proposed for the cofactor of TDP dependent enzymes.36,51–54 Key atoms have been given names for reference purposes throughout this article.
The rate of activation and turnover of TDP shows a substantial increase when bound to an enzyme rather than in solution.35 Several factors leading to the increase in activity have been proposed. One factor includes the strained conformation the cofactor adopts upon binding. This conformation places the 4′-amino group in close proximity to the thiazolium C2 atom, both introducing strain and lowering the pKa of the C2 hydrogen from 14 to 19 (depending on solvent) to approximately 9.47,60 The electronics of the pyrimidine ring of TDP would also undoubtedly change during the activation process. These changes could lead to a disruption of the aromaticity and more indirectly influence the energetics of TDP activation. The link between aromaticity and TDP activation has not been investigated previously. Uncovering such a link will provide better understanding of DXS and raise the question if this phenomenon is general for all TDP dependent enzymes (e.g., TK, PDH, etc.).
Herein, a hybrid quantum mechanical/molecular mechanical (QM/MM) study is carried out using the D. radiodurans DXS crystal structure.32 The energy profiles of the DHM and WMM are computed to determine the most favorable activation mechanism. Active site electrostatics are also probed to elucidate the stabilizing/destabilizing effects that govern this process. Further, two metrics of aromaticity are employed to quantify this effect and determine its role as a possible driving force in activation of TDP dependent enzymes. In addition to the computational work, a kinetics study, utilizing a coupled enzyme assay, was performed on mutant and wild-type forms of the D. radiodurans DXS enzyme. As part of the mutagenesis work, we have re-examined the H434A mutation, which is of particular interest in this study. Our study focuses on the KM and kcat due to their relationship with substrate affinity and reaction turnover, respectively. The combination of computational and experimental results helps bridge the gap between bulk behavior and atomistic understanding, ultimately leading to new insights into this unique enzyme.
METHODS
Computational Methods
The DXS active homodimer structure was used throughout this study. The crystal structure for the D. radiodurans DXS (PDB code 2O1X)32 enzyme with TDP bound was processed and parsed via www.charmming.org.61 A TDP molecule can be broken down into three moieties: a thymine-like pyrimidine ring, a pyrophosphate (residue name utilized in the topology file), and a thiazolium ring (Figure 2). Parameters for all three of these have been developed as part of the CHARMM general force field (CGenFF).62 Final TDP parameters were thus obtained by connecting the respective components and modifying charges (see Supporting Information). Parameter validation was done with respect to the TDP crystal conformation based on the rmsd (see Supporting Information).
Structural modifications were performed to ensure the active site Glu373 was protonated in agreement with experimental evidence.63,64 CGenFF and CHARMM22 protein (C22) force fields65 were used throughout. The system was solvated in a rhombododechedron crystal structure and neutralized with KCl salt to a final concentration of 0.15 M. The system was heated from 110 to 310 K over 100 ps and equilibrated for 200 ps at constant pressure (1 atm) and temperature (310 K). The total system size was then reduced by removing all of the water and salt ions beyond 12 Å from the surface of the protein. The reduced structure was then QM/MM minimized, without applying cutoffs, to a gradient tolerance of 0.002 kcal·mol−1·Å−1. All QM/MM calculations employed the Q-Chem4.0/CHARMM66–68 interface at the B3LYP/6–31G* level of theory.69–71 Additionally, the single link atom scheme was used to account for truncation of the QM region and employed group electrostatic exclusions to prevent over polarization of the QM region.
Reaction pathway calculations employed a combination of the replica path method (RPATh) and harmonic distance restraints (RESDi).68,72,73 The RPATh method permits the user to divide the system into discrete subsystems (replicas), which are allowed to change independently of the remaining “environment”. A subsystem was defined to be 6.5 Å around the QM region, which comprised TDP, Glu373, His120, and His434 (Figure 4a, 98 QM atoms). The QM region for the WMM included a water molecule (Wat9709) coordinating with the 4′-amino group of the pyrimidine ring of TDP and the Nε of His434. Wat9709 was removed from the initial structure prior to QM/MM minimization (vide supra) for the DHM. Two replicas of the subsystem were used to model successive steps along the reaction coordinate (δ), which was defined as a linear combination of the bond being broken and the bond being formed (Figure 4b, 101 QM atoms). The δ values were defined incrementally for each mechanism starting from the reactant state. The DHM scanned a range of −3.0 Å to 3.0 Å in increments of 0.3 Å with smaller increments of 0.1 Å used around the transition state (i.e., −1.0 Å to 1.0 Å) to provide finer detail. The WMM scanned a range of −1.1 Å to 1.1 Å in increments of 0.1 Å.
Figure 4.

Representations of the RS for DHM (a) and WMM (b). The dashed black lines illustrate the proton transfer reaction.
The charge perturbation analysis (CPA)74–77 technique involves QM/MM single point energy calculations where a single residue’s classical charge is scaled to zero to probe its electrostatic contribution. ΔE is computed by taking the difference of the modified (zero-charge residue) and the full QM/MM electronic energy: CPA calculations were performed for the reactant state (RS) and transition state (TS) of the DHM and WMM as determined by RPATh + RESDi calculations. The reactant state was the starting δ value for each mechanism, while the TS corresponded to the point along the path with the highest energy. ΔΔECPA = ΔECPA(RS) − ΔECPA(TS) provides insight into stabilizing/destabilizing electrostatic effects with respect to RS and TS. CPA was performed on all 82 residues found within 5 Å of the QM region for both mechanisms. To further characterize long-range electrostatic changes, the QM/MM dipole moments were calculated around the QM region for the RS and TS of each mechanism. The QM/MM dipoles take into account the external charge contributions of the MM region on the QM region. The calculations were carried out using Q-Chem 4.067 and initially visualized in IQmol with final rendering using PyMOL.
Two metrics were employed to quantify aromaticity and gauge the level of significance of it as a possible driving force of ylide formation: nucleus-independent chemical shifts (NICSs)78 and aromatic stabilization energy (ASE).79,80 NICS directly measures the aromatic character of a compound,78,79,81,82 while ASE reveals the stabilization/destabilization that arises from the aromaticity of a compound.83 Due to computational limitations, NICS calculations were performed on reduced versions of the RS and TS subsystems. The reduced subsystems contained 232 or 235 atoms for the DHM or WMM, respectively, the difference of three atoms being the absence of Wat9709 from the DHM. Ghost atoms were placed perpendicular to the plane of TDP’s pyrimidine ring. Due to the nonsymmetric protein active site, the NICS(0) (ring center), NICS(1), and NICS(−1) (atoms 1 Å above and below the plane of the ring) will be reported herein.80,82,84,85 All NICS calculations were performed using Q-Chem 4.067 at the B3LYP/6-31G* level of theory.69–71 ASE is typically computed via a reference homodesmotic reaction.83,86 A homodesmotic reaction must be defined such that equal numbers of each type of atom (sp3, sp2, sp) and bond (sp3−sp3, sp3−sp2, sp2−sp2, etc.) exist in both reactants and products.86 All structures used in ASE calculations were optimized at the B3LYP/6-311+G** level of theory. Energies for each molecule were corrected by subtracting out their respective zero point energy obtained from subsequent frequency calculations.
Experimental Methods
Materials
TDP, pyruvate, G3P, DXP sodium salt, bovine serum albumin, and LB broth were purchased from Sigma-Aldrich. NADPH was purchased from Alexis Biochemical, Ni-NTA resin was purchased from Invitrogen, and β-mercaptoethanol (β-Me) was purchased from Fisher. E. coli XL-10 cells, deoxynucleotide mix PCR grade, pfuUltra Hotstart DNA polymerase, QuikChange II site directed mutagenesis kit, and acetonitrile (HPLC grade) were purchased from Agilent. The DNA vectors pET28a(+) and pET15b(+) and E. coli BL-21 B(DE3) cells were purchased from EMD Biosciences. DNA sequencing services and primers were purchased from MWG operon. All the other reagents were of the highest quality commercially available.
Cloning of D. radiodurans DXS and E. coli DXR
A synthetic, codon optimized D. radiodurans dxs gene with 5′-NdeI and 3′-XhoI restriction sites in a pMK vector was purchased from Geneart (Germany). The dxs gene was excised from the pMK vector and cloned into the NdeI and XhoI sites of a pET28a(+) vector (kanamycin resistance) with an N-terminal His6-tag to yield the pET28a(+)-DXS plasmid. Successful cloning of the D. radiodurans dxs gene was confirmed by DNA sequencing at MWG Operon.
A synthetic, codon optimized E. coli dxr gene with 5′-NdeI and 3′-BamHI restriction sites in a pMK vector was purchased from Geneart (Germany). The dxr gene was excised from pMK vector and cloned into NdeI and BamHI restriction sites of pET15b(+) vector with a C-terminal His6 tag to yield the pET15b(+)-DXR plasmid. Gene insertion was confirmed by DNA sequencing.
Production of the D. radiodurans DXS Mutants
Site-directed mutagenesis was carried out using the QuikChange II site-directed mutagenesis kit. Briefly, the mutagenesis mixture consists of 50–100 ng of plasmid pET28a(+)-DXS as a template, 1× PCR reaction buffer, 0.4 mM each of the forward and reverse primer, 0.25 mM dNTP mixture, 5 μL of Quik solution, and 2.5 units of pfuUltra Hotstart polymerase in a 50 μL reaction. The overlap extension method was used to produce the DXS mutants that were difficult to create via site directed mutagenesis.87 The sequence of the mutant DNA was confirmed by DNA sequencing.
Assays for DXS Activity
We employed a DXS-DXR coupled assay to determine the wild-type and mutant DXS enzyme activities. In this way, the DXS-dependent production of DXP is ultimately coupled to the oxidation of NADPH to NADP+ via the DXR enzyme. The solution for the DXS-DXR coupled contained 100 mM HEPES, pH 8.0, 100 mM NaCl, 1 mg/mL BSA, 1 mM TDP, 1.5 mM MnCl2, 2 mM β-Me, 0.15 mM NADPH, 0.2 mg/mL DXR, and varying concentrations of pyruvate or G3P.88 Steady-state kinetic experiments were performed by varying pyruvate or G3P at a fixed saturating concentration of the cosubstrate. A DXS-DXR reaction solution was incubated at 37 °C for 5 min, the reaction was initiated by addition of 358 nM DXS, and the progress of the reaction monitored spectrophotometrically at 340 nm for the oxidation of NADPH. The DXS and DXR employed in this assay were overexpressed and purified based on the methods presented in the Supporting Information. Each sample was stored at −80 °C until used for the assay. The steady state initial velocity for DXS measured at various concentrations of pyruvate and G3P were fit to eq 1 (see Supporting Information for plots) using nonlinear regression analysis in Sigma-Plot 12.0.
| (1) |
RESULTS AND DISCUSSION
A central aim of this investigation is to determine and characterize the mechanism of TDP activation in DXS (Figure 2). There are two hypothesized mechanisms acting by a different GB: WMM (Wat9709) and DHM (His434). Though most TK enzymes are thought to rely on a histidine residue as the GB, key structural differences and mutagenesis results suggest DXS might diverge from the majority of TK enzymes.31,38,42,54,89 The reactant state QM/MM minimized structures (Figure 4a, Figure 4b) provides some initial insight into this process. Coordination of the water oxygen to the Hn (2.0 Å, Figure 4b) suggests water could act as the GB. Further, the distance between Ho and Nε (1.8 Å, Figure 4b) suggests that this could be the final destination of this proton. Alternatively, in the absence of Wat9709, His434 directly interacts with TDP albeit more distantly (4.2 Å, Figure 4a).32
To determine each mechanism’s feasibility, the RPATh + RESDi technique was employed and respective minimum energy pathways were computed. A plot of ΔE with respect to δ (Figure 5) values illustrates the energetic favorability of the WMM over the DHM. A δ value of 0.3 Å corresponds to the TS of both mechanisms. The difference between barriers can partially be explained by a conformational change that occurs during the DHM (Figure 6). This involves the movement of His434 into a conformation more favorable for deprotonation of the 4′-amino moiety. His434’s movement induces a strain in the protein backbone and perturbs the configuration of the local environment. This change in configuration accounts for a portion of the energetic differences between the WMM and DHM but does not provide a complete explanation. Further, a ΔE‡WMM value of 22.7 kcal·mol−1 is considerably higher than one might expect for an enzyme catalyzed proton transfer and cannot be explained by a simple conformational change.90
Figure 5.

Minimum energy profiles computed for the WMM and DHM. The different x-axes are used because of differences in the reaction coordinate ranges for WMM vs DHM; both are associated with the same y-axis. The ΔE‡ are 22.7 kcal·mol−1 and 33.7 kcal·mol−1 for the WMM (gray circles) and DHM (black squares), respectively.
Figure 6.

Representative conformational changes between the RS (yellow) and TS (green) of the DHM.
The reaction pathway calculations applied a restraint to the proton transfer involved in the DHM or WMM. No other restraints were applied to the system. Upon examination of structural changes during the reaction, a second proton was observed to spontaneously transfer from E373 to the N1 atom of TDP’s AP ring in both mechanisms (Figure 7). Since E373 was included in the QM region, the proton transfer occurred in response to electronic changes encountered during each mechanism. The combination of the restrained reaction path proton transfer and unrestrained E373 to N1 atom proton transfer represents the tautomerization of the AP to IP state (Figure 3). The formation of the ylide state is dependent upon first forming the IP state. There is some debate in the literature over the exact details of the IP state formation (vide supra).51–54 Most studies propose an equilibrium between the AP, APH+, and IP TDP states (Figure 3) particularly for apo enzymes.55 As highlighted in the Introduction, the pKa values of TDP’s N1 atom and E373 residue (see Supporting Information and Introduction) are approximated to be close to one another using experimental and empirically based computational techniques. The combination of the pKa values and observed responses from QM/MM calculations suggests a concerted mechanism as previously thought. Additional studies are underway to more fully address this unresolved question.
Figure 7.

Illustration of the proton transfer from E373 to TDP’s AP ring during the tautomerization reaction. (a) and (b) represent the reactant and product states, respectively. While this figure only depicts the structures of the WMM, a similar response was observed during the DHM.
The CPA method, which approximates electrostatic contributions of a single active site residue, was used to determine the stabilizing/destabilizing effects of active site residues as a function of both states (i.e., RS vs TS) and mechanisms (i.e., WMM vs DHM). Negative ΔΔE values indicate that a particular residue is more stabilizing toward the TS, whereas positive ΔΔE values show stabilization of the RS. From the 82 active site residues examined, there were 4 that showed substantive differences (Table 1). Residues found stabilizing the TS were D430, K289, H51, and Wat10307. K101 was found to preferentially stabilize the RS. K101, H51, and D430 were found in a catalytic triad-like configuration in the active site (Figure 8). It is unlikely that they play a direct role in this reaction due to their distance from the site of activity (7.2 Å). A cluster of water molecules were found to span the distance between the reaction site and triad, which suggests an electrostatic role. K289 coordinates to the negatively charged phosphate tail of TDP (Figure 8) and is highly conserved in D. radiodurans, as well as other TDP dependent enzymes. In fact, most TDP dependent enzymes are found to require a divalent metal ion and positive residues near the phosphate tail to anchor the cofactor.
Table 1.
ΔΔE Values for Four Residues of Interest in the WMM and DHMa
| ΔΔEDHM | ΔΔEWMM | |
|---|---|---|
| K101 | 2.0 | 7.5 |
| H51 | −10.1 | −1.2 |
| K289 | −12.3 | −2.5 |
| D430 | −21.3 | −10.8 |
Negative ΔΔE values indicate preferential stabilization of the TS, while positive ΔΔE values show stabilization of the RS preferentially. All values are in kcal·mol−1.
Figure 8.

Active site conformation of the residues discussed in the CPA results. Images show both the RS (yellow) and TS (green). (a) illustrates the DHM, while (b) shows the WMM.
The magnitudes of ΔΔEDHM values were consistently larger than the magnitudes of ΔΔEWMM values. This behavior is attributed to the structural change that the DHM TS must adopt in order to position His434 for deprotonation of TDP’s 4′-amino group. The increased TS stabilization for this mechanism suggests the enzyme is tuned to accommodate alternative activation routes, although they may not be the most favorable. For example, active site mutations are a common way that bacteria and other lower life forms (i.e., those that rely on MEP pathway) can adapt to changes in chemical environments. By tuning the DXS active site to stabilize TDP activation via varying general bases, evolutionary fitness is maximized.
To better characterize long-range electrostatic effects, QM/MM dipole moments for the RS and TS for each mechanism were computed and visualized (Figure 9). The RS dipole moments of both the WMM and DHM were essentially the same. Further, WMM dipoles, both RS and TS, are indistinguishable (Figure 9b) whereas the DHM TS dipole moment is significantly perturbed (Figure 9a). Again, this effect is attributed to the conformation change His434 undergoes during the DHM and appears to be the underlying source of DXS’s ability to stabilize non-water-mediated TDP activation.
Figure 9.

Illustrated above are the computed RS (yellow) and TS (green) dipoles of the WMM and DHM. (a) is the DHM, and (b) is the WMM.
Herein, we also report experimental kinetics studies of pyruvate and G3P binding and reaction in DXS and several DXS mutants (Table 2). For H434A, there exists negligible increase in catalytic rate for pyruvate as well as G3P in comparison to wild-type, respectively. KM values also slightly increased by 6.1- and 4.6-fold, respectively. G3P’s negatively charged phosphate tail is thought to bind in a positively charged region of the active site, which contains the polar H434 residue. Additionally, the negatively charged pyruvate is thought to interact with the same positive region but not as strongly.32 Therefore, the mutagenesis results suggest that the electrostatic effects that accompany the H434A mutation have a clear destabilizing effect on substate binding while enhancing turnover. This behavior is contrary to what would be expected if H434 is required for initial TDP activation. Thus, the H434A mutant supports the conclusion favoring a WMM for TDP activation.
Table 2.
DXS Steady-State Kinetics Data (Wild-Type and Mutants) for Both Pyruvate and G3Pa
| Pyruvate | ||||
|---|---|---|---|---|
| KM (mM) | kcat/KM (s−1 M−1) | kcat (s−1) | % WT | |
| wild-type | 0.28 ± 0.03 | 2.6 × 104 | 7.4 ± 0.3 | 100 |
| H82A | 0.23 ± 0.02 | 1.7 × 103 | 0.38 ± 0.01 | 5.1 |
| H304A | 1.7 ± 0.5 | 5.8 × 102 | 0.90 ± 0.01 | 12.1 |
| D430A | 0.52 ± 0.5 | 1.4 × 104 | 7.2 ± 0.2 | 97.3 |
| H434A | 1.7 ± 0.1 | 5.9 × 103 | 9.9 ± 0.2 | 133.8 |
|
| ||||
| G3P | ||||
| KM (mM) | kcat/KM (s−1 M−1) | kcat (s−1) | % WT | |
|
| ||||
| wild-type | 0.05 ± 0.01 | 1.5 × 105 | 7.9 ± 0.4 | 100 |
| H82A | 0.03 ± 0.01 | 1.3 × 104 | 0.37 ± 0.02 | 4.7 |
| H304A | 0.08 ± 0.02 | 1.1 × 104 | 0.90 ± 0.1 | 11.4 |
| D430A | 0.12 ± 0.01 | 6.6 × 104 | 7.7 ± 0.2 | 97.5 |
| H434A | 0.23 ± 0.01 | 4.2 × 104 | 9.6 ± 0.3 | 121.5 |
% WT was determined by comparing the mutant kcat to the wild-type kcat.
Another interesting correlation between CPA and mutagenesis results is related to the D430A mutant. As previously discussed, D430 is found in an electrostatic triad of residues that includes K101 and H51 (Figure 8). While the kcat for D430A mutant remains relatively unchanged, the KM for pyruvate and G3P increases 1.9 and 2.4 times, respectively. This behavior indicates a role in substrate binding rather than catalysis, similar to H434. The corresponding residue in yeast TK (D477) has been studied previously.91 D477 was shown to a have a rather large effect on activity and substrate binding. In comparison, DXS shows only a 50% loss of activity that is caused by decreased substrate affinity. This speaks to the difference between DXS and other TK enzymes and highlights the need to study this unique subclass of enzyme.
Two histidine residues are in close proximity to each other (3.7 Å between Nε atoms for H82 and H304) and the center of activity (5.1 and 5.7 Å from the thiazolium C2 atom, respectively, for H82 and H304) of DXS. Table 2 shows that the H82A and H304A mutants produce catalytically defective enzymes resulting in only 2–12% kcat and kcat/KM values when compared to wild-type. The loss of activity can be explained by their proximity to the thiazolium C2 atom. These residues can assist in stabilizing the α-carbanion/enamine intermediate following pyruvate decarboxylation (Figure 2, step 5). While activity in these mutants is significantly retarded, detectable levels of activity are retained. This retention might be explained by the proximity of these two residues to one another. Upon the loss of one histidine, it is possible for the other His residue to recover partial functionality. There is one noticeable difference in the results of these two mutants. The H304A KM for pyruvate has increased compared to the wild type, while KM value for H82A remains similar to the wild-type value. This indicates that while both residues are clearly catalytically important, H304A protrudes into the pyruvate binding site and, therefore, plays a role in binding, which cannot be replaced by H82, thus accounting for observed differences in mutant KM values for pyruvate.
With these mutagenesis results, it became apparent that a longer simulation was required to examine active site conformational dynamics. Thus, the 2O1X structure was simulated for an additional 20 ns with the first 2 ns discarded (details found in Supporting Information). The trajectories were compared to the QM/MM minimized RS. The distance between the 4′-amino group and the Nε atom of H434 revealed two major conformations (Figure 10a). The first conformation lasts for ~5.0 ns and has H434 3.5 Å from the AP ring on average. The second conformation has H434 7.1 Å from the AP ring on average and remains throughout the simulation. The fact that the second conformation is stable for the majority of the simulation and places the histidine beyond the range of direct deprotonation of its 4′-amino group provides further support for a water mediated mechanism. Additionally, the backbone and side chain fluctuations were calculated for significant CPA residues (e.g., E373 and H434, Figure 10b). The conformational change of H434 to a position proximal to K101 and D430 accounts for the larger side chain fluctuations of K101 and D430 (Figure 10). The introduction of H434’s imidazole would force K101 and D430 to move in order to accommodate the bulky polar side chain. The combination of the motion of these residues with H434 being the final resting place of the proton abstracted from TDP’s amino group suggests a possible regulatory role for H434. H434 could act as a shuttle involved in regenerating the TDP-ylide for further reactions by displacing the abstracted proton onto D430. This perfectly aligns with experimental results showing that the removal of this residue (H434A) slightly increases kcat, allowing any proton transfer from TDP’s amino group to D430 to occur more rapidly via a water mediated process (picosecond time scale) rather than the H434 side chain motion that likely occurs on the nanosecond time scale.
Figure 10.

Analysis for 18 ns of the unrestrained simulation of the 2O1X DXS structure utilized in this investigation. (a) shows the distances over time for Nε of H434 to N4′ of TDP’s amino group. (b) shows the fluctuations for backbone (black), and side chains (gray) for residues H51, K101, H124, K289, E373, D430, and H434. These residues represent the QM region and key CPA residues previously discussed. (c) is a snapshot from the 18 ns trajectory with H434 in proximity to the 4′-amino group. (d) is representative of H434 in the second conformation.
TDP reactivity is clearly dependent on the surrounding environment, e.g., rate of reaction increases a billion-fold when bound to an enzyme35. Several attempts to determine the underlying energetics have attributed this behavior to the strained “V” shape TDP adopts upon binding.33,34,47 Given the pKa changes this conformation induces, it is surprising that the barrier to activation (i.e., proton transfer) is significantly higher than expected: ΔE‡ = 22.7 kcal·mol−1 vs 5–10 kcal·mol−1 for typical proton transfers90. One possible cause of this is the loss of aromaticity that occurs during ylide formation. Additionally, overestimation of the barrier may be due to the inability to carry out free energy simulations; nevertheless, the energy barrier difference is a more meaningful quantity when seeking to differentiate two possible mechanisms.
To examine the former, i.e., aromaticity, effects, both NICS and ASE were computed. NICS calculations estimate the aromaticity of a molecule, negative NICS values indicating aromaticity and positive NICS values antiaromaticity. Table 3 reports NICS results for RS and TS of TDP activation via DHM and WMM. An average of the ΔNICS values was used to quantify the relative change in aromatic character. Results indicate the AP ring is aromatic in both the RS and TS with values close to those published for similar pryimidine analogs.83 However, the TS consistently shows lower aromatic character than the RS, which supports our hypothesis of aromaticity regulating ylide formation.
Table 3.
Calculated NICS Values for the WMM and DHM RS and TSa
| DHM-RS | DHM-TS | ΔNICS | |
|---|---|---|---|
| NICS(1) | −7.1 | −5.7 | 1.4 |
| NICS(0) | −5.0 | −2.7 | 2.3 |
| NICS(−1) | −9.3 | −6.7 | 2.6 |
| average | 2.1 | ||
|
| |||
| WMM-RS | WMM-TS | ΔNICS | |
|
| |||
| NICS(1) | −6.8 | −4.7 | 2.1 |
| NICS(0) | −4.6 | −2.2 | 2.5 |
| NICS(−1) | −8.8 | −6.3 | 2.5 |
| average | 2.4 | ||
The NICS(0) values are taken from the center of the pyrimidine ring. The NICS(1) and NICS(−1) values are points away and toward a proximal phenylalanine (F398), respectively. A comparison set of benzene (−9.8) and cyclobutadiene (27.6) were computed to show reference aromatic and antiaromatic values, respectively. The average ΔNICS values represent a 29.4% and 35.6% decrease in aromaticity for DHM and WMM, respectively.
Calculating the ASE for TDP’s AP state should provide additional information about the importance of aromaticity in ylide formation. A homodesmotic reaction (Figure 11) provides a reference for determining ASE. Thiamin serves as a model compound for this purpose and represents the key components (e.g., 4′-amino and thiazolium moieties) of TDP. Systems with positive values of ASE are considered to be aromatic, whereas those with negative values are antiaromatic. ASE values are determined as the difference in energies between both halves of the reference reaction (Figure 11). Thiamin has an ASE of 37.6 kcal·mol−1, which is again in close agreement with previously published results of similar pyrimidine derivatives.83
Figure 11.
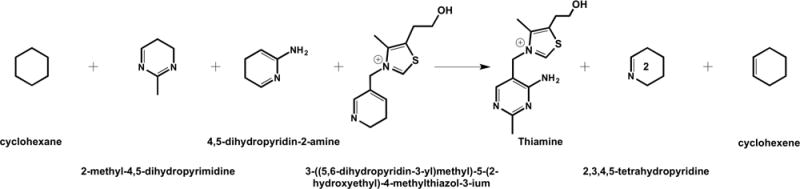
Homodesmotic reaction used in evaluating the aromatic stabilization energy for a model TDP.
Combining ASE values with the average decrease in aromaticity (i.e., ΔNICS, Table 3), we approximate the stabilization lost at the TS of each mechanism. Aromaticity losses of 13.4 kcal·mol−1 and 11.1 kcal·mol−1 for WMM and DHM were computed, respectively. Interestingly, we again observe the DHM TS being less destabilized when compared to the WMM TS. This provides further evidence that DXS is well adapted to stabilizing alternative mechanisms of TDP activation. Finally, when the total barrier heights are considered, it becomes clear that the loss of aromaticity plays a major role in TDP activation and the initial step of isoprenoid biosynthesis.
CONCLUSION
The ΔΔE‡ of 10.0 kcal·mol−1 difference between the WMM and DHM mechanisms indicates the WMM is the energetically favorable route for ylide formation in DXS. The RPATh + RESDi results seem to suggest the mechanism of proton transfer acts in a concerted fashion proceeding via the tautomeric route between the AP and IP state. Further investigation is ongoing to confirm the relative energetics of a stepwise versus concerted mechanism.
CPA results were indicative of H434 playing a role in longrange electrostatic stabilization, which is more clearly illustrated upon examination of the RS and TS active site dipole moments. Mutagenesis studies performed reveal H434 to play a role in substrate binding but not likely a direct role in catalysis. The H434A mutant results reinforce the CPA results. Additionally, a D430A mutant revealed a lower catalytic significance for DXS in comparison to the corresponding yeast TK mutant,91 again illustrating mechanistic differences. Furthermore, H82A and H304A DXS mutants showed significant decreases in activity (2–12% of wild-type). Given their proximity and retention of measurable activity, it is likely these residues function as backups to each other. This comports nicely with computational results that indicate DXS is well suited to functioning via alternative mechanisms (i.e., different general bases), something that would offer a significant evolutionary advantage.
ΔE‡ WMM is significantly higher than what might be expected for a relatively simple proton transfer.90 CPA results could not account for such behavior. However, upon examination of the 4′-amino moiety, it was evident from structural changes that aromaticity may be changing. The results of NICS and ASE calculations showed that indeed the AP ring was losing aromaticity. If the aromatic contribution is taken into consideration, the new ΔE‡WMM would be closer to 8.5 kcal·mol−, which is in the range of similar reactions. This clearly shows that loss of aromaticity plays a key role in controlling activation of TDP in DXS. Further, restoration of this aromaticity upon intramolecular proton transfer from the C2 of the thiazolium ring to the 4′-amino group should ultimately drive the final ylide formation.
Supplementary Material
Acknowledgments
H.L.W. acknowledges NIH (Grants 1K22HL088341-01A1, 4K22311045-02), NSF (Grant CHE-1464946), and the University of South Florida (start-up) for funding. Additionally, D.J.M. acknowledges funding from two NIH grants (Grants RO3-DA034323 and R15-GM107864). Computations were performed at the USF Research Computing Center (NSF Grant CHE-1531590). This research was supported in part by a seed grant from the Florida Center of Excellence for Biomolecular Identification and Targeted Therapeutics (FCoE-BITT) to D.J.M. and a Graduate Multidisciplinary Scholar (GMS) award from FCoE-BITT to S.H.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.6b07248.
PDB information for the transition states of the WMM and DHM; link atom details for QM/MM reaction path calculations; CPA results; topology and parameter files for TDP; methods for the overexpression and purification of DXS and DXR; steady-state initial velocity plots with varying concentrations of G3P and pyruvate; details about 20 ns simulations; results of PROPKA3.1 calculations on DXS chain A (PDF)
The pKa for the N1 atom of DXS was determined via pH rate profile studies. The E373 pKa was estimated using PROPKA3.1 with TDP but without pyruvate and G3P. The complete output of the PROPKA3.1 calculation can be found in the Supporting Information.
Notes
The authors declare no competing financial interest.
References
- 1.Sacchettini JC, Poulter CD. Biochemistry: Creating Isoprenoid Diversity. Science. 1997;277:1788–1789. doi: 10.1126/science.277.5333.1788. [DOI] [PubMed] [Google Scholar]
- 2.Kuzuyama T, Seto H. Diversity of the Biosynthesis of the Isoprene Units. Nat Prod Rep. 2003;20:171–183. doi: 10.1039/b109860h. [DOI] [PubMed] [Google Scholar]
- 3.Rohmer M. The Mevalonate-Independent Methylerythritol 4-Phosphate (MEP) Pathway for Isoprenoid Biosynthesis, Including Carotenoids. Pure Appl Chem. 1999;71:2279–2284. [Google Scholar]
- 4.Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN. Identification, Evolution, and Essentiality of the Mevalonate Pathway for Isopentenyl Diphosphate Biosynthesis in Gram-Positive Cocci. J Bacteriol. 2000;182:4319–4327. doi: 10.1128/jb.182.15.4319-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaykin S, Law J, Phillips AH, Tchen TT, Bloch K. Phosphorylated Intermediates in the Synthesis of Squalene. Proc Natl Acad Sci U S A. 1958;44:998–1004. doi: 10.1073/pnas.44.10.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloch K, Chaykin S, Phillips AH, Dewaard A. Mevalonic Acid Pyrophosphate and Isopentenylpyrophosphate. J Biol Chem. 1959;234:2595–2604. [PubMed] [Google Scholar]
- 7.Rohmer M, Knani M, Simonin P, Sutter B, Sahm H. Isoprenoid Biosynthesis in Bacteria: A Novel Pathway for the Early Steps Leading to Isopentenyl Diphosphate. Biochem J. 1993;295:517–524. doi: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenreich W, Schwarz M, Cartayrade A, Arigoni D, Zenk MH, Bacher A. The Deoxyxylulose Phosphate Pathway of Terpenoid Biosynthesis in Plants and Microorganisms. Chem Biol. 1998;5:R221–R233. doi: 10.1016/s1074-5521(98)90002-3. [DOI] [PubMed] [Google Scholar]
- 9.Lichtenthaler HK. The 1-Deoxy-D-Xylulose-5-Phosphate Pathway of Isoprenoid Biosynthesis in Plants. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:47–65. doi: 10.1146/annurev.arplant.50.1.47. [DOI] [PubMed] [Google Scholar]
- 10.Eisenreich W, Bacher A, Arigoni D, Rohdich F. Biosynthesis of Isoprenoids via the Non-Mevalonate Pathway. Cell Mol Life Sci. 2004;61:1401–1426. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Handa S, Ramamoorthy D, Spradling TJ, Guida WC, Adams JH, Bendinskas KG, Merkler DJ. Production of Recombinant 1-Deoxy-D-Xylulose-5-Phosphate Synthase from Plasmodium Vivax in Escherichia Coli. FEBS Open Bio. 2013;3:124–129. doi: 10.1016/j.fob.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramamoorthy D, Handa S, Merkler D, Guida W. Plasmodium Vivax 1-Deoxy-D-Xylulose-5-Phosphate Synthase: Homology Modeling, Domain Swapping, and Virtual Screening. J Data Min Genomics Proteomics. 2014 doi: 10.4172/2153-0602.S1-003. [DOI] [Google Scholar]
- 13.Jomaa H, et al. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 14.Kuzuyama T, Shimizu T, Takahashi S, Seto H. Fosmidomycin, a Specific Inhibitor of 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase in the Nonmevalonate Pathway for Terpenoid Biosynthesis. Tetrahedron Lett. 1998;39:7913–7916. [Google Scholar]
- 15.Zeidler J, Schwender J, Muller C, Wiesner J, Weidemeyer C, Beck E, Jomaa H, Lichtenthaler HK. Inhibition of the Non-Mevalonate 1-Deoxy-D-Xylulose-5-Phosphate Pathway of Plant Isoprenoid Biosynthesis by Fosmidomycin. Z Naturforsch, C: J Biosci. 1998;53:980–986. [Google Scholar]
- 16.Heuston S, Begley M, Gahan CGM, Hill C. Isoprenoid Biosynthesis in Bacterial Pathogens. Microbiology. 2012;158:1389–1401. doi: 10.1099/mic.0.051599-0. [DOI] [PubMed] [Google Scholar]
- 17.Dairi T, Kuzuyama T, Nishiyama M, Fujii I. Convergent Strategies in Biosynthesis. Nat Prod Rep. 2011;28:1054–1086. doi: 10.1039/c0np00047g. [DOI] [PubMed] [Google Scholar]
- 18.Sprenger GA, Schorken U, Wiegert T, Grolle S, DeGraaf AA, Taylor SV, Begley TP, BringerMeyer S, Sahm H. Identification of a Thiamin-Dependent Synthase in Escherichia Coli Required for the Formation of the 1-Deoxy-D-Xylulose 5-Phosphate Precursor to Isoprenoids, Thiamin, and Pyridoxol. Proc Natl Acad Sci U S A. 1997;94:12857–12862. doi: 10.1073/pnas.94.24.12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harker M, Bramley PM. Expression of Prokaryotic 1-Deoxy-D-Xylulose-5-Phosphatases in Escherichia Coli Increases Carotenoid and Ubiquinone Biosynthesis. FEBS Lett. 1999;448:115–119. doi: 10.1016/s0014-5793(99)00360-9. [DOI] [PubMed] [Google Scholar]
- 20.Matthews PD, Wurtzel ET. Metabolic Engineering of Carotenoid Accumulation in Escherichia Coli by Modulation of the Isoprenoid Precursor Pool with Expression of Deoxyxylulose Phosphate Synthase. Appl Microbiol Biotechnol. 2000;53:396–400. doi: 10.1007/s002530051632. [DOI] [PubMed] [Google Scholar]
- 21.Bouvier F, d’Harlingue A, Suire C, Backhaus RA, Camara B. Dedicated Roles of Plastid Transketolases during the Early Onset of Isoprenoid Biogenesis in Pepper Fruits. Plant Physiol. 1998;117:1423–1431. doi: 10.1104/pp.117.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Estevez JM, Cantero A, Reindl A, Reichler S, Leon P. 1-Deoxy-D-Xylulose-5-Phosphate Synthase, a Limiting Enzyme for Plastidic Isoprenoid Biosynthesis in Plants. J Biol Chem. 2001;276:22901–22909. doi: 10.1074/jbc.M100854200. [DOI] [PubMed] [Google Scholar]
- 23.Lange BM, Wildung MR, McCaskill D, Croteau R. A Family of Transketolases That Directs Isoprenoid Biosynthesis via a Mevalonate-Independent Pathway. Proc Natl Acad Sci U S A. 1998;95:2100–2104. doi: 10.1073/pnas.95.5.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lois LM, Campos N, Putra SR, Danielsen K, Rohmer M, Boronat A. Cloning and Characterization of a Gene from Escherichia Coli Encoding a Transketolase-Like Enzyme That Catalyzes the Synthesis of D-1-Deoxyxylulose 5-Phosphate, a Common Precursor for Isoprenoid, Thiamin, and Pyridoxol Biosynthesis. Proc Natl Acad Sci U S A. 1998;95:2105–2110. doi: 10.1073/pnas.95.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lois LM, Rodriguez-Concepcion M, Gallego F, Campos N, Boronat A. Carotenoid Biosynthesis during Tomato Fruit Development: Regulatory Role of 1-Deoxy-D-Xylulose 5-Phosphate Synthase. Plant J. 2000;22:503–513. doi: 10.1046/j.1365-313x.2000.00764.x. [DOI] [PubMed] [Google Scholar]
- 26.Begley TP, et al. Thiamin Biosynthesis in Prokaryotes. Arch Microbiol. 1999;171:293–300. doi: 10.1007/s002030050713. [DOI] [PubMed] [Google Scholar]
- 27.Hill RE, Himmeldirk K, Kennedy IA, Pauloski RM, Sayer BG, Wolf E, Spenser ID. The Biogenetic Anatomy of Vitamin B-6 - a C-13 NMR Investigation of the Biosynthesis of Pyridoxol in Escherichia Coli. J Biol Chem. 1996;271:30426–30435. doi: 10.1074/jbc.271.48.30426. [DOI] [PubMed] [Google Scholar]
- 28.Bailey AM, Mahapatra S, Brennan PJ, Crick DC. Identification, Cloning, Purification, and Enzymatic Characterization of Mycobacterium Tuberculosis 1-Deoxy-D-Xylulose 5-Phosphate synthase. Glycobiology. 2002;12:813–820. doi: 10.1093/glycob/cwf100. [DOI] [PubMed] [Google Scholar]
- 29.Schenk G, Leeper FJ, England R, Nixon PF, Duggleby RG. Investigation of the Mechanistic Functions of Residues HIS113 and HIS114 in Pyruvate Decarboxylase from Zymomonas Mobilis: A Proposed Model in the Binding of the Substrate Pyruvate. FASEB J. 1997;11:A1135–A1135. [Google Scholar]
- 30.Costelloe SJ, Ward JM, Dalby PA. Evolutionary Analysis of the TPP-Dependent Enzyme Family. J Mol Evol. 2008;66:36–49. doi: 10.1007/s00239-007-9056-2. [DOI] [PubMed] [Google Scholar]
- 31.Frank RAW, Leeper FJ, Luisi BF. Structure, Mechanism and Catalytic Duality of Thiamine-Dependent Enzymes. Cell Mol Life Sci. 2007;64:892–905. doi: 10.1007/s00018-007-6423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiang S, Usunow G, Lange G, Busch M, Tong L. Crystal Structure of 1-Deoxy-D-Xylulose 5-Phosphate Synthase, a Crucial Enzyme for Isoprenoids Biosynthesis. J Biol Chem. 2007;282:2676–2682. doi: 10.1074/jbc.M610235200. [DOI] [PubMed] [Google Scholar]
- 33.Breslow R. The Mechanism of Thiamine Action 0.2. Rapid Deuterium Exchange in Thiazolium Salts. J Am Chem Soc. 1957;79:1762–1763. [Google Scholar]
- 34.Breslow R. On the Mechanism of Thiamine Action 0.4. Evidence from Studies on Model Systems. J Am Chem Soc. 1958;80:3719–3726. [Google Scholar]
- 35.Jordan F, Li HJ, Brown A. Remarkable Stabilization of Zwitterionic Intermediates May Account for a Billion-Fold Rate Acceleration by Thiamin Diphosphate-Dependent Decarboxylases. Biochemistry. 1999;38:6369–6373. doi: 10.1021/bi990373g. [DOI] [PubMed] [Google Scholar]
- 36.Meyer D, Neumann P, Ficner R, Tittmann K. Observation of a Stable Carbene at the Active Site of a Thiamin Enzyme. Nat Chem Biol. 2013;9:488–490. doi: 10.1038/nchembio.1275. [DOI] [PubMed] [Google Scholar]
- 37.Singleton CK, Wang JJL, Shan L, Martin PR. Conserved Residues Are Functionally Distinct within Transketolases of Different Species. Biochemistry. 1996;35:15865–15869. doi: 10.1021/bi9616920. [DOI] [PubMed] [Google Scholar]
- 38.Eubanks LM, Poulter CD. Rhodobacter Capsulatus 1-Deoxy-D-Xylulose 5-Phosphate Synthase: Steady-State Kinetics and Substrate Binding. Biochemistry. 2003;42:1140–1149. doi: 10.1021/bi0205303. [DOI] [PubMed] [Google Scholar]
- 39.Brammer LA, Meyers CF. Revealing Substrate Promiscuity of 1-Deoxy-D-Xylulose 5-Phosphate Synthase. Org Lett. 2009;11:4748–4751. doi: 10.1021/ol901961q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsue Y, Mizuno H, Tomita T, Asami T, Nishiyama M, Kuzuyama T. The Herbicide Ketoclomazone Inhibits 1-Deoxy-D-Xylulose 5-Phosphate Synthase in the 2-C-Methyl-D-Erythritol 4-Phosphate Pathway and Shows Antibacterial Activity against Haemophilus Influenzae. J Antibiot. 2010;63:583–588. doi: 10.1038/ja.2010.100. [DOI] [PubMed] [Google Scholar]
- 41.Sisquella X, de Pourcq K, Alguacil J, Robles J, Sanz F, Anselmetti D, Imperial S, Fernandez-Busquets X. A single-molecule force spectroscopy nanosensor for the identification of new antibiotics and antimalarials. FASEB J. 2010;24:4203–4217. doi: 10.1096/fj.10-155507. [DOI] [PubMed] [Google Scholar]
- 42.Brammer LA, Smith JM, Wade H, Meyers CF. 1-Deoxy-D-Xylulose 5-Phosphate Synthase Catalyzes a Novel Random Sequential Mechanism. J Biol Chem. 2011;286:36522–36531. doi: 10.1074/jbc.M111.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brandt GS, Nemeria N, Chakraborty S, McLeish MJ, Yep A, Kenyon GL, Petsko GA, Jordan F, Ringe D. Probing the Active Center of Benzaldehyde Lyase with Substitutions and the Pseudosubstrate Analogue Benzoylphosphonic Acid Methyl Ester. Biochemistry. 2008;47:7734–43. doi: 10.1021/bi8004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maraite A, Schmidt T, Ansorge-Schumacher MB, Brzozowski AM, Grogan G. Structure of the ThDP-Dependent Enzyme Benzaldehyde Lyase Refined to 1.65 a Resolution. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2007;63:546–548. doi: 10.1107/S1744309107028576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaplun A, Binshtein E, Vyazmensky M, Steinmetz A, Barak Z, Chipman DM, Tittmann K, Shaanan B. Glyoxylate Carboligase Lacks the Canonical Active Site Glutamate of Thiamine-Dependent Enzymes. Nat Chem Biol. 2008;4:113–118. doi: 10.1038/nchembio.62. [DOI] [PubMed] [Google Scholar]
- 46.Brammer Basta LA, Patel H, Kakalis L, Jordan F, Freel Meyers CL. Defining Critical Residues for Substrate Binding to 1-Deoxy-D-Xylulose 5-Phosphate Synthase - Active Site Substitutions Stabilize the Predecarboxylation Intermediate C2α-Lactylthiamin Diphosphate. FEBS J. 2014;281:2820–2837. doi: 10.1111/febs.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jordan F. Current Mechanistic Understanding of Thiamin Diphosphate Dependent Enzymatic Reactions. Nat Prod Rep. 2003;20:184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- 48.Querol-Audi J, Boronat A, Centelles JJ, Imperial S. Catalytically Important Residues in E. coli 1-Deoxy-D-Xylulose 5-Phosphate Synthase. J Biosci Med. 2014;02:30–35. [Google Scholar]
- 49.Nikkola M, Lindqvist Y, Schneider G. Refined Structure of Transketolase From Saccharomyces Cerevisiae at 2 Angstrom Resolution. J Mol Biol. 1994;238:387–404. doi: 10.1006/jmbi.1994.1299. [DOI] [PubMed] [Google Scholar]
- 50.Wikner C, Meshalkina L, Nilsson U, Nikkola M, Lindqvist Y, Sundstrom M, Schneider G. Analysis of an Invariant Cofactor-Protein Interaction in Thiamin Diphosphate-Dependent Enzymes by Site-Directed Mutagenesis: Glutamic-Acid-418 in Transketolase Is Essential for Catalysis. J Biol Chem. 1994;269:32144–32150. [PubMed] [Google Scholar]
- 51.Balakrishnan A, Gao Y, Moorjani P, Nemeria NS, Tittmann K, Jordan F. Bifunctionality of the Thiamin Diphosphate Cofactor: Assignment of Tautomeric/Ionization States of the 4′-Aminopyrimidine Ring When Various Intermediates Occupy the Active Sites during the Catalysis of Yeast Pyruvate Decarboxylase. J Am Chem Soc. 2012;134:3873–3885. doi: 10.1021/ja211139c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balakrishnan A, Paramasivam S, Chakraborty S, Polenova T, Jordan F. Solid-State Nuclear Magnetic Resonance Studies Delineate the Role of the Protein in Activation of Both Aromatic Rings of Thiamin. J Am Chem Soc. 2012;134:665–672. doi: 10.1021/ja209856x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nemeria NS, Chakraborty S, Balakrishnan A, Jordan F. Reaction Mechanisms of Thiamin Diphosphate Enzymes: Defining States of Ionization and Tautomerization of the Cofactor at Individual Steps. FEBS J. 2009;276:2432–2446. doi: 10.1111/j.1742-4658.2009.06964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel H, Nemeria NS, Brammer LA, Freel Meyers CL, Jordan F. Observation of Thiamin-Bound Intermediates and Microscopic Rate Constants for Their Interconversion on 1-Deoxy-D-Xylulose 5-Phosphate Synthase: 600-Fold Rate Acceleration of Pyruvate Decarboxylation by D-Glyceraldehyde-3-Phosphate. J Am Chem Soc. 2012;134:18374–18379. doi: 10.1021/ja307315u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jordan F, Nemeria NS. Progress in the Experimental Observation of Thiamin Diphosphate-Bound Intermediates on Enzymes and Mechanistic Information Derived from These Observations. Bioorg Chem. 2014;57:251–262. doi: 10.1016/j.bioorg.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 56.Bas DC, Rogers DM, Jensen JH. Very Fast Prediction and Rationalization of pKa Values for Protein-Ligand Complexes. Proteins: Struct, Funct Genet. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Robertson AD, Jensen JH. Very Fast Empirical Prediction and Rationalization of Protein pKa Values. Proteins: Struct, Funct Genet. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 58.Olsson MHM, Sondergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 59.Sondergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. J Chem Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 60.Guo FS, Zhang DQ, Kahyaoglu A, Farid RS, Jordan F. Is a Hydrophobic Amino Acid Required to Maintain the Reactive v Conformation of Thiamin at the Active Center of Thiamin Diphosphate-Requiring Enzymes? Experimental and Computational Studies of Isoleucine 415 of Yeast Pyruvate Decarboxylase. Biochemistry. 1998;37:13379–13391. doi: 10.1021/bi9807097. [DOI] [PubMed] [Google Scholar]
- 61.Miller BT, Singh RP, Klauda JB, Hodoscek M, Brooks BR, Woodcock HL. III HARMMing: A New, Flexible Web Portal for CHARMM. J Chem Inf Model. 2008;48:1920–1929. doi: 10.1021/ci800133b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vanommeslaeghe K, et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kern D, Kern G, Neef H, Tittmann K, KillenbergJabs M, Wikner C, Schneider G, Hubner G. How Thiamine Diphosphate Is Activated in Enzymes. Science. 1997;275:67–70. doi: 10.1126/science.275.5296.67. [DOI] [PubMed] [Google Scholar]
- 64.Lindqvist Y, Schneider G, Ermler U, Sundstrom M. 3-Dimensional Structure of Transketolase, a Thiamine Diphosphate Dependent Enzyme, at 2.5 Angstrom Resolution. EMBO J. 1992;11:2373–2379. doi: 10.1002/j.1460-2075.1992.tb05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacKerell AD, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 66.Brooks BR, et al. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shao Y, et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys Chem Chem Phys. 2006;8:3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 68.Woodcock HL, Hodoscek M, Brooks BR. Exploring SCC-DFTB Paths for Mapping QM/MM Reaction Mechanisms. J Phys Chem A. 2007;111:5720–5728. doi: 10.1021/jp0714217. [DOI] [PubMed] [Google Scholar]
- 69.Becke AD. Density-Functional Thermochemistry 0.3. The Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 70.Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys Rev B: Condens Matter Mater Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 71.Hariharan PC, Pople JA. Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theor Chim Acta. 1973;28:213–222. [Google Scholar]
- 72.Chu JW, Trout BL, Brooks BR. A Super-Linear Minimization Scheme for the Nudged Elastic Band Method. J Chem Phys. 2003;119:12708–12717. [Google Scholar]
- 73.Woodcock HL, Hodoscek M, Gilbert ATB, Gill PMW, Schaefer HF, Brooks BR. Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations. J Comput Chem. 2007;28:1485–1502. doi: 10.1002/jcc.20587. [DOI] [PubMed] [Google Scholar]
- 74.Bash PA, Field MJ, Davenport RC, Petsko GA, Ringe D, Karplus M. Computer-Simulation and Analysis of the Reaction Pathway of Triosephosphate Isomerase. Biochemistry. 1991;30:5826–5832. doi: 10.1021/bi00238a003. [DOI] [PubMed] [Google Scholar]
- 75.Cui Q, Elstner M, Kaxiras E, Frauenheim T, Karplus M. A QM/MM Implementation of the Self-Consistent Charge Density Functional Tight Binding (SCC-DFTB) Method. J Phys Chem B. 2001;105:569–585. [Google Scholar]
- 76.Lee YS, Worthington SE, Krauss M, Brooks BR. Reaction Mechanism of Chorismate Mutase Studied by the Combined Potentials of Quantum Mechanics and Molecular Mechanics. J Phys Chem B. 2002;106:12059–12065. [Google Scholar]
- 77.Hargis JC, Vankayala SL, White JK, Woodcock HL. Identification and Characterization of Non-covalent Interactions that Drive Binding and Specificity in DD-Peptidases and β-Lactamases. J Chem Theory Comput. 2014;10:855–864. doi: 10.1021/ct400968v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.von Schleyer PR, Jiao HJ. What Is Aromaticity? Pure Appl Chem. 1996;68:209–218. [Google Scholar]
- 79.Cyranski MK, Krygowski TM, Katritzky AR, Schleyer PV. To What Extent Can Aromaticity Be Defined Uniquely? J Org Chem. 2002;67:1333–1338. doi: 10.1021/jo016255s. [DOI] [PubMed] [Google Scholar]
- 80.Cyranski MK, Schleyer PV, Krygowski TM, Jiao HJ, Hohlneicher G. Facts and Artifacts about Aromatic Stability Estimation. Tetrahedron. 2003;59:1657–1665. [Google Scholar]
- 81.Corminboeuf C, Heine T, Seifert G, Schleyer PV, Weber J. Induced Magnetic Fields in Aromatic n-Annulenes - Interpretation of NICS Tensor Components. Phys Chem Chem Phys. 2004;6:273–276. [Google Scholar]
- 82.Chen ZF, Wannere CS, Corminboeuf C, Puchta R, Schleyer PV. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem Rev. 2005;105:3842–3888. doi: 10.1021/cr030088+. [DOI] [PubMed] [Google Scholar]
- 83.Alonso M, Miranda C, Martin N, Herradon B. Chemical Applications of Neural Networks: Aromaticity of Pyrimidine Derivatives. Phys Chem Chem Phys. 2011;13:20564–20574. doi: 10.1039/c1cp22001b. [DOI] [PubMed] [Google Scholar]
- 84.Bao P, Yu Z-H. New Procedure to Evaluate Aromaticity at the Density Functional Theory, Hartree-Fock, and Post-Self-Consistent Field Levels. J Comput Chem. 2011;32:248–259. doi: 10.1002/jcc.21614. [DOI] [PubMed] [Google Scholar]
- 85.Krygowski TM, Cyranski MK, Czarnocki Z, Hafelinger G, Katritzky AR. Aromaticity: A Theoretical Concept of Immense Practical Importance. Tetrahedron. 2000;56:1783–1796. [Google Scholar]
- 86.Wheeler SE, Houk KN, Schleyer PVR, Allen WD. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J Am Chem Soc. 2009;131:2547–2560. doi: 10.1021/ja805843n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-Directed Mutagenesis by Overlap Extension using the Polymerase Chain Reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 88.Humnabadkar V, Jha RK, Ghatnekar N, De Sousa SM. A High-Throughput Screening Assay for Simultaneous Selection of Inhibitors of Mycobacterium Tuberculosis 1-Deoxy-D-Xylulose-5-Phosphate Synthase (DXS) or 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (DXR) J Biomol Screening. 2011;16:303–312. doi: 10.1177/1087057110394845. [DOI] [PubMed] [Google Scholar]
- 89.Mitschke L, Parthier C, Schroder-Tittmann K, Coy J, Ludtke S, Tittmann K. The Crystal Structure of Human Transketolase and New Insights into Its Mode of Action. J Biol Chem. 2010;285:31559–31570. doi: 10.1074/jbc.M110.149955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perrin CL, Nielson JB. “Strong” hydrogen bonds in chemistry and biology. Annu Rev Phys Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 91.Nilsson U, Meshalkina L, Lindqvist Y, Schneider G. Examination of Substrate Binding in Thiamin Diphosphate-Dependent Transketolase by Protein Crystallography and Site-Directed Mutagenesis. J Biol Chem. 1997;272:1864–1869. doi: 10.1074/jbc.272.3.1864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.