

Significance
Malaria parasites develop within red blood cells inside a membrane-enclosed parasitophorous vacuole. An essential step in their life cycle is the exit of mature parasites from the blood cell, a multistage process termed egress. To do this, the parasites orchestrate a highly regulated sequence of membrane permeabilization and breakage steps culminating in the explosive release of parasites for a new round of infection. Here, we describe a previously unidentified permeabilization of the vacuolar membrane at the start of egress, preceding membrane rupture, suggesting a new initiation step in egress. We also show that, in the final minutes of egress, the blood cell membrane abruptly loses its structural rigidity and collapses around the parasites, showing a precise timing for cytoskeletal breakdown.
Keywords: malaria, egress, electron tomography, soft X-ray microscopy, electron energy loss spectroscopy
Abstract
In the asexual blood stages of malarial infection, merozoites invade erythrocytes and replicate within a parasitophorous vacuole to form daughter cells that eventually exit (egress) by sequential rupture of the vacuole and erythrocyte membranes. The current model is that PKG, a malarial cGMP-dependent protein kinase, triggers egress, activating malarial proteases and other effectors. Using selective inhibitors of either PKG or cysteine proteases to separately inhibit the sequential steps in membrane perforation, combined with video microscopy, electron tomography, electron energy loss spectroscopy, and soft X-ray tomography of mature intracellular Plasmodium falciparum parasites, we resolve intermediate steps in egress. We show that the parasitophorous vacuole membrane (PVM) is permeabilized 10–30 min before its PKG-triggered breakdown into multilayered vesicles. Just before PVM breakdown, the host red cell undergoes an abrupt, dramatic shape change due to the sudden breakdown of the erythrocyte cytoskeleton, before permeabilization and eventual rupture of the erythrocyte membrane to release the parasites. In contrast to the previous view of PKG-triggered initiation of egress and a gradual dismantling of the host erythrocyte cytoskeleton over the course of schizont development, our findings identify an initial step in egress and show that host cell cytoskeleton breakdown is restricted to a narrow time window within the final stages of egress.
The major cause of severe human malaria is Plasmodium falciparum, and its asexual blood cycle is the source of all clinical disease (1). Egress is an important step in the blood life cycle, as it allows daughter merozoites produced by intracellular parasite replication to escape and invade new erythrocytes, thereby continuing and amplifying the infection. Merozoites develop within a parasitophorous vacuole (PV), a membrane-bound compartment that forms during invasion (2–4), so the daughter parasites have two compartments to escape (5, 6).
Blood-stage malaria parasites replicate by schizogony, in which several rounds of nuclear division form a multinucleated syncytium called a schizont. Individual merozoites are then produced by an unusual form of cytokinesis called budding or segmentation, which involves invagination of the single plasma membrane of the schizont. Minutes before egress, the segmented schizont suddenly transforms from an irregular to a relatively symmetrical structure with the merozoites arranged around the central digestive vacuole (5). This process, referred to as “flower formation” or rounding up, is usually accompanied by noticeable swelling of the PV and apparent shrinkage of the host cell (4, 5, 7–9). The first membrane to rupture at egress is the parasitophorous vacuole membrane (PVM) (5, 6, 8). When the PV does not occupy the entire infected cell, the individual merozoites can be seen to be expelled into the blood cell cytosol seconds before they escape from the erythrocyte (8–10). Erythrocyte membrane rupture involves formation of a single pore, followed by blebbing and vesiculation at the site of release (4, 5, 7, 8, 11). High-speed video microscopy and modeling of the erythrocyte membrane has suggested that the ruptured blood cell membrane then spontaneously curls open and inverts to facilitate merozoite dispersal (7, 12). However, the single pore is not the only disruption to the membrane as both membrane vesicles and ghosts generally remain after egress (5, 13). There is currently no corresponding model for escape from the PVM.
The precise biochemical mechanisms mediating egress are not fully elucidated, but multiple factors play a role. Minutes before initiation of egress, the malarial cGMP-dependent protein kinase G (PKG) is activated to regulate release of a parasite serine protease called SUB1 from specialized organelles called exonemes into the PV (14). SUB1 has several targets, including a multiprotein merozoite surface complex called MSP1/6/7 and a set of soluble papain-like PV proteins called the SERA family (15–18) that have been implicated in egress in the blood, liver, and mosquito stages of the parasite life cycle (19–22). Processing of the MSP1/6/7 complex is necessary for efficient escape from the erythrocyte through interaction of SUB1-processed MSP1 on the surface of the merozoites with spectrin of the host erythrocyte cytoskeleton (10). Proteolytic processing by SUB1 of the SERA proteins is temporally associated with egress (20, 23–27), and at least one P. falciparum SERA family member, SERA6, is likely a cysteine protease that is activated by SUB1 (16). As well as PKG, a parasite calcium-dependent kinase called CDPK5 has been implicated in egress (28). However, in CDPK5-deficient parasites, SUB1 discharge and MSP1/6/7 and SERA processing are unaffected, suggesting that CDPK5 action is either independent from or downstream of the PKG/SUB1/MSP/SERA pathway (28).
Events downstream of SUB1 activation and MSP/SERA processing are less well understood. SUB1 has multiple substrates in addition to the SERAs and MSP1/6/7 (29), and other proteases have been proposed to play a role in egress, including the erythrocyte protease calpain-1 (30–35). Proteomic studies based on analysis of parasite populations synchronized by physical techniques have indicated that erythrocyte membrane and cytoskeletal proteins are proteolysed during or before egress (13, 36). It has been suggested on the basis of that work and atomic force microscopy (AFM) that the host cell cytoskeleton is progressively degraded over several hours of the ∼48 h-long erythrocytic life cycle of P. falciparum (13). However, in these studies, the developmental status (time from egress) of the individual parasitized cells examined was unknown. Other effector proteins involved in membrane breakdown are not fully characterized. In the related apicomplexan parasite Toxoplasma gondii, the pore-forming perforin-like protein 1 (PLP1) mediates rupture of vacuole and host cell membranes (37), whereas in Plasmodium the perforin-like proteins PLP2 and PLP1 have been implicated in egress of gametes (38, 39) and asexual blood forms, respectively (40).
Despite the above insights into the molecules that regulate egress, many details remain obscure concerning the timing, order, and nature of the membrane perturbations involved. The selective PKG inhibitors compound 1 (C1) and compound 2 (C2) reversibly inhibit egress before the rounding up stage (10, 14, 28, 41). In contrast, treatment with the broad-spectrum cysteine protease inhibitor E64 allows PVM rupture but selectively prevents erythrocyte membrane rupture, resulting in merozoites trapped in the blood cell after rupture of the PVM (30, 42). Here we use single cell tracking by video microscopy, electron tomography, and X-ray tomography of C1- or C2-treated or E64-treated P. falciparum schizonts to capture and discriminate intermediate stages in egress to an unprecedented degree of temporal accuracy. We show that although the PKG inhibitors prevent PVM rupture, there is a previously undetected, initial step in egress in which permeabilization of the PVM occurs 10–30 min before its complete rupture into multilayered vesicles. Just before or at the point of PVM rupture, the blood cell cytoskeleton undergoes a sudden breakdown causing the red blood cell membrane to collapse around the intracellular parasites. Finally, the blood cell membrane becomes permeable seconds before merozoite escape.
Results
Electron Tomography of P. falciparum Schizonts Reveals the Fate of the PVM at Different Steps in Egress.
As asexual blood stages of P. falciparum invariably replicate asynchronously in vitro, resulting in “mixed” cultures that contain parasites at various stages of maturation, we enriched mature schizonts by centrifugation over cushions of Percoll. We then cultured these preparations for 4–6 h in the presence of the PKG inhibitors C1 or C2 to obtain parasite cultures containing a high proportion of schizonts arrested at a very late stage of maturation. Alternatively, we used E64 to obtain schizonts stalled in the process of egress (Fig. 1A). The schizonts were then vitrified by high-pressure freezing and freeze substituted into plastic resin for sectioning. Examination by electron tomography showed that the parasites in the C1-treated preparation fell into three broad groups: those in which segmentation (merozoite budding) had not begun (as indicated by a single multinucleated parasite, with a single membrane underlying the PVM), those at a stage of partial segmentation (in which the daughter merozoites are partially separated but still attached), and those (the majority) in which segmentation was complete, producing fully separated daughter merozoites. Importantly, all of the C1-blocked schizonts examined displayed an intact PVM, in accord with previous evidence that PKG inhibitors potently stall egress at a stage before rounding up and PVM rupture (10, 14, 28, 41). Consistent with this, in all partially segmented schizonts we observed a clear contrast difference between the material in the vacuole and the material in the blood cell cytosol, with the red cell cytosol displaying a darker, more electron-dense appearance than the contents of the PV (Fig. 1B, Top). This difference in staining indicated that the contents of these two compartments differed markedly in composition. Strikingly, many tomograms of fully segmented schizonts from these same C1-stalled preparations showed an equalization of contrast across the PVM (Fig. 1B, Bottom). This contrast equalization suggested that—despite the apparently intact PVM in these cells—the contents of the blood cell cytosol and the vacuole had undergone mixing. Further examples are shown in Fig. S1.
Fig. 1.

Tomography of C1 and E64 stalled schizonts reveals the fate of the PVM at different stages in egress. (A) Schematic indicating the steps at which egress is stalled by the inhibitors C1/C2 and E64. (B) Life cycle stage-specific equalization of vacuole and erythrocyte contents in C1-treated schizonts. Tomogram slices from freeze-substituted sections of a partially segmented schizont showing a contrast difference across the PVM (Top) and a fully segmented schizont with an equalization of contrast across the PVM (Bottom). Each image is the average of 10 central slices from a tomogram and is shown with and without outlining of key features. Red, blood cell membrane; yellow, PVM; cyan, merozoite plasma membrane; green, merozoite nuclei; purple, apical organelles. (Scale bar, 500 nm.) (C) Slices from tomograms of freeze-substituted sections of schizonts arrested by E64. Each slice is shown with and without outlining of key features. Brown, digestive vacuole; other colors as above. In Top, an asterisk marks a knob structure on the erythrocyte membrane. (Scale bar, 500 nm.) (D) Slices in the xy, xz, and yz planes from tomograms of membrane whorls in E64 stalled schizonts, demonstrating that they are spherical multilamellar vesicles. Lines in the xy image show the positions of the other planes. (Scale bar, 100 nm.)
Fig. S1.

Further examples of schizonts with and without equalization of contrast across the vacuole membrane. Tomogram slices of nonleaky (A) and leaky (B) schizonts with representative regions shown at increasing zoom to give an overview of the cells and show that the PVM is present. Arrows indicate PVM, and asterisks indicate knob structures. [Scale bar, (Left) 250 nm and (Middle and Right) 50 nm.]
To directly assess the composition of the two compartments, we applied electron energy loss spectroscopy (EELS) to analyze their elemental compositions (Fig. S2). Different elements have characteristic electron energy loss spectra, and the EELS method maps this information over the image. This analysis confirmed that the contrast difference corresponded to a difference in composition of the PV lumen and blood cell cytosol and that this difference disappeared in cells showing equalized contrast by EM.
Fig. S2.

EELS analysis confirms mixing of vacuole lumen and erythrocyte cytosol in C1-treated, mature schizonts. (A and B) Overview TEM image of a freeze-dried, vitrified cell section with a contrast difference between the vacuole and blood cell. Darkfield (DF) and nitrogen maps collected from the boxed region are shown on Right. The PVM is outlined in yellow, merozoite plasma membrane in cyan and erythrocyte membrane in red. (C and D) TEM overview image from a cell showing equalization of contrast across the PVM. DF and nitrogen maps collected from the boxed region are shown on Right. Outlining colors as in A and B. In contrast to A and B, C and D show no difference between nitrogen levels either side of the PVM, which is visible as a line of pixels with lower nitrogen content in C. (E) Plot of mean electron counts per pixel in the vacuole and blood cell cytosol from nitrogen maps from cells that do/do not appear leaky. Cells with a contrast difference contain approximately twice as much nitrogen in the blood cell cytosol than in the PV. In cells displaying contrast equalization, nitrogen levels were identical across the PVM, consistent with permeabilization of the PVM. Each paired score was from a different schizont (n = 7). *P < 0.001.
To quantify the stage specificity of the equalization of contrast across the PVM, the life cycle stage and presence or absence of a contrast difference were assessed and quantified in electron micrographs of thin sections of C1-treated as well as untreated schizonts (Table S1). Analysis of the C1-treated schizonts showed that contrast equalization was evident in ∼50% of fully segmented parasites but was only observed in one example of a partially segmented schizont out of 80 analyzed. Importantly, examination of schizonts prepared in the absence of PKG inhibitors also revealed contrast equalization in 16% of segmented schizonts. These results suggested that contrast equalization occurs largely or entirely following merozoite segmentation and moreover confirmed that the observed contrast equalization before PVM rupture was not an artifact of C1 treatment.
Table S1.
Equalization of red cell cytosol and PV contents across the PVM in compound 1-treated and untreated schizonts
| Development stage | Leakage state | Compound 1 treated | Untreated |
| Unsegmented schizonts | 113 | 79 | |
| Partially segmented schizonts | Nonleaky | 79 (99%) | 42 (100%) |
| Leaky | 1 (1%) | 0 (0%) | |
| Total | 80 | 42 | |
| Fully segmented schizonts | Nonleaky | 67 (49%) | 51 (84%) |
| Leaky | 71 (51%) | 10 (16%) | |
| Total | 138 | 61 | |
| Total schizonts | 331 | 182 | |
Compound 1-treated and untreated schizonts were imaged and assessed for their life cycle stage and the presence of leakage across the PVM. Percentages in parentheses refer to the percentage of cells with/without an equalization of contrast across the PVM at that life cycle stage. Unsegmented schizonts could not be assessed for leakage, as in most cases the parasites filled the entire vacuolar volume, with no gap between parasite membrane and PVM.
In contrast to the PKG inhibitors, the broad-spectrum cysteine protease inhibitor E64 does not prevent PVM rupture but potently prevents final rupture of the erythrocyte membrane. Consistent with this, tomography of schizonts arrested by E64 showed a single membrane surrounding the parasites and loss of the PV and blood cell contents (Fig. 1C). The presence of knob structures on the remaining enclosing membrane (Fig. 1C, Top) and staining under the membrane consistent with the presence of the erythrocyte cytoskeleton confirmed that this was the blood cell membrane. The empty appearance of the blood cell cytosol in the E64-arrested cells was consistent with previous descriptions of erythrocyte membrane poration preceding rupture (9, 40). In contrast to earlier parasite stages, the erythrocyte membrane shape was observed to closely follow the contours of the parasites in these cells (Fig. 1C). Also present were whorls of membrane vesicles, presumably the remains of the ruptured PVM. Tomography of the E64-treated schizonts enabled 3D visualization of the extensive, multilamellar nature of these vesicles (Fig. 1D).
Time Course of Membrane Permeabilization and Breakage During Egress.
To monitor the vacuole and blood cell cytosol compartments during the course of egress in live schizonts, we used a transgenic P. falciparum parasite line (called 3D7_mCherryEXP1) that expresses mCherry fused to the secretory signal sequence of the PV protein EXP1. This targets the fluorescent fusion protein to the PV lumen. Examination of mature, C1-treated mCherryEXP1 schizonts revealed that although in some cells the fluorescence signal was restricted to the PV, in others it extended into the erythrocyte cytosol (Fig. 2A), suggesting permeability of the PVM to proteins of the molecular mass of the mCherryEXP1 fusion (∼35.2 kDa) and consistent with the equalization of contrast observed by EM. These observations suggest that the PVM becomes permeable before activation of PKG, previously thought to be the first step in egress.
Fig. 2.

The PVM becomes permeable before its rupture. (A) DIC and fluorescence images of C1-treated schizonts expressing mCherry in the PV. Schizonts in which the fluorescence signal is restricted to the vacuole were classified as nonleaky (∼50% of 561 cells analyzed) or leaky when the fluorescence signal extended into erythrocyte cytosol (∼35%). In ∼15% of the schizonts, the vacuole occupied most of the volume of the infected cell, precluding detection of leakiness; these were classified as ambiguous. Note that the brightest feature in fluorescence is the food vacuole; the PV (dotted line) and erythrocyte (dashed line) are indicated on the DIC panels. (Scale bar, 2 μm.) (B) Graph showing the proportion of leaky cells plotted according to whether they egressed or not during a 30-min recording (total 234 cells from three separate recordings). Error bars, SD. More egressed than nonegressed cells were classified ambiguous as it was not possible to determine leakage, likely due to the PVM being less clearly visible in leaky cells. (C) Selected frames from a time-lapse video of egress. Fluorescence leaks from the PV into the blood cell between the first two depicted frames, ∼20 min before rounding up. Time after removal of the C2 block is indicated. (Scale bar, 2 μm.) Brightness and contrast have been linearly adjusted to the same levels for each of the frames shown. The progressive loss of fluorescence is due to photobleaching.
To further investigate the temporal relationship between PVM permeabilization and egress, mature mCherryEXP1 schizonts cultured for 3–4 h in the presence of C2 were washed to remove the inhibitor, allowing the cells to progress to egress. The schizonts were imaged during this process by time-lapse video microscopy to monitor changes in the distribution of fluorescence. Movies S1 and S2 and Fig. 2 B and C show that PVM rupture and egress were always preceded by leakage of the fluorescence into the host cell cytosol. Of the cells that egressed during the time-lapse experiments, half were already leaky at the start or became leaky during recording. Of the cells that did not egress, only 18% were observed to be leaky during the recording. Furthermore, 73% of these leaky cells that did not rupture rounded up, which occurs a few minutes before egress (5), indicating that they would have likely have undergone egress shortly after the 30-min recording time. All of the cells that were not leaky at the start of the videos but egressed during the recording became leaky 10–30 min before final escape of parasites (tracking of 14 cells). Leakage occurred several minutes before rounding up and while the PVM still appeared intact (Fig. 2C and Movies S1 and S2). These observations confirmed the EM and EELS data and showed that permeabilization of the PVM occurs only 10–30 min before PVM rupture and egress.
Soft X-Ray Cryotomography and Scanning Electron Microscopy Reveal Dramatic Changes in Membranes and Cell Shape During Egress.
To obtain further insights into the fate of the membranes in intact, C2-, or E64-treated cells without dehydration or sectioning, we used soft X-ray cryotomography, as the greater penetration depth of soft X-rays compared with electrons makes it possible to generate 3D reconstructions of intact, frozen-hydrated cells (reviewed in ref. 43). Although the resolution of this method is lower than for EM, the individual parasites and some of their organelles were clearly visible in the images (Fig. 3 A–F). In the E64-arrested schizonts, most of the contents of the erythrocyte cytosol were absent, confirming the tomographic data (Figs. 1 B and C and 3 B and E). In addition, the 3D overview of whole cells provided by these data revealed striking differences in shape and mechanical properties between the C2-blocked and E64-arrested schizonts. In the former case, with the intact vacuole present, the erythrocyte membrane formed a globular enclosure around the vacuole with an extending remnant of the original biconcave shape (Fig. 3C). In contrast, after PV breakdown in the E64-arrested schizonts, the erythrocyte membrane had collapsed around the parasites, following their contours (Fig. 3F). This collapse was also clearly present in the electron tomography sections (Fig. 1 B and C) and was seen in optical microscopy as a compaction and rounding up of the schizont (Fig. 2C, compare 6- and 26-min time points). Scanning EM of these cells confirmed the change in shape and membrane collapse (Fig. 3 G–J). In C2, 74% of the cells (790 cells examined) retained a vestige of the original biconcave disk shape, whereas in E64 48% (of a total of 759) showed the erythrocyte membrane collapsed around the parasites (Table S2). Because the rounding up and collapse occurred only a few minutes before final egress (Fig. 2C and Movies S1 and S2), these findings show that loss of mechanical integrity in the red cell membrane and cytoskeleton takes place only in the final stage of the egress pathway.
Fig. 3.

Soft X-ray tomography and SEM shows loss of mechanical integrity of the red cell membrane in the final stages of egress. (A) Slice from tomogram of C2-arrested schizont. (B) Outline of erythrocyte membrane (red), PVM (yellow), and parasites (cyan) in the tomogram slice in A. (C) 3D rendering of the schizont. The vacuole (yellow) is densely packed with merozoites (cyan) that have been collectively rather than individually rendered, for clarity. The overall height of the cell is ∼5 μm. (D) Tomogram slice from an E64-arrested schizont, shown with outlining of membranes in E. Remnants of the PVM are visible. (F) 3D rendering of the schizont. The top and bottom of the 3D structures appear truncated because of the missing data at tilt angles >60°. Nevertheless, the reconstructions show that the C2-blocked schizont has a rounded shape but that progression to the E64-arrested stage results in collapse of the host cell to a flattened sac ∼2 μm in height on the grid surface. (G–J) SEM of C2-blocked (G and H) and E64-blocked (I and J) schizonts shows collapse of the erythrocyte membrane just before egress. In the C2-blocked condition, the infected cells have a globular shape with a remnant of the original biconcave disk. In E64, the membrane collapses down over the parasites. Occasional uninfected erythrocytes can be seen (asterisk), providing an internal control for sample processing. [Scale bar, (A–F, H, and J) 1 μm and (G and I) 10 μm.]
Table S2.
Quantification of C2 and E64 blocked schizont morphologies by SEM
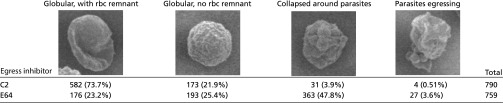 |
Cells were classified and counted according to their overall morphology. Representative images are given in the top row of the table. Numbers in parentheses refer to percentages for each of C2/E64.
Discussion
We have used selective pharmacological inhibition of steps in egress coupled with tomographic and live cell imaging to show that the PVM becomes permeable before its lysis, allowing contents of the PV lumen and erythrocyte cytosol to mix. Importantly, the occurrence of PVM poration in C1-arrested cells shows that it occurs upstream of or independent of PKG signaling and SUB1 discharge into the PV. Our observation reveals a previously unidentified prior step in egress, in which the PVM is permeabilized before being disrupted. Intriguingly, our findings mirror those of Sturm et al., who noted gradual permeabilization of the PVM before its complete rupture in liver stage schizonts of the rodent malarial species Plasmodium berghei (44). This suggests similarities in the fate of the PVM in these otherwise very different developmental stages of the parasite life cycle.
The fact that the leaky PVM is visibly intact in tomograms and contains no breakages or disruptions visible by electron tomography suggests that permeabilization is not due to major membrane breakage but rather to smaller perturbations such as formation of localized pores. The mechanism underlying PVM poration is unknown. In the related apicomplexan T. gondii, the pore-forming perforin-like protein PLP1 plays an important role in egress from the host cell by disrupting both the vacuole and the host cell membrane (37). The evidence for an analogous role for a PLP1 ortholog in malarial egress is less clear. Although PLP1 has been indirectly implicated in poration of the erythrocyte membrane in P. falciparum (40), genetic knockout of either the PLP1 or PLP2 homolog in P. berghei produced no phenotype in the asexual blood stages (39, 45), casting doubts on the essentiality of PLP1 or PLP2 function in these developmental stages.
Live cell video microscopy revealed that, after PVM poration in blood-stage schizonts, there follows a 10–20-min period without further evident structural changes, followed by the sudden rounding up of the blood cell and increased merozoite motility (Movies S1 and S2). EM of E64-stalled schizonts showed a total breakdown of the PVM into multilamellar vesicles (Fig. 1D). EM and X-ray tomography of parasites arrested with E64 at the post-PVM rupture stage showed that the erythrocyte membrane then becomes permeable, indicated by the complete loss of the residual hemoglobin and PV contents and consistent with the findings of others (9, 40). In the live cell video experiments, we observed this loss of blood cell cytosolic contents in about one third of the schizonts just before parasite escape. Given the 5-s increment between frames in our time-lapse movies (a compromise between time resolution and photobleaching), this fraction of detected events is consistent with a previous report that the interval between erythrocyte membrane poration and its rupture is of the order of a few seconds (9).
The second key finding of this study is that a major change in erythrocyte structure occurs rapidly after release of the C2 block (Fig. 3), showing that the erythrocyte membrane in E64-blocked schizonts loses its rigidity and collapses around the parasites. The rounded cells appear smaller in diameter (Fig. 2C). This loss of cell shape and alteration in mechanical properties, both of which have been observed in optical and AFM studies (46), implies that the erythrocyte cytoskeleton undergoes a very sudden, rapid loss of its mechanical integrity just before egress. Importantly, the observation is not in accord with a previous suggestion that the host cell cytoskeleton is progressively dismantled over several hours during schizont maturation (13, 36). In those earlier studies, the timing of cytoskeleton breakdown in individual cells could not be distinguished, as the analyses were of populations of cells in which egress was not blocked. Under these conditions, cells would gradually egress, accounting for the observed accumulation of cytoskeleton breakdown products. AFM analysis of schizonts from those populations showed cytoskeleton expansion but not loss of erythrocyte mechanical integrity, and does not provide the temporal resolution reported here, as the status of the individual parasites examined was unknown (13). The high degree of parasite synchrony afforded by the PKG inhibitors exploited in the present study, along with the much higher resolution and better cell preservation afforded by the microscopy methods used, gives us a much more accurate picture of these events. Our observations suggest that the effector mechanisms that degrade the host cell cytoskeleton to enable erythrocyte membrane rupture act very rapidly in the final stages of the egress pathway. A few seconds after cell collapse, the erythrocyte membrane breaks, allowing the final, explosive release of parasites. The fact that a similar, two-step breakdown (poration followed by rupture) occurs successively in the PVM and erythrocyte membrane raises the possibility that the effector molecules mediating poration of both membranes may be similar or identical.
In conclusion, our results suggest that poration of the PVM is the first detectable step in egress of the asexual blood-stage malaria parasite (Fig. 4). This occurs after parasite segmentation but before PKG-mediated SUB1 discharge. The mechanism underlying PVM poration is unknown, but as the leaky PVM appears intact, permeation is likely to arise from small, localized perforations such as membrane pores, potentially allowing small molecules such as calcium ions and parasite proteins into the residual erythrocyte where they can access the erythrocyte cytoskeleton and membrane. About 10 min after the first detection of PVM poration, the red blood cell rounds up, likely due to loss of mechanical integrity of the erythrocyte cytoskeleton mediated by protease activity. Around the same time, the PVM ruptures into multilamellar vesicles. In the following seconds, the blood cell membrane becomes permeable, rapidly followed by the explosive exit of the parasites. Identification of the effector molecules involved in PVM poration and cytoskeleton collapse may lead to new antimalarial therapies designed to interfere with progression of the malaria parasite blood stage life cycle.
Fig. 4.

Model of the stages of egress. The PVM undergoes poration before its rupture to form multilamellar vesicles. The cytoskeleton loses mechanical integrity and the erythrocyte membrane becomes porous before final parasite escape.
Methods
Parasite Culture and Egress Inhibitor Treatment.
P. falciparum clone 3D7 and parasite line 3D7_mCherryEXP1 (SI Methods for a description of generation of the latter) were cultured in human red blood cells in RPMI 1640 medium supplemented with Albumax (Invitrogen) as described previously (47, 48). Parasites were synchronized by centrifugation on cushions of 63% (vol/vol) Percoll (GE Healthcare) (47). Where inhibitor treatments were required, schizonts were cultured in medium containing either C1/C2 (2 μM) or E64 (50 μM) for 3–4 h before preparing the cells for imaging.
Live Cell Imaging.
Video and fluorescence microscopy were carried out on a Nikon Eclipse Ni-E microscope equipped with a Plan Apo 100×/1.45 oil immersion differential interference contrast (DIC) objective, a C11440 ORCA-Flash 4.0 digital CMOS camera (Hamamatsu), and a temperature-controlled stage held at 37 °C. Schizont suspensions in gassed medium were sealed into prewarmed viewing chambers constructed as described previously (14), immediately transferred to the microscope stage, and images and videos acquired using NIS Elements software (Nikon). Videos were started 5 min after the removal of the C1/2 block and were acquired over 30 min at 5-s intervals. Images and videos were analyzed using FIJI (49).
Electron Tomography.
Mature schizonts were pelleted by centrifugation, mixed with 20% (wt/vol) dextran in RPMI medium with C1/C2 (2 μm) or E64 (50 μm) where necessary and baker’s yeast, and frozen using a HPM010 high-pressure freezer (Baltec). Vitrified cells were freeze-substituted into Lowicryl HM20 resin with uranyl acetate stain and cut into thin sections. Tilt series were collected on a Tecnai F20 200 keV field emission gun microscope (FEI) or a Tecnai T12 120 keV tungsten filament microscope (FEI), equipped with a US4000 CCD camera (Gatan). Dual axis tilt series were typically collected from –66° to +66° with an increment of 2° using the acquisition software SerialEM (50).
Soft X-Ray Cryotomography.
C2- and E64-treated schizonts were fixed in 2% (vol/vol) paraformaldehyde in PBS, added to electron microscopy grids coated with 0.01% (wt/vol) poly-L-lysine (Sigma) and 250 nm gold fiducial markers (BBI solutions), and plunge frozen in liquid ethane. Tilt series were collected on the UltraXRM-S/L220c X-ray microscope (Zeiss, previously Xradia) at the B24 beamline of the Diamond synchrotron with a 1024 PixisB CCD camera (Princeton instruments) and a 40 nm zone plate with X-rays of 500 eV. Tilt series were typically collected from –60° to +60° with an increment of 0.5°.
Tomographic Reconstructions.
Electron and X-ray tomograms were reconstructed using etomo, part of the IMOD package (51). Postreconstruction, tomograms were denoised by nonlinear anisotropic diffusion filtering in IMOD. Segmentation of key features was done manually in IMOD.
Scanning EM.
Schizonts arrested with either C2 or E64 were fixed in 2.5% glutaraldehyde, washed, osmicated (1% OsO4 for 16 h), dehydrated, critical point dried, and sputter coated with 5 nm gold for scanning EM. Images were collected on a JEOL JSM 7610F with 2.6 kV accelerating voltage.
SI Methods
Generation of Parasite Line 3D6_mCherryEXP1.
Parasite line 3D7_mCherryEXP1 was generated by transfecting 3D7-AttB parasites (52) with the plasmids pINT (52) and pBLD432, which contain an expression cassette for the EXP1-mCherry fusion. To produce pBLD432, sequence encoding the EXP1 signal sequence was amplified by PCR using primers CVO075 (GGACcctaggATGAAAATCTTATCAGTATTTTTTCTTGCTC) and CVO077 (GGACagatctGTGTTCAGTGCCACTTACGAGGTTG) using genomic 3D7 DNA as a template. The PCR product was cloned into the AvrII and BglII sites of pA406 (a kind gift of A. Osborne, University College London, London) to form a fusion gene downstream of the CAM promoter. 3D7-AttB parasites were transfected as previously described (53). Transfectants were selected with 2.5 nM WR and 300 μg/mL G418 (Life Technologies).
EELS.
The 100 nm-thick cryosections were directly cut from high-pressure frozen cells, which were then freeze-dried and coated with 4 nm carbon. EELS data were collected on a JEOL 2100 200 keV transmission electron microscope with a LaB6 filament and a post column energy filter (Gatan GIF quantum) and Digital Micrograph (Gatan). Cell overview images were collected in EFTEM mode before switching to STEM mode for collecting EELS datasets. Spectrum images were typically collected using an exposure of 5–10 s per pixel, collected as five separate exposures, and summed. Typical pixel size was 20 nm × 20 nm × 0.5 eV.
EELS data were processed in Digital Micrograph (Gatan). Spectra were aligned and shifted to the correct energy onsets using the carbon and nitrogen edges, whose presence and position had been confirmed in the first spectra that were collected shortly after aligning the zero loss peak. In the spectra, nitrogen gave a strong signal and so was used to track the differences in composition between compartments. Nitrogen signal was extracted using a 30 eV window at 400 eV and a 25 eV background window at 373 eV and using the digital micrograph power law model for background subtraction. For semiquantitative analysis on material either side of the PVM, the darkfield image was used to determine the positions of the PV and blood cell cytosol, the electron counts for those pixels were extracted from the nitrogen maps, and the mean and SD for each region were calculated.
Supplementary Material
Acknowledgments
We thank Kirsty McLellan-Gibson, Daniel Clare, Giulia Zanetti, Carolyn Moores, Luchun Wang, Calum Dickinson, and David Houldershaw for help and discussions. We thank JEOL for SEM facilities. We acknowledge the support of Gatan BBSRC CASE PhD Studentship BB/F016948/1 (to V.L.H.), MRC Project Grant G1100013 (to H.R.S., M.J.B., and R.A.F.), and Wellcome Equipment Grants 101488, 079605, and 086018 (to H.R.S., M.J.B., and R.A.F.). This work was also supported by the Francis Crick Institute, MRC Grant U117532063 (to M.J.B.), Wellcome Trust ISSF2 funding to the London School of Hygiene and Tropical Medicine, and a Wellcome Trust Career Re-Entry Fellowship 095836/Z/1/Z (to C.v.O.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the EM Database for EM tomograms (accession nos. EMD-3586, EMD-3587, EMD-3606, and EMD-3610) and EMPIAR for X-ray tomograms (accession no. EMPIAR-10087).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1619441114/-/DCSupplemental.
References
- 1.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415(6872):673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 2.Aikawa M, Miller LH, Johnson J, Rabbege J. Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite. J Cell Biol. 1978;77(1):72–82. doi: 10.1083/jcb.77.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dluzewski AR, et al. Origins of the parasitophorous vacuole membrane of the malaria parasite, Plasmodium falciparum, in human red blood cells. J Cell Sci. 1992;102(Pt 3):527–532. doi: 10.1242/jcs.102.3.527. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak JA, Miller LH, Whitehouse WC, Shiroishi T. Invasion of erythrocytes by malaria merozoites. Science. 1975;187(4178):748–750. doi: 10.1126/science.803712. [DOI] [PubMed] [Google Scholar]
- 5.Glushakova S, Yin D, Li T, Zimmerberg J. Membrane transformation during malaria parasite release from human red blood cells. Curr Biol. 2005;15(18):1645–1650. doi: 10.1016/j.cub.2005.07.067. [DOI] [PubMed] [Google Scholar]
- 6.Wickham ME, Culvenor JG, Cowman AF. Selective inhibition of a two-step egress of malaria parasites from the host erythrocyte. J Biol Chem. 2003;278(39):37658–37663. doi: 10.1074/jbc.M305252200. [DOI] [PubMed] [Google Scholar]
- 7.Abkarian M, Massiera G, Berry L, Roques M, Braun-Breton C. A novel mechanism for egress of malarial parasites from red blood cells. Blood. 2011;117(15):4118–4124. doi: 10.1182/blood-2010-08-299883. [DOI] [PubMed] [Google Scholar]
- 8.Gilson PR, Crabb BS. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int J Parasitol. 2009;39(1):91–96. doi: 10.1016/j.ijpara.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Glushakova S, et al. New stages in the program of malaria parasite egress imaged in normal and sickle erythrocytes. Curr Biol. 2010;20(12):1117–1121. doi: 10.1016/j.cub.2010.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das S, et al. Processing of Plasmodium falciparum merozoite surface protein MSP1 activates a spectrin-binding function enabling parasite egress from RBCs. Cell Host Microbe. 2015;18(4):433–444. doi: 10.1016/j.chom.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winograd E, Clavijo CA, Bustamante LY, Jaramillo M. Release of merozoites from Plasmodium falciparum-infected erythrocytes could be mediated by a non-explosive event. Parasitol Res. 1999;85(8-9):621–624. doi: 10.1007/s004360050606. [DOI] [PubMed] [Google Scholar]
- 12.Callan-Jones A, Albarran Arriagada OE, Massiera G, Lorman V, Abkarian M. Red blood cell membrane dynamics during malaria parasite egress. Biophys J. 2012;103(12):2475–2483. doi: 10.1016/j.bpj.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millholland MG, et al. The malaria parasite progressively dismantles the host erythrocyte cytoskeleton for efficient egress. Mol Cell Proteomics. 2011;10(12):M111.010678. doi: 10.1074/mcp.M111.010678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins CR, et al. Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 2013;9(5):e1003344. doi: 10.1371/journal.ppat.1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koussis K, et al. A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J. 2009;28(6):725–735. doi: 10.1038/emboj.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruecker A, et al. Proteolytic activation of the essential parasitophorous vacuole cysteine protease SERA6 accompanies malaria parasite egress from its host erythrocyte. J Biol Chem. 2012;287(45):37949–37963. doi: 10.1074/jbc.M112.400820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeoh S, et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell. 2007;131(6):1072–1083. doi: 10.1016/j.cell.2007.10.049. [DOI] [PubMed] [Google Scholar]
- 18.Arastu-Kapur S, et al. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4(3):203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- 19.Aly ASI, Matuschewski K. A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts. J Exp Med. 2005;202(2):225–230. doi: 10.1084/jem.20050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delplace P, et al. Protein p126: A parasitophorous vacuole antigen associated with the release of Plasmodium falciparum merozoites. Biol Cell. 1988;64(2):215–221. doi: 10.1016/0248-4900(88)90080-9. [DOI] [PubMed] [Google Scholar]
- 21.Pang XL, Mitamura T, Horii T. Antibodies reactive with the N-terminal domain of Plasmodium falciparum serine repeat antigen inhibit cell proliferation by agglutinating merozoites and schizonts. Infect Immun. 1999;67(4):1821–1827. doi: 10.1128/iai.67.4.1821-1827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt-Christensen A, Sturm A, Horstmann S, Heussler VT. Expression and processing of Plasmodium berghei SERA3 during liver stages. Cell Microbiol. 2008;10(8):1723–1734. doi: 10.1111/j.1462-5822.2008.01162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aoki S, et al. Serine repeat antigen (SERA5) is predominantly expressed among the SERA multigene family of Plasmodium falciparum, and the acquired antibody titers correlate with serum inhibition of the parasite growth. J Biol Chem. 2002;277(49):47533–47540. doi: 10.1074/jbc.M207145200. [DOI] [PubMed] [Google Scholar]
- 24.Delplace P, Fortier B, Tronchin G, Dubremetz JF, Vernes A. Localization, biosynthesis, processing and isolation of a major 126 kDa antigen of the parasitophorous vacuole of Plasmodium falciparum. Mol Biochem Parasitol. 1987;23(3):193–201. doi: 10.1016/0166-6851(87)90026-0. [DOI] [PubMed] [Google Scholar]
- 25.Knapp B, Nau U, Hundt E, Küpper HA. A new blood stage antigen of Plasmodium falciparum highly homologous to the serine-stretch protein SERP. Mol Biochem Parasitol. 1991;44(1):1–13. doi: 10.1016/0166-6851(91)90215-r. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Mitamura T, Fox BA, Bzik DJ, Horii T. Differential localization of processed fragments of Plasmodium falciparum serine repeat antigen and further processing of its N-terminal 47 kDa fragment. Parasitol Int. 2002;51(4):343–352. doi: 10.1016/s1383-5769(02)00042-9. [DOI] [PubMed] [Google Scholar]
- 27.Miller SK, et al. A subset of Plasmodium falciparum SERA genes are expressed and appear to play an important role in the erythrocytic cycle. J Biol Chem. 2002;277(49):47524–47532. doi: 10.1074/jbc.M206974200. [DOI] [PubMed] [Google Scholar]
- 28.Dvorin JD, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328(5980):910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silmon de Monerri NC, et al. Global identification of multiple substrates for Plasmodium falciparum SUB1, an essential malarial processing protease. Infect Immun. 2011;79(3):1086–1097. doi: 10.1128/IAI.00902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandramohanadas R, et al. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science. 2009;324(5928):794–797. doi: 10.1126/science.1171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Bonniec S, et al. Plasmepsin II, an acidic hemoglobinase from the Plasmodium falciparum food vacuole, is active at neutral pH on the host erythrocyte membrane skeleton. J Biol Chem. 1999;274(20):14218–14223. doi: 10.1074/jbc.274.20.14218. [DOI] [PubMed] [Google Scholar]
- 32.Dua M, Raphael P, Sijwali PS, Rosenthal PJ, Hanspal M. Recombinant falcipain-2 cleaves erythrocyte membrane ankyrin and protein 4.1. Mol Biochem Parasitol. 2001;116(1):95–99. doi: 10.1016/s0166-6851(01)00306-1. [DOI] [PubMed] [Google Scholar]
- 33.Raphael P, et al. A cysteine protease activity from Plasmodium falciparum cleaves human erythrocyte ankyrin. Mol Biochem Parasitol. 2000;110(2):259–272. doi: 10.1016/s0166-6851(00)00283-8. [DOI] [PubMed] [Google Scholar]
- 34.Hanspal M, Dua M, Takakuwa Y, Chishti AH, Mizuno A. Plasmodium falciparum cysteine protease falcipain-2 cleaves erythrocyte membrane skeletal proteins at late stages of parasite development. Blood. 2002;100(3):1048–1054. doi: 10.1182/blood-2002-01-0101. [DOI] [PubMed] [Google Scholar]
- 35.Dhawan S, Dua M, Chishti AH, Hanspal M. Ankyrin peptide blocks falcipain-2-mediated malaria parasite release from red blood cells. J Biol Chem. 2003;278(32):30180–30186. doi: 10.1074/jbc.M305132200. [DOI] [PubMed] [Google Scholar]
- 36.Bowyer PW, Simon GM, Cravatt BF, Bogyo M. Global profiling of proteolysis during rupture of Plasmodium falciparum from the host erythrocyte. Mol Cell Proteomics. 2011;10(5):M110.001636. doi: 10.1074/mcp.M110.001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kafsack BFC, et al. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science. 2009;323(5913):530–533. doi: 10.1126/science.1165740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deligianni E, et al. A perforin-like protein mediates disruption of the erythrocyte membrane during egress of Plasmodium berghei male gametocytes. Cell Microbiol. 2013;15(8):1438–1455. doi: 10.1111/cmi.12131. [DOI] [PubMed] [Google Scholar]
- 39.Wirth CC, et al. Perforin-like protein PPLP2 permeabilizes the red blood cell membrane during egress of Plasmodium falciparum gametocytes. Cell Microbiol. 2014;16(5):709–733. doi: 10.1111/cmi.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garg S, et al. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat Commun. 2013;4:1736. doi: 10.1038/ncomms2725. [DOI] [PubMed] [Google Scholar]
- 41.Taylor HM, et al. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot Cell. 2010;9(1):37–45. doi: 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glushakova S, Mazar J, Hohmann-Marriott MF, Hama E, Zimmerberg J. Irreversible effect of cysteine protease inhibitors on the release of malaria parasites from infected erythrocytes. Cell Microbiol. 2009;11(1):95–105. doi: 10.1111/j.1462-5822.2008.01242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carzaniga R, Domart MC, Collinson LM, Duke E. Cryo-soft X-ray tomography: A journey into the world of the native-state cell. Protoplasma. 2014;251(2):449–458. doi: 10.1007/s00709-013-0583-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sturm A, et al. Alteration of the parasite plasma membrane and the parasitophorous vacuole membrane during exo-erythrocytic development of malaria parasites. Protist. 2009;160(1):51–63. doi: 10.1016/j.protis.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Ishino T, Chinzei Y, Yuda M. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell Microbiol. 2005;7(2):199–208. doi: 10.1111/j.1462-5822.2004.00447.x. [DOI] [PubMed] [Google Scholar]
- 46.Chandramohanadas R, et al. Biophysics of malarial parasite exit from infected erythrocytes. PLoS One. 2011;6(6):e20869. doi: 10.1371/journal.pone.0020869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris PK, et al. Molecular identification of a malaria merozoite surface sheddase. PLoS Pathog. 2005;1(3):0241–0251. doi: 10.1371/journal.ppat.0010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blackman MJ. 1994. Purification of Plasmodium falciparum merozoites for analysis of the processing of merozoite surface protein-1. Methods in Cell Biology, Microbes as Tools for Cell Biology, ed Russell DG (Academic Press, San Diego), Vol 45, pp 213–220.
- 49.Schindelin J, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152(1):36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 51.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 52.Nkrumah LJ, et al. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods. 2006;3(8):615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collins CR, et al. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol Microbiol. 2013;88(4):687–701. doi: 10.1111/mmi.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.