

Abstract
LipL and Cpr19 are non-heme, mononuclear Fe(II)-dependent, α-ketoglutarate (αKG):UMP oxygenases that catalyze the formation of CO2, succinate, phosphate, and uridine-5′-aldehyde, the last of which is a biosynthetic precursor for several nucleoside antibiotics that inhibit bacterial translocase I (MraY). To better understand the chemistry underlying this unusual oxidative dephosphorylation and establish a mechanistic framework for LipL and Cpr19, we report herein the synthesis of two biochemical probes – [1′,3′,4′,5′,5′-2H]UMP and the phosphonate derivative of UMP – and their activity with both enzymes. The results are consistent with a reaction coordinate that proceeds through the loss of one 2H atom of [1′,3′,4′,5′,5′-2H]UMP and stereospecific hydroxylation geminal to the phosphoester to form a cryptic intermediate, (5′R)-5′-hydroxy-UMP. Thus, these enzyme catalysts can additionally be assigned as UMP hydroxylase-phospholyases.
Keywords: Biosynthesis, non-heme iron, α-ketoglutarate, oxygenase, antibiotic, nucleoside, translocase I
1. Introduction
Non-heme, mononuclear Fe(II)- and α-ketoglutarate (αKG)-dependent oxygenases are recognized for their ability to catalyze a wide array of chemical transformations including hydroxylation, demethylation, epoxidation, hetero- and carbocyclic ring expansion/closure, desaturation, and halogenation [1,2]. The consensus reaction coordinate of this large and functionally diverse superfamily [3] begins by sequential binding of an Fe(II) cofactor, cosubstrates αKG and the primary substrate undergoing oxidative modification, and, lastly, O2. The chemistry, which is reviewed in great detail elsewhere [3–7], is then initiated by oxidative decarboxylation of αKG to yield CO2, succinate, and a transient Fe(IV)-oxo species. For most enzymes of the superfamily, the subsequent step is utilization of the Fe(IV)-oxo – a strong oxidizing agent – to modify the primary substrate by (i) hydrogen atom abstraction followed by radical rebound resulting in hydroxylation or (ii) sequential abstraction of two hydrogen atoms resulting in a desaturated product. Whether a given enzyme catalyzes hydroxylation—hence functioning as a dioxygenase; desaturation—hence functioning as a monooxygenase; or, less commonly, a distinct chemical transformation such as epimerization is likely guided by precise substrate positioning; the conformational flexibility of enzyme-substrate complex during catalysis; and the inherent chemical nature of the primary substrate and enzyme-Fe(IV)-oxo complex [8–12]. However, the determinants/selection rules for the substrate and mechanistic partitioning are not entirely understood despite being the subject of numerous investigations with model systems and enzymes of the superfamily.
LipL and Cpr19 are relatively new members of the superfamily that catalyze the O2, Fe(II), and αKG-dependent conversion of UMP to uridine-5′-aldehyde (U5′A; Fig. 1A) [13–15], the latter of which is a biosynthetic precursor for several highly modified uridine-containing antibiotics including the 5′-C-glycyluridine-containing liponucleosides represented by A-90289 A, caprazamycins, and liposidomycins (Fig. 1B) and the uridine-5′-carboxamide-containing monosaccharidyl nucleosides represented by A-102395, A-503083 B, and capuramycin (Fig. 1C) [16]. The activity of LipL, and soon thereafter Cpr19, was initially discovered based in part on the sequence similarity to putative proteins annotated as clavaminic acid synthase (CAS), which was one of the first discovered bacterial members of the non-heme, mononuclear Fe(II)- and αKG-dependent oxygenase superfamily. CAS alone highlights the catalytic versatility of this superfamily by catalyzing three non-sequential and unique oxidative transformations – hydroxylation, ring closure, and desaturation – during clavam biosynthesis [17–20]. Previous biochemical characterization of LipL and Cpr19, however, revealed intriguing differences in substrate selectivity and chemical outcome to not only CAS but other members of the superfamily as well. Notably, LipL and Cpr19 were shown to (i) recognize a free nucleotide as the primary substrate and (ii) catalyze a net oxidative dephosphorylation reaction. LipL and Cpr19 were not, however, the first members of the superfamily identified to generate an aldehyde functionality. Two noteworthy examples of aldehyde-generating members are αKG:taurine dioxygenase (TauD), which catalyzes the conversion of taurine to aminoacetaldehyde and sulfite and is perhaps the best studied enzyme of the superfamily [21–25], and alkylsulfatase (AtsK), which converts alkylsulfates to an aliphatic aldehyde and sulfate [26,27]. For TauD and AtsK, it has been postulated that the reaction coordinate proceeds through geminal hydroxylation to the sulfite and sulfate, respectively, although this hypothetical, hydroxylated intermediate has never been directly detected, nor has the fate of the second atom of O2 been tracked for either enzyme due to inherent technical challenges.
Fig. 1.
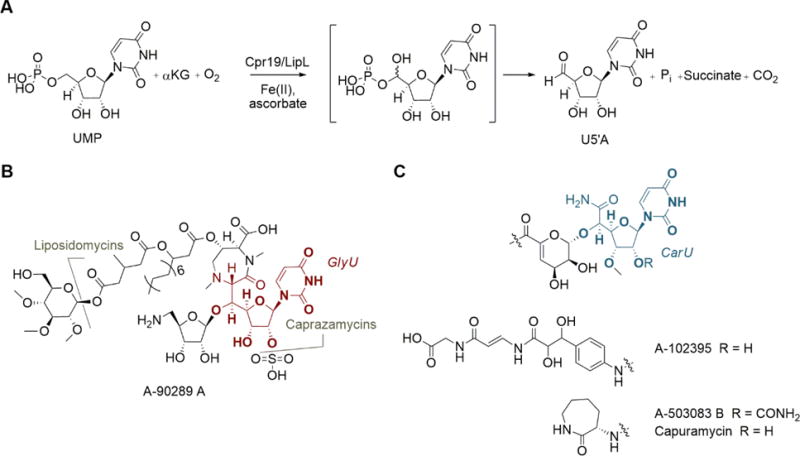
Function of LipL and Cpr19. (A) The oxidative dephosphorylation reaction catalyzed by LipL and Cpr19. (B) Structures of representative liponucleoside antibiotics containing a 5′-C-glycyluridine core (GlyU, highlighted in red). (C) Structures of representative monosaccharidyl nucleoside antibiotics containing a uridine-5′-carboxamide core (CarU, highlighted in blue).
As predicted, the reaction catalyzed by LipL and Cpr19 was previously shown to result in the incorporation of one atom of O2 into C-2 of αKG during the formation of the co-products succinate and CO2 [13]. Similar to TauD and AtsK, however, the destination of the other O atom was not established. Nonetheless, considering the precedent within the superfamily and the reported biochemical characteristics of LipL and Cpr19, we hypothesized that oxidative dephosphorylation occurs through 5′-hydroxylation using the putative Fe(IV)-oxo species, which would equate to the loss of one hydrogen atom from the primary substrate UMP and incorporation of one atom of oxygen into the product U5′A. The possibility remained, however, that LipL and Cpr19 catalyze a desaturation of the primary substrate – not unlike one reaction catalyzed by CAS and a few other members of the superfamily – with concomitant hydrolysis and tautomerization, which would contrastingly equate to a sequential loss of two hydrogen atoms from UMP (Supplementary Fig. S1). To better understand the chemistry behind this novel oxidative dephosphorylation reaction and establish a mechanistic framework for LipL and Cpr19, we report the synthesis and enzymatic conversion of two biochemical probes to aid in tracking the atoms of the substrates. As described herein, the results suggest these new enzyme catalysts can be additionally assigned as UMP hydroxylase-phospholyases.
2. Materials and Methods
2.1. Chemicals, reagents, and instrumentation
UMP, [U-2H]glucose, α-KG, β–NADP+ (tetrasodium salt), ATP (disodium salt), phospho(enol)pyruvate trisodium salt (PEP), uracil, and ascorbic acid were purchased from Sigma-Aldrich (St. Louis, MO) or Promega (Madison, WI). Buffers, salts, organic solvents and media components were purchased from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA). Synthetic oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Malachite green binding assay was performed with a colorimetric-based Sensolyte MG Phosphatase Assay Kit from AnaSpec, Inc. (Fremont, CA). UV/Vis spectroscopy was performed with a Bio-Tek μQuant microplate reader using Microtest™ 96-well plates (BD Biosciences) or a Shimadzu UV/Vis-1800 Spectrophotometer. HPLC was performed with a Waters Alliance 2695 separation module (Milford, MA) equipped with a Waters 2998 diode array detector and an analytical Apollo C18 column (250 mm × 4.6 mm, 5 μm) or a semi-preparative Apollo C18 column (250 mm × 10 mm, 5 μm) purchased from Grace (Deerfield, IL). Electrospray ionization-MS was performed using an Agilent 6120 Quadrupole MSD mass spectrometer (Agilent Technologies, Santa Clara, CA) equipped with an Agilent 1200 Series Quaternary LC system and an Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm, 80 Å). High-resolution mass spectroscopy (HRMS) was obtained from either the University of Kentucky Mass Spectrometry Core Facility or from the University of Minnesota, Department of Chemistry Mass Spectrometry Facility. NMR data were collected using a Varian Unity Inova 400 or 500 MHz Spectrometer (Varian, Inc., Palo Alto, CA) at the University of Kentucky, and a Bruker Avance III 600 MHz spectrometer equipped with a 1.7 mm 1H(13C/15N) cryoprobe at the University of Wisconsin, Madison.
2.2 Chemical syntheses
U5′A was synthesized as previously reported [13], and the identity was confirmed by MS and NMR spectroscopic analysis. U5′A: 1H NMR (D2O, 500 MHz): δ 4.00 (dd, 1H, J = 4.0, 3.5 Hz), 4.32–4.26 (1H, m), 4.37 (dd, 1H, J = 6.0, 5.5 Hz), 5.17 (d, 1H, J = 4.0 Hz), 5.88 (d, 1H, J = 8.0 Hz), 5.96 (d, 1H, J = 6.0 Hz), 7.88 (d, 1H, J = 8.0 Hz); 13C NMR (D2O, 125 MHz): δ 69.6, 73.3, 86.2, 88.5, 102.4, 141.9, 151.7, 166.1. The detailed procedure and spectroscopic data for the synthesis of 5′-deoxyuridine-5′-methylphosphonate (UMcP) is provided in the supporting information available online. The 5′-hydroxy epimers of UMcP were prepared as previously reported [28], and the identity confirmed by MS and NMR spectroscopic analysis. (5′S)-5′-hydroxy-UMcP: 1H NMR (300MHz, D2O) δ 1.7 – 1.95 (m, 2H), 4.03 (t, 1H), 4.10 (m, 1H), 4.2 – 4.3 (m, 2H), 5.83 (d, 1H), 5.87 (d, 1H), 7.83 (d, 1H); 13C NMR (300MHz, D2O) δ 31.6, 67.2, 68.7, 73.5, 87.5, 87.9, 102.6, 141.9, 151.9, 166.1. HRMS (ESI+) calcd. for C10H16N2O9P [M − Na + 2H]+ 339.0593; found 339.0592. (5′R)-5′-hydroxy-UMcP: 1H NMR (300MHz, D2O) δ 1.70 – 1.90 (m, 2H), 4.01 (dd, 1H), 4.02 – 4.18 (m, 1H), 4.21 (dd, 1H), 4.3 (dd, 1H), 5.84 (d, 1H), 5.89 (d, 1H), 7.95 (d, 1H); 13C NMR (300MHz, D2O) δ 32.0, 67.3, 70.3, 73.7, 87.0, 88.6, 102.4, 142.0, 151.8, 166.2. HRMS (ESI+) calcd. for C10H16N2O9P [M – Na + 2H]+ 339.0593; found 339.0592.
2.3 Enzymatic synthesis of [1′,3′,4′,5′,5′-2H]UMP
Genes for phosphoribosyl pyrophosphate synthetase (prps) from Salmonella typhimirium, ribose-5-phosphate isomerase (rpi) from Escherichia coli, and uracil phosphoribosyltransferase (uprt) from E. coli were amplified by PCR using the Expand Long Template PCR system from Roche with supplied buffer 2, 200 μM dNTPs, 5% dimethyl sulfoxide, 10 ng of DNA template, 5 units of DNA polymerase, and 10 μM each of the following primer pairs: Stprps (forward) 5′-GGTATTGAGGGTCGCATGCCTGATATCAAGCTTTTTGCTGG-3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTCAATGCTCGAACATGGCGGAAATC-3′; Ecrpi (forward) 5′- GGTATTGAGGGTCGCATGACGCAGGATGAATTGAAAAAAG-3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTCATTTCACAATGGTTTTGACACC-3′; and Ecuprt (forward) 5′- GGTATTGAGGGTCGCATGAAGATCGTGGAAGTCAAAC-3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTTATTTCGTACCAAAGATTTTGTC-3′. DNA templates for PCR cloning were either E. coli DH5α genomic DNA (EcrpiA, Ecuprt) or plasmid pBRS11R (Stprps; from Dr. Vern L. Schramm, Albert Einstein University, New York). The thermocycler program included an initial hold at 94 °C for 10 s, 56 °C for 15 s, and 68 °C for 50 s. The DNA fragments of the expected size were purified by 1% agarose gel and the purified PCR products were inserted into pET30-Xa/LIC using ligation-independent cloning following the provided protocol to yield pET30-Stprps, pET30-EcripA, and pET30- Ecuprt. Cloned DNA was sequenced to confirm PCR fidelity. Protein production, purification, and assessment was performed using routine conditions as previously described [14]. His6-tagged proteins were utilized without further modifications.
A single reaction mixture (1 ml) consisted of 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 5 mM uracil, 20 mM PEP, 1 mM NADP+, 1 mM ATP, 2.5 mM αKG, 1 mM NH4Cl, 1 mM D-[U-2H]glucose, 80 U of hexokinase, 160 U of pyruvate kinase, 100 U of glucose-6-phosphate dehydrogenase, 160 U of glutamate dehydrogenase, 8 U of 6-phosphogluconate dehydrogenase, 25 μg of EcRPI, 100 μg of StPRPS, 25 μg of EcRpiA, 80 U of myokinase and 5 U of inorganic pyrophosphatase. The reaction was carried out at 30 °C overnight and terminated by ultracentrifugation using a Microcon YM-3. Following removal of protein by centrifugation, the reaction components were analyzed by HPLC using a C-18 reverse-phased column under ion-pairing conditions (monitored at 254 nm). A gradient from 40 mM acetic acid-triethylamine pH 6.5 (A) to 20% methanol (B) was used in the following manner (time range and linear increase to % B: 0–4 min, 0%; 4–24 min, 50%; 24–26 min 100%; 26–32 min, 100%; 32–35 min, 0%. The flow rate was kept constant at 1 mL/min. A peak with the identical retention time as authentic UMP was collected, freeze-dried, and analyzed by LC-MS using a linear gradient from 0.1% formic acid in water to 0.1% formic acid in acetonitrile over 20 min. The flow rate was kept constant at 0.4 mL/min, and elution was monitored at 254 nm.
2.4 HPLC-based activity assays of LipL and Cpr19
The cloning of lipL and cpr19 and the production of purified proteins were previously reported [13–15]. Routine reactions with LipL consisted of 50 mM Tris-HCl (pH 7.5), 1 mM UMP, 1.25 mM αKG, 200 μM ascorbate, 100 μM FeCl2, and 100 nM LipL at 30 °C. Routine reactions with Cpr19 consisted of 50 mM Tris-HCl (pH 7.5), 1 mM UMP, 1.25 mM αKG, 1 mM ascorbate, 500 μM FeCl2, and 100 nM Cpr19 at 30 °C. Reactions were terminated by ultracentrifugation using a Microcon YM-3. Following removal of protein by centrifugation, the reaction components were analyzed by HPLC using a C-18 reverse-phased column under ion-pairing conditions and a linear gradient as described above for the isolation of [1′,3′,4′,5′,5′-2H]UMP. Reactions using [1′,3′,4′,5′,5′-2H]UMP as a substrate consisted of 5 mM HEPES (pH 7.5), 300 μM of [1′,3′,4′,5′,5′-2H]UMP, 250 μM αKG, 100 μM ascorbate, 100 μM FeCl2, and 50 nM LipL or Cpr19 at 30 °C. Reactions were carried out at 30 °C overnight and terminated by ultracentrifugation for removal of protein. Samples were analyzed by LC-MS for identification of products.
2.5. Kinetic analysis
The activity of LipL and Cpr19 was detected by monitoring the formation of inorganic phosphate with the malachite green binding assay. For kinetic analyses involving LipL, reactions consisted of 50 mM Tris-HCl (pH 7.5), 1 mM ascorbate, 500 μM FeCl2, 100 nM LipL, near saturating αKG (1 mM), variable UMP (100 μM – 10 mM), and variable UMcP (5 μM – 1 mM). The reactions were performed at 30 °C for 3 min and analyzed under initial velocity conditions. Each data point represents a minimum of three replicate end point assays. The Lineweaver-Burke plot was indicative of competitive inhibition, thus data were fitted using nonlinear regression with global curve fitting (GraphPad Prism) to the following equation for competitive inhibition: .
2.6. Enzyme-catalyzed production of (5′S)-5′-hydroxy-UMcP
Large scale isolation of the Cpr19 product (5′S)-5′-hydroxy-UMcP starting from UMcP was carried out with HPLC using a C18 reverse-phase semipreparative column using ion-pairing conditions as described above with a flow rate of 3.5 mL/min. The peak corresponding to the product was collected and freeze-dried prior to HRMS, 1D and 2D NMR spectroscopic analysis. 1H NMR (600 MHz, D2O) δ 1.89 (m, 2H), 4.09 (app t,1H), 4.14 (m,1H), 4.29 – 4.30 (m, 2H), 5.86 (d, 1H), 5.93 (d, 1H), 7.90 (d, 1H); 13C NMR (600MHz, D2O) δ 27.5, 67.5, 68.7, 73.5, 87.5, 102.2, 141.5, 151.9, 165.7. HRMS (ESI−) calcd. for C10H15N2O9P [M-H]- 337.05152; found 337.04652.
3. Results
3.1. Tracking the H atoms of the prime substrate UMP
To track the loss of H atoms of the ribosyl moiety of UMP, deuterated substrate was prepared and reacted with LipL or Cpr19. A one-pot, total enzymatic synthesis of [1′,3′,4′,5′,5′-2H]UMP starting from D-[U-2H]glucose was used (Fig. 2A), a strategy that was based on a previously reported method for preparing site-specifically labelled nucleotides for assay development and measurement of kinetic isotopic effects for unrelated nucleotide metabolizing enzymes [29]. The synthesis utilizes 10 enzymes (7 commercial proteins and 3 recombinant proteins produced and purified from Escherichia coli) from glycolysis, pentose phosphate, and nucleotide salvage pathways. HPLC analysis of the reaction mixture after an overnight incubation revealed the formation of a new peak with an identical retention time and UV-VIS spectrum as authentic UMP (Fig. 2B). LC-MS analysis revealed an [M-H]- ion at m/z = 327.9, which was 5.2 amu greater than UMP isolated using unlabeled glucose as a control (Fig. 2C). The loss of two 2H from D-[U-2H]glucose was expected based upon prior in depth biochemical studies of the enzymes used in the total synthesis; additionally, [1′,3′,4′,5′,5′-2H]UMP was the expected regiochemistry of deuterium incorporation based upon the established stereochemical selectivity of 6-phosphogluconate dehydrogenase and ribose-5-phosphate isomerase [30,31].
Fig. 2.
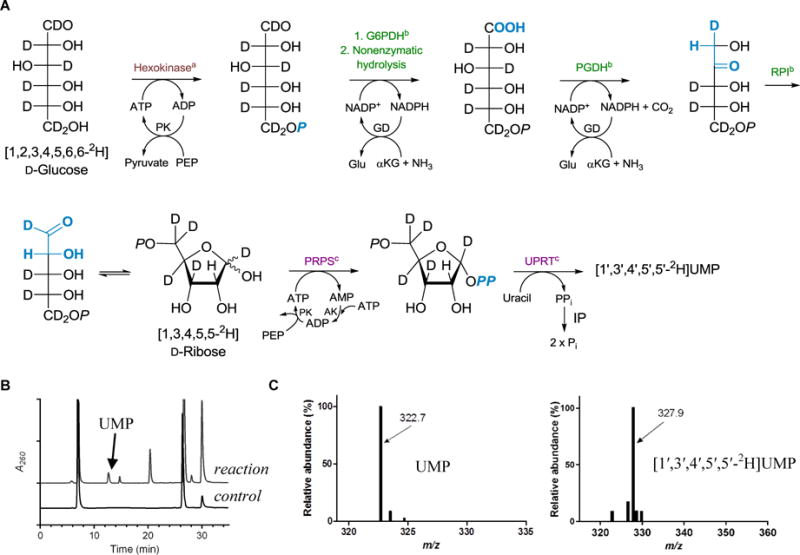
Enzymatic synthesis and analytical analysis of [1′,3′,4′,5′,5′-2H]UMP. (A) One-pot, total enzymatic synthesis of UMP from D-[U-2H]glucose. The primary metabolic pathway for highlighted enzymes are aglycolysis, bpentose phosphate, and cnucleotide metabolism. Abbreviations are G6PDH, glucose-6-phosphate dehydrogenase; PGDH, 6-phosphogluconate dehydrogenase; RPI, ribose-5-phosphate isomerase; PRPS, phosphoribosylpyrophosphate synthetase; UPRT, uracil phosphoribosyltransferase; PK, pyruvate kinase; GD, glutamate dehydrogenase; AK, adenylate kinase (myokinase); IP inorganic pyrophosphatase; PEP, phosphoenolpyruvate; and αKG, α-ketoglutarate. (B) HPLC analysis of the one-pot enzymatic synthesis of UMP. A260, absorbance at 260 nm. (C) Negative mass spectra for the peak corresponding to UMP starting with unlabeled glucose (left) or D-[U-2H]glucose (right).
With the deuterated substrate in hand, Cpr19 and LipL reactions were performed using conditions that facilitated complete conversion to product U5ʹA. For Cpr19 LC-MS analysis of the reaction components in comparison to the appropriate controls revealed a new peak that co-eluted with authentic U5ʹA and had an [M-H]- ion at m/z = 244.8, which was 4.0 amu greater than U5′A generated from unlabeled UMP (Fig. 3). An identical result was obtained with LipL (data not shown). Thus, 4 of the 5 deuterium atoms from [1′,3′,4′,5′,5′-2H]UMP are retained during the conversion to U5ʹA, corresponding to the overall loss of one H atom from the primary substrate during the transformation.
Fig. 3.
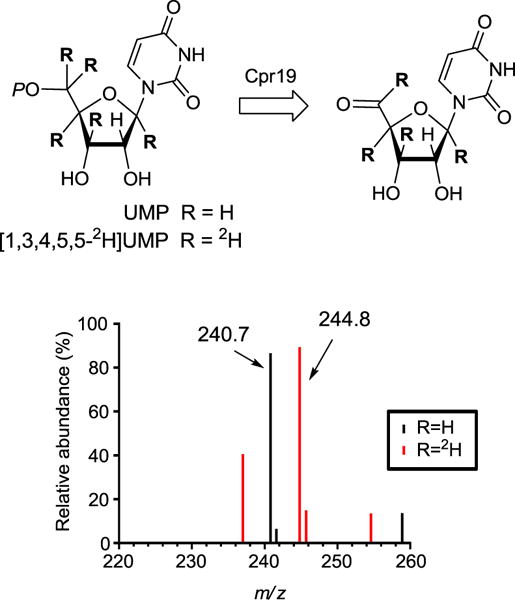
Monitoring the loss of H atoms for the Cpr19-catalyzed reaction. The expected reaction catalyzed by Cpr19, and mass spectra (positive-ion mode) for the peak corresponding to U5ʹA starting with unlabeled UMP or D-[1ʹ,3ʹ,4ʹ,5ʹ,5ʹ-2H]UMP.
3.2. Evidence for a mechanism proceeding with stereospecific hydroxylation
With the goal of trapping a hydroxylated product, the phosphonate derivative of UMP (UMcP) was targeted as a potential surrogate substrate for LipL and Cpr19 (Fig. 4A). The derivative was synthesized from uridine and diethyl(hydroxymethyl)phosphonate using a described procedure with slight modifications [32]. The identity of UMcP was confirmed by HRMS and NMR spectroscopic analysis and comparison with the published data (Supplementary Fig. S2–S4). Reactions with UMcP and LipL, however, did not yield any products as judged by HPLC and LC-MS analyses, and hence UMcP was not a surrogate substrate for LipL. Instead, kinetic characterization revealed UMcP was a strong competitive inhibitor with respect to UMP, yielding a Ki = 800 nM (Fig. 4B). In contrast to LipL, reactions of Cpr19 with UMcP revealed a new, small peak eluting just prior to the unreacted substrate (Fig. 4C), and this new peak had a UV-VIS spectrum characteristic of a uracil-containing product. Despite a maximum conversion of ~1–2% based on HPLC, enough material was collected for preliminary characterization by HRMS to reveal an [M-H]- ion of m/z = 337.04652 (Supplementary Fig. S5), consistent with the molecular formula C10H15N2O9P for a hydroxylated product, uridine-5′-C-methylphosphonate (5′-OH-UMcP) (expected m/z = 337.05152). A peak with a mass corresponding to a desaturated product was not detected. Interpretation of both the 1D and 2D NMR spectra of the product was also consistent with the assignment of a C-5′ hydroxylated product (Supplementary Table S1 and Supplementary Fig. S6–S9). However, the low yields and degradation of the purified product—particularly to uracil and an unidentified polyhydroxylated molecule in the style of the degradation of synthetic nucleoside phosphonates that was previously reported [33]—hindered the analytical characterization and hence conclusive identification of the product.
Fig. 4.
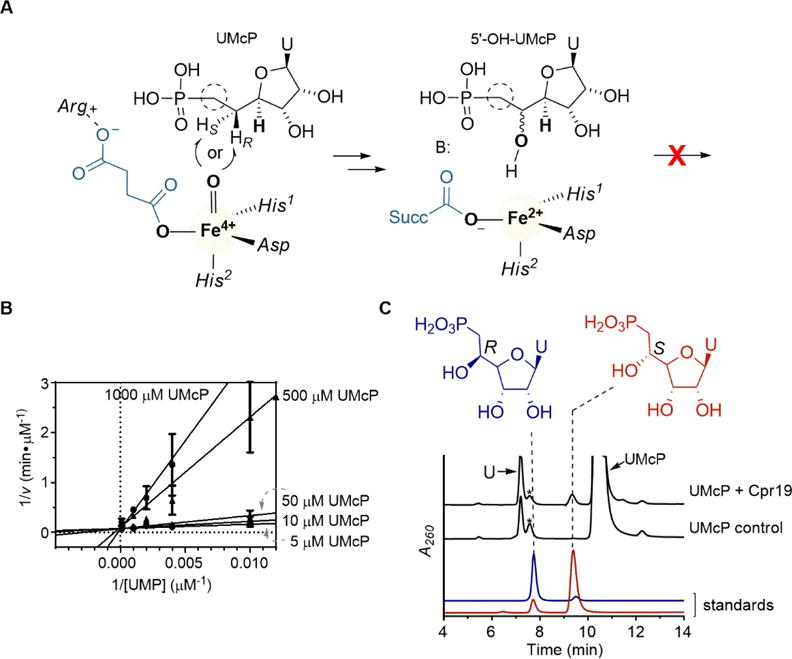
Reaction of Cpr19 and LipL with the phosphonate derivative of UMP. (A) Proposed outcome for the reaction of 5′-deoxyuridine-5′-methylphosphonate (UMcP) with LipL and Cpr19. Amino acid ligands denote protein side chains used to bind the iron and co-product succinate. (B) Lineweaver-Burk plot of UMcP inhibition with respect to variable UMP with LipL. (C) HPLC analysis of the Cpr19-catalyzed reaction starting with UMcP in comparison to synthetic standards (5′S)-hydroxy-UMcP and (5′R)-hydroxy-UMcP and control reactions without enzyme. *denotes an unidentified contaminant of synthetic 5′-deoxyuridine-5′-methylphosphonate with a distinct UV-VIS profile compared to uracil-containing compounds; U denotes uracil. A260, absorbance at 260 nm.
To overcome the scalability challenge for satisfactory evidence for the existence of hydroxylated UMcP, authentic standards were synthesized following our prior report using concise, stereoselective syntheses of (5′S)-uridine-5′-C-methylphosphonate [(5′S)-OH-UMcP] and (5′R)-uridine-5′-C-methylphosphonate [(5′S)-OH-UMcP] [28]. The sodium salts of these two epimers, each in 9:1 diastereoisomeric excess [28], were readily separated by HPLC using the ion-pairing conditions routinely used to track the activity of LipL and Cpr19 (Fig. 4C). Subsequent HPLC analysis revealed the product of Cpr19 with the surrogate substrate coeluted with the 5′S epimer, which was further confirmed by coinjection and MS analysis (Fig. 4C).
4. Discussion
Enzymes of the non-heme, mononuclear Fe(II)- and αKG-dependent oxygenase superfamily catalyze a wide array of oxidative transformations on a structurally diverse range of prime substrates. LipL and Cpr19, relatively new members of the superfamily, are the first to catalyze a net oxidative dephosphorylation and recognize a free nucleotide as a substrate, hence expanding upon this diversity of transformations and substrate spectrum. As part of the initial functional assignment, which was primarily motivated by the requirement for an enzyme to generate U5′A and the lack of any other candidate to catalyze such oxidative chemistry, LipL and Cpr19 were demonstrated to have characteristics that are shared by other enzymes in the superfamily, including the requirement for Fe(II), αKG, and O2 for activity, the incorporation of one O atom into the coproduct succinate, and the ability of ascorbic acid to stimulate activity [13,15]. Therefore, it was logically speculated that the reaction coordinate – like other members of the superfamily – proceeds through an Fe(IV)-oxo species, which is likely used to hydroxylate UMP at C-5′ leading to phosphate elimination concomitant with product aldehyde formation.
To test this hypothesis, we first set out to monitor the fate of the H atoms of UMP by enzymatically synthesizing [1′,3′,4′,5′,5′-2H]UMP, reacting the purified, labeled substrate with Cpr19 or LipL, and identifying the extent of deuterium retention in the product. Interestingly, di-deuterated taurine (1-[2H]taurine) has been used for several mechanistic studies with TauD to reveal a large pre-steady state and steady state kinetic isotope effect, consistent with H abstraction by an Fe(IV)-oxo species [23–25,34–37]. Both 1-[2H]taurine and U-[2H]taurine have also been used to define the structural orientation of taurine relative to the iron center [38,39]. However, to our knowledge, neither the fate of the deuteriums atoms in the final organic product nor regioselectivety of H abstraction has been reported. For both Cpr19 and LipL, the mass of the product from reactions starting with [1′,3′,4′,5′,5′-2H]UMP was clearly indicative of the retention of four deuterium atoms during transformation to the product U5ʹA. Thus, this result supported a mechanism involving the loss of a single H atom, which clearly disfavors a desaturation event (Supplementary Fig. S1).
We next hoped to provide compelling evidence that a hydroxylation-phospholyase mechanism was employed by detecting a hydroxylated product upon substituting the substrate phosphate functionality with a poor leaving group. A similar strategy has been used to probe the mechanism of the non-heme, mononuclear Fe(II)- and αKG-dependent N-demethylase AlkB, which – although specific for dsDNA – has a remarkable substrate flexibility in that the enzyme is able to catalyze N-demethylation of all four bases [40]. The 3-deaza derivative of 3-methylcytosine-DNA was able to be converted in vitro to the monohydroxylated product 3-deaza-3-hydroxymethylcytosine by AlkB in low yields, which was also confirmed within the enzyme-substrate co-crystal structure when exposed to O2. Although very low yields of product were likewise realized with Cpr19, we were able to identify a monohydroxylated product based on spectroscopic analyses and comparisons to synthetic standards. Additionally, and perhaps of highest significance, the stereochemistry of this hydroxylation was established, as the product coeluted with authentic (5′S)-OH-UMcP. Assuming the hydroxyl is installed at the identical position occupied by the abstracted hydrogen, the results suggest the proR hydrogen atom of UMcP is initially removed by the Fe(IV)-oxo intermediate. Due to the change in priorities based on the Cahn Ingold Prelog rules, this would suggest that the proS hydrogen atom of UMP is abstracted to form a 5′ carbon-centered radical and an Fe(III)-hydroxo center, and radical rebound generates an unstable (5′R)-hydroxy-UMP (Fig. 5). Although the proS hydrogen atom is likely abstracted to retain the relative stereochemistry following rebound, it is noteworthy that other enzymes of this superfamily are known to integrate epimerization during the catalytic cycle (for example, carbapenem synthase CarC and CAS) [41–43], and thus remains a possibility for LipL and Cpr19.
Fig. 5.
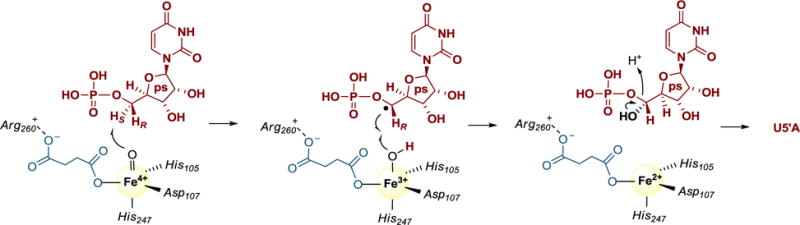
Proposed mechanism following the αKG-dependent formation of the Fe(IV)-oxo species for UMP:α-ketoglutarate dioxygenases using Cpr19 as a model. ps, primary substrate.
A major goal moving forward is to identify and understand the role of unique residues of LipL/Cpr19 that guide nucleotide binding and oxidation, of which the mechanistic framework for the latter has now been established. In several instances within the superfamily, the substrate reactivity has been shown to be dictated by a single active site residue that appears unique to a particular enzyme activity. Among other notable examples [10,44,45], an active site Tyr residue in CarC is essential for epimerization [41,42] and a Phe residue in TauD is a critical determinant of precise reactivity [36]. In other cases, it appears that the lack of an active site residue is important; for example, the lack of a carboxylate ligand as part of the facial triad is a diagnostic feature for halogenases of this superfamily [9]. Although it is tempting to propose a model for substrate binding and catalysis for LipL and Cpr19 based on the known structures of primary substrate-bound TauD and AtsK [27,46], the overall low sequence to these enzymes and the diverse modes and orientations of primary substrate binding throughout the entire superfamily precludes such a predictive or generalized model with acceptable accuracy [47]. Given that this is the first example of a free nucleotide-utilizing oxygenase of the superfamily, a bona fide structure is necessary to better explain the mechanistic results reported here.
In summary, we have provided evidence that LipL and Cpr19 function as true dioxygenases, incorporating one atom of O2 into αKG and the other into C-5′ of UMP to form a cryptic, hydroxylated intermediate. Furthermore, the hydroxylation appears to occur with defined stereochemistry based on the isolation of (5′S)-OH-UMcP upon using the surrogate substrate, the phosphonate of UMP. Thus, LipL and Cpr19, and by extension other homologues that have been identified in gene clusters for related nucleoside antibiotics, can be additionally described as UMP hydroxylase-phospholyases.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health Grant AI087849 and the National Center for Advancing Translational Sciences Grant UL1TR000117.
Abbreviations
- U5′A
uridine-5ʹ-aldehyde
- UMP
uridine-5′-monophoshate
- αKG
α-ketoglutarate
- CAS
clavaminic acid synthase
- UMcP
5′-deoxyuridine-5′-methylphosphonate
- (5′S)-OH-UMcP
(5′S)-uridine-5′-C-methylphosphonate
- (5′S)-OH-UMcP
(5′R)-uridine-5′-C-methylphosphonate
- HRMS
high-resolution mass spectroscopy
Appendix A. Supplementary Data
Supplementary data including synthetic procedures, Supplementary Table S1, and Supplementary Fig. S1–S9 can be found in the online version at XXXXXX.
References
- 1.Hausinger RP. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 2.Wu LF, Meng S, Tang GL. Ferrous iron and α-ketoglutarate-dependent dioxygenases in the biosynthesis of microbial natural products. Biochim Biophys Acta. 2016;1864:453–470. doi: 10.1016/j.bbapap.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Schofield C, Hausinger R. RSC Metatallobiology: 2-Oxoglutarate-Dependent Oxygenases. Royal Society of Chemistry; 2015. [Google Scholar]
- 4.Martinex S, Hausinger RP. Catalytic mechanisms of Fe(II)- and 2-oxoglutarate-dependent oxygenases. J Biol Chem. 2015;290:20702–20711. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollinger JM, Krebs C. Stalking intermediates in oxygen activation by iron enzymes: Motivation and method. J Inorg Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Hangasky JA, Taabazuiing CY, Valliere MA, Knapp JA. Imposing function down a (cupin)-barrel: secondary structure and metal stereochemistry in the αKG-dependent oxygenases. Metallomics. 2013;5:287–301. doi: 10.1039/c3mt20153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schofield CJ, Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr Opin Struct Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 8.Srnec M, Wong SD, Matthews ML, Krebs C, Bollinger JM, Solomon EI. Electronic structure of the ferryl intermediate in the α-ketoglutarate depedent non-heme iron halogenase SyrB2: Contributions to H atom abstraction reactivity. J Am Chem Soc. 2016;138:5110–5122. doi: 10.1021/jacs.6b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci USA. 2009;42:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Usharani D, Janardanan D, Shaik S. Does the TauD enzyme always hydroxylate alkanes, while an analogous synthetic non-heme reagent always desaturates them? J Am Chem Soc. 2010;133:176–179. doi: 10.1021/ja107339h. [DOI] [PubMed] [Google Scholar]
- 11.Kulik HJ, Drennan CL. Substrate placement influences reactivity in non-heme halogenases and hydroxylases. J Biol Chem. 2013;288:11233–11241. doi: 10.1074/jbc.M112.415570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solomon EI, Light KM, Liu LV, Srnec M, Wong SD. Geometric and electronic structure contributions to function in non-heme iron enzymes. Acc Chem Res. 2013;46:2725–2739. doi: 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Chi X, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen JM, Elliott GI, Rohr J, Van Lanen SG. Characterization of LipL as a non-heme, Fe (II)-dependent α-ketoglutarate: UMP dioxygenase that generates uridine-5′-aldehyde during A-90289 biosynthesis. J Biol Chem. 2011;286:7885–7892. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z, Unrine J, Nonaka K, Van Lanen SG. Fe(II)-dependent, uridine-5′-monophosphate:α-ketoglutarate dioxygenases in the synthesis of 5′-modified nucleosides. Methods Enzymol. 2012;516:153–168. doi: 10.1016/B978-0-12-394291-3.00031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai W, Goswami A, Yang Z, Liu X, Green KD, Barnard-Britson S, Baba S, Funabashi M, Nonaka K, Sunkara M, Morris AJ, Garneau-Tsodikova S, Van Lanen SG. The biosynthesis of capuramycin-type antibiotics: Identification of the A-102395 biosynthetic gene cluster, mechanism of self-resistance, and formation of uridine-5′-carboxamide. J Biol Chem. 2015;290:13710–13724. doi: 10.1074/jbc.M115.646414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winn M, Goss RJ, Kimura KI, Bugg TD. Antimicrobial nucleoside antibiotics targeting cell wall assembly: Recent advances in structure–function studies and nucleoside biosynthesis. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 17.Salowe SP, Marsh EN, Townsend CA. Purification and characterization of clavaminate synthase from Streptomyces clavuligerus: an unusual oxidative enzyme in natural product biosynthesis. Biochemistry. 1990;29:6499–6508. doi: 10.1021/bi00479a023. [DOI] [PubMed] [Google Scholar]
- 18.Busby RW, Townsend CA. A single monomeric iron center catalyzes three nonsuccessive oxidative transformations. Bioorg Med Chem. 1996;4:1059–1064. doi: 10.1016/0968-0896(96)00088-0. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Ren J, Stammers DK, Baldwin JE, Harlos K, Schofield CJ. Structural origins of the selectivity of the trifunctional oxygenase clavaminic acid synthase. Nat Struct Biol. 2000;7:127–133. doi: 10.1038/72398. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Ren JS, Harlos K, McKinnon CH, Clifton IJ, Schofield CJ. Crystal structure of a clavaminate synthase–Fe (II)–2-oxoglutarate–substrate–NO complex: Evidence for metal centred rearrangements. FEBS Lett. 2002;517:7–12. doi: 10.1016/s0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- 21.Eichhorn E, van der Ploeg JR, Kertesz MA, Leisinger T. Characterization of α-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J Biol Chem. 1997;272:23031–23036. doi: 10.1074/jbc.272.37.23031. [DOI] [PubMed] [Google Scholar]
- 22.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe (IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 23.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/α-ketoglutarate dioxygenase. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 24.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Krebs C. EXAFS spectroscopic evidence for an Fe O unit in the Fe (IV) intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 25.Grzyska PK, Appelman EH, Hausinger RP, Proshlyakov DA. Insight into the mechanism of an iron dioxygenase by resolution of steps following the FeIV═ O species. Proc Natl Acad Sci. 2010;107:3982–3987. doi: 10.1073/pnas.0911565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahnert A, Kertesz MA. Characterization of a sulfur-regulated oxygenative alkylsulfatase from Pseudomonas putida S-313. J Biol Chem. 2000;275:31661–31667. doi: 10.1074/jbc.M005820200. [DOI] [PubMed] [Google Scholar]
- 27.Müller I, Kahnert A, Pape T, Sheldrick GM, Meyer-Klaucke W, Dierks T, Kertesz M, Usón I. Crystal structure of the alkylsulfatase AtsK: Insights into the catalytic mechanism of the Fe(II) α-ketoglutarate-dependent dioxygenase superfamily. Biochemistry. 2004;43:3075–3088. doi: 10.1021/bi035752v. [DOI] [PubMed] [Google Scholar]
- 28.Meurillon M, Chaloin L, Périgaud C, Peyrottes S. Synthesis of pyrimidine‐containing nucleoside β‐(R/S)‐hydroxyphosphonate analogues. Eur J Org Chem. 2011:3794–3802. [Google Scholar]
- 29.Parkin DW, Leung HB, Schramm VL. Synthesis of nucleotides with specific radiolabels in ribose. Primary 14C and secondary 3H kinetic isotope effects on acid-catalyzed glycosidic bond hydrolysis of AMP, dAMP, and inosine. J Biol Chem. 1984;259:9411–9417. [PubMed] [Google Scholar]
- 30.Lienhard GE, Rose IA. The mechanism of action of 6-phosphogluconate dehydrogenase. Biochemistry. 1964;3:190–195. doi: 10.1021/bi00890a009. [DOI] [PubMed] [Google Scholar]
- 31.Malaisse W, Zahner D, Malaisse Lagae F, Verbruggen I, Kayser F, Biesemans M, Willem R. Diastereotopic specificity of phosphoriboisomerase. Med Sci Res. 1994;22:247–249. [Google Scholar]
- 32.Xu Y, Flavin MT, Xu ZQ. Preparation of new Wittig reagents and their application to the synthesis of α, β-unsaturated phosphonates. J Org Chem. 1996;61:7697–7701. doi: 10.1021/jo9608275. [DOI] [PubMed] [Google Scholar]
- 33.Yengoyan L, Rammler DH. Nucleoside phosphonic acids. I. The synthesis of 5′-deoxyuridine 5′-phosphonic acids and derivatives. Biochemistry. 1966;5:3629–3638. doi: 10.1021/bi00875a036. [DOI] [PubMed] [Google Scholar]
- 34.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD) J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 35.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM. Kinetic dissection of the catalytic mechanism of taurine: α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 36.McCusker KP, Klinman JP. Modular behavior of TauD provides insight into the origin of specificity in α-ketoglutarate-dependent nonheme iron oxygenases. Proc Natl Acad Sci USA. 2009;106:19791–19795. doi: 10.1073/pnas.0910660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCusker KP, Klinman JP. Facile synthesis of 1, 1-[2 H 2]-2-methylaminoethane-1-sulfonic acid as a substrate for taurine α ketoglutarate dioxygenase (TauD) Tetrahedron Lett. 2009;50:611–613. [Google Scholar]
- 38.Muthukumaran RB, Grzyska PK, Hausinger RP, McCracken J. Probing the iron-substrate orientation for taurine/α-ketoglutarate dioxygenase using deuterium electron spin echo envelope modulation spectroscopy. Biochemistry. 2007;46:5951–5959. doi: 10.1021/bi700562t. [DOI] [PubMed] [Google Scholar]
- 39.Casey TM, Grzyska PK, Hausinger RP, McCracken J. Measuring the orientation of taurine in the active site of the non-heme Fe (II)/α-ketoglutarate-dependent taurine hydroxylase (TauD) using electron spin echo envelope modulation (ESEEM) spectroscopy. J Phys Chem. 2013;117:10384–10394. doi: 10.1021/jp404743d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi C, Jia G, Hou G, Dai Q, Zhang W, Zheng G, Jian X, Yang Cai-Guang, Cui Q, He C. Iron-catalyzed oxidation intermediates captured in a DNA repair dioxygenase. Nature. 2010;468:330–333. doi: 10.1038/nature09497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang WC, Guo Y, Wang C, Butch SE, Rosenzweig AC, Boal AK, Krebs C, Bollinger JM. Mechanism of the C5 stereoinversion reaction in the biosynthesis of carbapenem antibiotics. Science. 2014;343:1140–1144. doi: 10.1126/science.1248000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clifton IJ, Doan LX, Sleeman MC, Topf M, Suzuki H, Wilmouth RC, Schofield CJ. Crystal structure of carbapenem synthase (CarC) J Biol Chem. 2003;278:20843–20850. doi: 10.1074/jbc.M213054200. [DOI] [PubMed] [Google Scholar]
- 43.Salowe SP, Krol WJ, Iwata-Reuyl D, Townsend CA. Elucidation of the order of oxidations and identification of an intermediate in the multistep clavaminate synthase reaction. Biochemistry. 1991;30:2281–2292. doi: 10.1021/bi00222a034. [DOI] [PubMed] [Google Scholar]
- 44.Nam W, Lee YM, Fukuzumi S. Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron (IV)-oxo complexes. Acc Chem Res. 2014;47:1146–1154. doi: 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]
- 45.Khaleeli N, Busby RW, Townsend CA. Site-directed mutagenesis and biochemical analysis of the endogenous ligands in the ferrous active site of clavaminate synthase. The His-3 variant of the 2-His-1-carboxylate model. Biochemistry. 2000;39:8666–8673. doi: 10.1021/bi000534c. [DOI] [PubMed] [Google Scholar]
- 46.Elkins JM, Ryle MJ, Clifton IJ, Dunning Hotopp JC, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, Roach PL. X-ray crystal structure of Escherichia coli taurine/α-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry. 2002;41:5185–5192. doi: 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- 47.Jaroszewski L. Protein structure prediction based on sequence similarity. Methods Mol Biol. 2009;569:129–156. doi: 10.1007/978-1-59745-524-4_7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.