

Abstract
Rationale
Hypertrophic cardiomyopathy (HCM) is a prototypic single gene disease caused mainly by mutations in genes encoding sarcomere proteins. Despite the remarkable advances, the causal genes in about 40% of the HCM cases remain unknown, typically in small families and sporadic cases, wherein co-segregation could not be established.
Objective
To test the hypothesis that the “missing causal genes” in HCM is, in part, because of an oligogenic etiology, wherein the pathogenic variants do not co-segregate with the phenotype.
Methods and Results
A clinically affected trio with HCM underwent clinical evaluation, electrocardiography, echocardiography, magnetic resonance imaging, and whole exome sequencing (WES). Pathogenic variants in the WES data were identified using established algorithms. Family members were genotyped by Sanger sequencing and co-segregation was analyzed.
The siblings had a severe while the mother had a mild course. Variant analysis showed that the trio shared 145 heterozygous pathogenic variants in 139 genes, including two in cardiomyopathy genes TTN and ALPK3. The siblings also had the pathogenic variant p.Ala13Thr variant in MYL2, a known gene for HCM. The sibling’s father also carried the p.Ala13Thr variant, in whom an unambiguous diagnosis of HCM could not be made because of concomitant severe aortic stenosis. The TTN variant segregated with HCM, except in a 7-year boy, who had a normal phenotype. The ALPK3 variant, shared by the affected trio, did not segregate with the phenotype.
Conclusions
We posit that a subset of HCM might be oligogenic caused by multiple pathogenic variants that do not perfectly co-segregate with the phenotype.
Keywords: Rare variants, cardiac hypertrophy, gene mutation, exome sequencing, genetic testing, cardiomyopathy, exome
Subject Terms: Genetics
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is considered a classic single gene disease, diagnosed clinically by cardiac hypertrophy in the absence of a discernible external cause 1. HCM is caused mainly by mutations in genes encoding sarcomere and sarcomere-associated proteins 1. Over a dozen causal genes for HCM have been identified, primarily through genetic linkage analysis in large families. Mutations in MYH7 and MYBPC3, encoding β-myosin heavy chain and myosin binding protein C, respectively, are the two most common causes of HCM, together accounting for about one-half of all HCM cases 1. Despite the remarkable recent genetic discoveries, the causal genes in approximately 40% of the HCM cases have remained unknown.
The causal genes in large families with HCM have been essentially identified. The so-called “missing causal genes” primarily if not exclusively pertains to HCM occurring in the sporadic cases or in small families, whereby causality can not be ascertained unambiguously, due to extreme genetic heterogeneity of the humans 2. Accordingly, each human genome contains several thousand pathogenic variants, including variants in genes encoding sarcomere proteins 3, 4. The genetic diversity also raises the possibility that multiple pathogenic variants in aggregate might be responsible for a fraction of the classically recognized single gene disorders, such as HCM. In accord with this notion, in one end of the spectrum, HCM is a classic single gene disorder, whereby a single genetic variant (GV) is sufficient to cause the disease (Figure 1). Such GV would exhibit a high penetrance and co-segregate with the phenotype in the family. On the end of the spectrum, multiple GVs, each exerting a modest effect size, collectively induce the HCM phenotype. In the latter case, each GV would show a low penetrance and not co-segregate with the phenotype. A prelude to this hypothesis is the previous reports of multiple mutations in patients with HCM, generally associated with a pronounced phenotype 5–9. In an oligogenic etiology, multiple pathogenic GVs would be distributed among the family members and would not show a perfect co-segregation with the phenotype. Discordant distributions of the GVs among the family members would contribute to variability in the phenotypic expression of HCM.
Figure 1. Hypothesis.
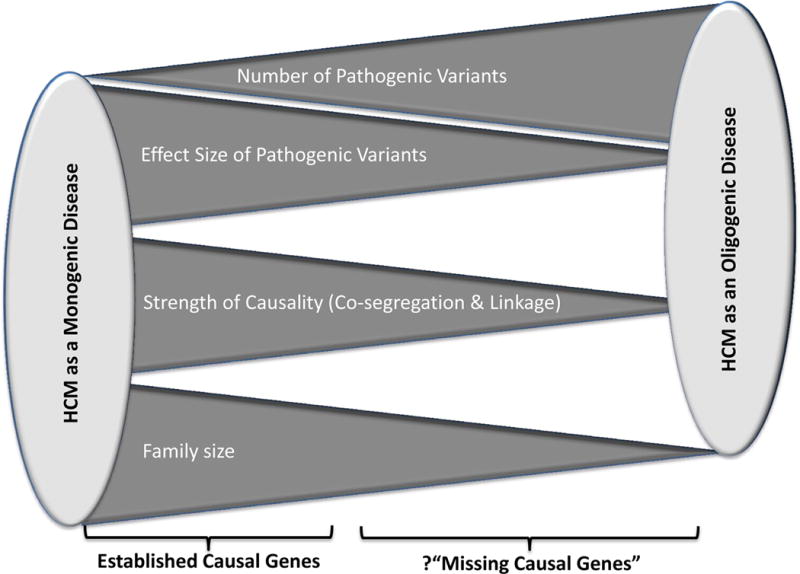
The spectrum of genetic etiology of HCM is illustrated. On one end of the spectrum, a single rare variant that exerts a large effect size leads to familial HCM with a high level of co-segregation. On the other end of the spectrum, HCM is caused by multiple pathogenic variants, each exerting a modest to moderate effect size and in aggregate cause HCM in sporadic cases and small families. In such scenario, the variants do not co-segregate with the phenotype. The latter group might explain, in part, the failure to identify the “missing causal genes” in HCM.
MYL2, encoding myosin light chain 2, was of the first few causal genes identified for HCM through family studies 10. Likewise, mutations in TTN, encoding giant sarcomere protein titin, are causal for hereditary cardiomyopathies, particularly dilated cardiomyopathy (DCM) 11. More recently, homozygous truncating mutations in ALPK3, encoding α-kinase 3, are also implicated as causes of pediatric cardiomyopathies 12. We describe a family in whom three rare variants in MYL2, TTN, and ALPK3 are likely pathogenic in HCM, and the aggregation of the three is associated with a severe phenotype.
Methods
Phenotypic characterization
Medical history and physical examination were completed in the family members. The participants underwent 12-lead electrocardiography (ECG), echocardiography (M-mode, 2D, and Doppler), and cardiac magnetic resonance imaging (CMR).
Exome sequencing and variant annotation
Whole exome sequencing (WES), comprising of ~ 20,000 genes, was performed on an Illumina 2500 platform to produce 2×100 bp reads as part of the clinical genetic testing. Sequencing reads were aligned to the human reference genome (hg19) using BWA-Mem (v0.7.8)13. To identify the GVs in each exome, BAM files, containing the sequence alignment data were analyzed using Platypus (v0.8.1), which is one of the first callers to integrate local assembly into its calling processes and is orders of magnitude faster than alternative programs 14. GVs were annotated using CASSANDRA (v2.0), which combines annovar output with other public data sources to output annotated variants (https://www.hgsc.bcm.edu/software) 15, 16. Then, the GV were filtered based on a minor allele frequency of <1%, as assessed from the ExAc data in the non-finish Caucasians http://exac.broadinstitute.org/). The variants were considered priority candidates if they met any of the following criteria:
Stop gain, splicing, and insertion or deletion variants. For splice variants, all variants in the splice regions (±5 bp of splice position) were called. However, GVs located ±2 bp of the splice junctions were considered more likely to be pathogenic than variants located in the splice region (±5 bp), in accord with the American College of Medical Genetics guidelines 17.
Pathogenic or likely pathogenic variants as annotated by ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/)
-
Nonsynonymous variants that met all the following criteria
CADD_PH (combined annotation-dependent depletion phred) score > 10
Medium or high likelihood of being deleterious as assessed by mutation assessor (http://mutationassessor.org/r3/),
PolyPhen2:HVAR (http://genetics.bwh.harvard.edu/pph2/) score > 0.9
Sanger sequencing
The candidate variants of interest were verified by Sanger sequencing and were typed in additional family members, as available.
RESULTS
Phenotypic data
Phenotypic findings in members of a nuclear family are described wherein the affected members show HCM with a variable degree of cardiac hypertrophy. The Pedigree is shown in Figure 2.
Figure 2. Pedigree.
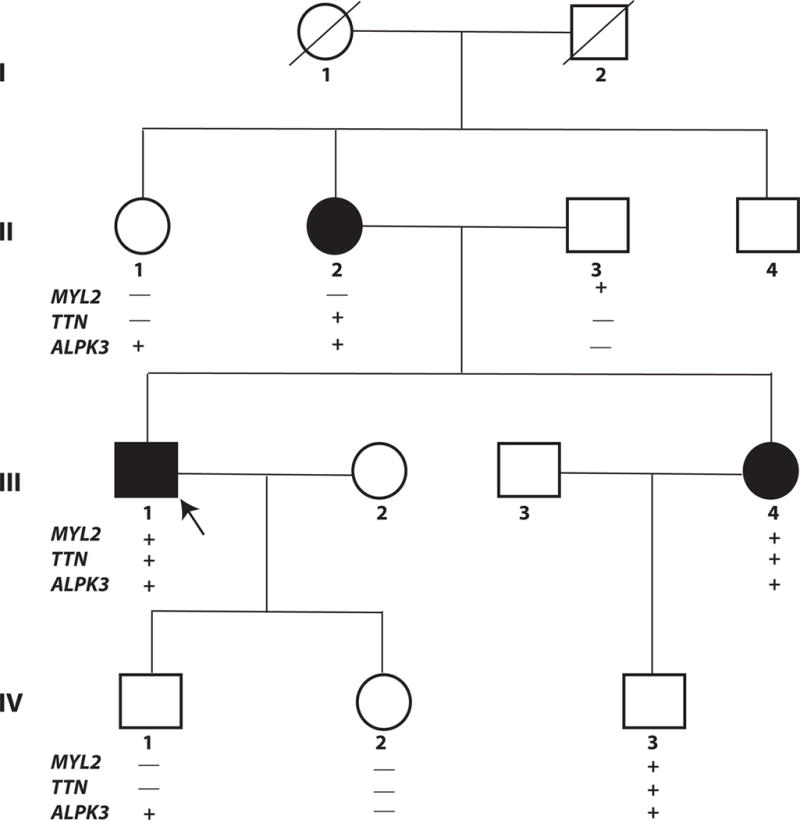
Three affected members are shown in full circles (females) or squares (males) and the unaffected members in open circles and squares. Generations are numbered I through IV and members in each generation are identified by Arabic numerals. Arrow points to the proband. Deceased individuals are identified by the ⁄ symbol.
The candidate genes are listed for each generation. Those carrying the variants are identified by the + and those who do not by the – signs.
III-I (proband)
The proband is a 44-year old male of European (non-Finnish) ancestry who presented 3 years ago with persistent but mild chest pain and dyspnea. Physical examination was remarkable for the presence of II/VI mid-systolic murmur that increased with the Valsalva maneuver. Cardiac enzymes were negative for acute coronary syndromes. A 12-lead ECG was notable for sinus tachycardia at the rate of 104 bpm and evidence of left ventricular hypertrophy (LVH) with repolarization abnormalities. Myocardial stress perfusion tomography showed homogeneous distribution of the radiotracer in the myocardium. An echocardiogram showed a hyperdynamic left ventricle with moderate asymmetric septal hypertrophy, a maximum wall thickness of 1.8 cm. There was a resting left ventricular outflow tract (LVOT) gradient of 20 mmHg that increased to 40 mmHg upon Valsalva maneuver. Rhythm monitoring did not show significant cardiac arrhythmias except for periods of sinus tachycardia at rest. CMR findings were notable for septal hypertrophy and diffuse patchy hyperenhancement, involving multiple myocardial walls. He was treated with a beta-blocker, and lipid lowering agents (Atorvastatin and Gemfibrozil) and showed symptomatic improvement.
Two years after the initial diagnosis, the proband returned with symptoms of increasing dyspnea with a mild level of physical activity and chest pain. Repeat evaluation was notable for evidence of LVH with repolarization abnormalities on a 12-lead ECG and a hyperdynamic left ventricle with a septal thickness of 2 cm, systolic anterior motion of mitral leaflets, mild to moderate mitral regurgitation, and an LVOT gradient of 100 mmHg on echocardiography (Figure 3A). He underwent surgical myectomy, which led to resolution of LVOT gradient and symptoms.
Figure 3. Hypertrophic cardiomyopathy (HCM) phenotype in the siblings.
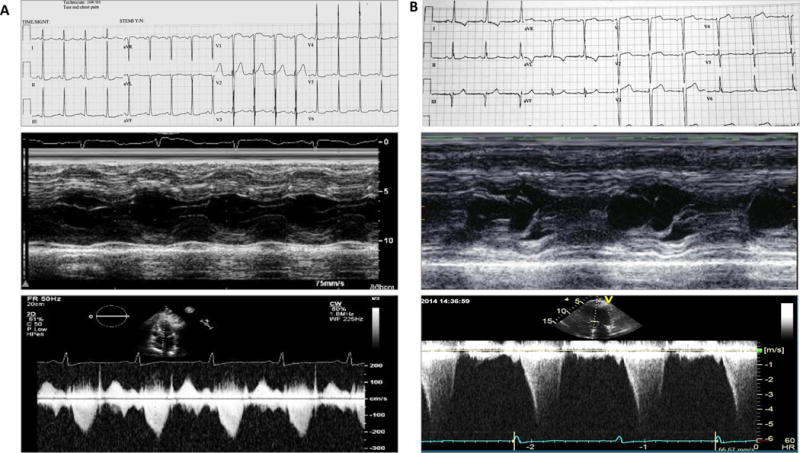
Panel A shows 12-lead electrocardiogram (upper panel), m mode image of the left ventricle, depicting the interventricular septal thickness (middle panel), and the recording of Doppler velocities at the left ventricular outflow tract (lower panel) in the proband (III-1).
Panel B depicts the same phenotypic data, as in Panel A, in individual III-4. Phenotypic data in both individuals indicate a severe phenotype.
II-2
The proband’s mother was diagnosed with HCM at age 63 upon family screening. She had the echocardiographic findings of a hyperdynamic left ventricle and an interventricular septal thickness of 19 mm. She had a history of brief episodes of palpitations and one episode of syncope a few years earlier, which was thought to be due to orthostatic hypotension. Medical history was otherwise unremarkable, as was her physical examination. Specifically, she had no history of systemic arterial hypertension, valvular disease, or other conditions to explain cardiac hypertrophy. A 12-lead ECG was notable for possible left atrial enlargement. A 30-day rhythm monitoring showed rare ventricular ectopic beats and episodes of sinus tachycardia. CMR showed a normal LV wall thickness and function and minimal patchy mid myocardial hyperenhancement in the basal inferior and infero-lateral wall. She had no LVOT obstruction. She has been treated with a low dose of a beta-blocker and has done well since.
II-3
Proband’s father, who is 73 years old, presented with dyspnea on exertion and easy fatigability. He was found to have severe aortic stenosis with an aortic valve area of 1 cm2 and a peak gradient of 68 mmHg. He had mild LVH with a wall thickness of 13 mm on an echocardiogram and a left ventricular ejection fraction (LVEF) of > 70%. A 12-lead ECG was unremarkable without evidence of LVH. He also had an 80% luminal diameter stenosis of left anterior descending coronary artery on coronary angiography. He underwent aortic valve replacement and a single vessel coronary artery bypass surgery. He has been asymptomatic since surgery.
III-4
Proband’s sister was also diagnosed with HCM at 43 years of age upon family screening by ECG and Echocardiography. She was asymptomatic at the time of diagnosis and her physical exam was notable for the presence of a II/VI mid-systolic ejection murmur at left sternal border that radiated toward base of the heart. She also had a mildly elevated arterial blood pressure. A 12-lead ECG showed evidence of LVH, interventricular conduction delay, and repolarization abnormalities. An echocardiogram showed severe asymmetric septal hypertrophy with a hyperdynamic LV systolic function, systolic anterior motion of the mitral leaflet, and a LVOT gradient of 60 to 70 mmHg at rest (Figure 3B). CMR findings were also notable for asymmetric septal hypertrophy and mild patchy hyperenhancement. A 30-day rhythm monitoring showed occasional episodes of sinus tachycardia and rare ventricular ectopic beats.
She has been treated with a beta-blocker, low doses of an angiotension II receptor blocker and a diuretic; the later two medications for treatment of systemic arterial hypertension, and a statin for a high plasma low-density lipoprotein-cholesterol level. She has remained asymptomatic with unchanged ECG and echocardiographic findings during the last 3 years.
Other family members
II-1 is a 72-year old woman with history of intermittent atrial flutter with a normal interventricular septal thickness (8 mm) and a normal LVEF (58%). Likewise, individual II-4 is an asymptomatic 70-year old male, who had normal ECG, echocardiogram, and myocardial stress perfusion tomography.
Proband’s children (IV-1 and IV-2) were asymptomatic and had normal ECGs and echocardiograms on the last evaluation. Likewise, individual IV-3 was a 7-year old asymptomatic boy who had a normal ECG and echocardiogram.
Whole exome sequencing data
Three clinically affected members of the family (II-2, III-1, and III-4) underwent clinical WES. The average coverage produced for each exome was in excess of 70X. Analysis of base-by-base coverage of the exons in 16 genes implicated as causes of HCM or HCM phenocopy (MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC1, MYL2, MYL3, TTN, ALPK3, FH11, PLN, GLA, LAMP2, PRKAG2, and NEXN), showed that 97 to 100% of the exons had a read coverage of at least 10X and 89 to 100% had at least 20X coverage (Online Table I). As an example, ~ 97% of the consensus coding sequence bases in the TTN gene had at least 20X redundant coverage. The coverage metric also included the first and last two intronic bases for each exon, which ensures identification of the splicing variants. No clear causal variant segregating with the phenotype was identified. Variant analysis showed that each exome had ~ 30,000 GVs, of which approximately 7,000 were shared among the three affected members.
Pathogenic variants shared by three affected members
After applying the filters for identification of the pathogenic variants, described in the Methods section, 145 heterozygous variants in 139 genes were shared among the three affected members (Online Table II). Among the shared pathogenic variants, the corresponding genes for the majority were expressed in the heart at an FPKM (fragments per kilo bases of mRNA for per million mapped reads) values > 10 and 38 at an FPKM of 50 or greater per GTEx database (http://www.gtexportal.org/).
Among the candidate genes that contained shared pathogenic variants only TTN and ALPK3 have previously been implicated in hereditary cardiomyopathies. TTN, encoding titin and ALPK3, encoding alpha kinase 3, contained missense mutation p.Lys22368Asn (MAF: 0.00007) and the non-synonymous p.Thr81Met (MAF: 0.0015), respectively. The TTN residue shows perfect conservation from humans to fish and resides in a constitutive exon, which has high transcriptional usage 18. Although the genomic region containing the ALPK3 variant shows low evolutionary conservation and lacks alignments from multiple species, it still met the criteria for being a pathogenic variant. None of remaining shared pathogenic variants is either implicated in cardiomyopathy or occurs in genes coding for the known sarcomere proteins.
Pathogenic variants shared by siblings
Considering that siblings had severe HCM phenotype with LVOT obstruction, as opposed to their mother, who had a mild phenotype, we analyzed shared pathogenic variants between the two siblings. The siblings shared 386 pathogenic coding variants, including the p.Ala13Thr variant (rs104894363, MAF in non-Finish European population: 0.0005) in MYL2 gene, which is a known gene for HCM. No other variant in biologically plausible genes for cardiomyopathies was shared between the siblings.
Extension of the variant analysis to additional family members
The three candidate pathogenic variants were typed in additional family members by Sanger sequencing (Online Figure 1). MYL2 p.Ala13Thr variant was present in the proband’s father’s DNA (II-3), who had mild LVH. However, unequivocal diagnosis of HCM could not be established because of the concomitant presence of severe aortic stenosis, which could explain cardiac hypertrophy in this family member. Individual II-3 did not carry the ALPK3 or the TTN variants. The MYL2 gene is a known causal gene for HCM 10 and the p.Ala13Thr variant is considered pathogenic or likely pathogenic in ClinVar by multiple submitters. The MYL2 variant was absent in other family members except for an asymptomatic 7-year old child (IV-3), who had a normal 12-lead ECG and echocardiogram. The absence of a phenotype in this 7-year old boy might reflect age-dependence of penetrance. This 7-year old child also had inherited the TTN and ALPK3 variants from his affected mother and hence, is at a higher risk of developing HCM. No other unaffected family member had the TTN variant.
The ALPK3 p.Thr81Met variant was present in proband’s maternal aunt (II-1), who had atrial flutter but a normal LV wall thickness and LVEF upon last evaluation at 72 years of age. In addition, an asymptomatic 10-year child (IV-1) who had inherited the ALKP3 variant from the proband had a normal ECG and echocardiogram.
DISCUSSION
The findings of the pathogenic variants in three genes, previously implicated as causes of HCM, put forward the hypothesis that a subset of HCM, which is traditionally considered an archetypical single gene disorder, might be oligogenic in etiology, The three affected family members share pathogenic variants in the TTN and ALKP3 genes and the two siblings, who had a severe phenotype, also carried an additional pathogenic variant in the MYL2, an established gene for HCM 10. A 7-year old child had inherited the above three pathogenic variants but was phenotypically normal. The pathogenic variants, with the exception of the TTN variant, do not show co-segregation with the phenotype, which would expected in an oligogenic etiology. The presence of multiple pathogenic variants renders segregation analysis and determination of genetic causality in the conventional manner inconclusive 2. Accordingly, such variants conventionally would not be considered as causal variants for single gene disorders. Consequently, we propose that a yet-to-be determined fraction of the so-called “missing causal genes” in HCM might be due to the presence of multiple pathogenic variants that might not co-segregate with the phenotype in small families with HCM.
Given the small size of the family, the causality of the pathogenic variants in TTN, MYL2, and ALPK3 in the present family with HCM cannot be unambiguously established. Nevertheless, all three variants, particularly the TTN and MYL2 variants, were considered pathogenic by several bioinformatics algorithms (Online Table III). These pathogenic variants might act alone or in combination with each other or other variants to influence the phenotype.
Amongst the three likely causal variants, the TTN missense variant (p.Lys22368Asn), which resides in a transcriptionally high usage exon, was present in the affected trio and seem to be the most pathogenic variant. It could the actual causal variant for HCM in the present family. In this scenario, the MYL2 variant, which was present in the siblings, and the ALPK3 variant, which was present in all three, might function as the modifiers of the phenotype. Against the possibility is the fact that TTN mutations typically cause DCM, and rarely HCM 11. In addition, given the enormous size of the TTN gene, encoding for a protein with > 34,000 amino acids, and the small size of the family, a random segregation of the TTN variant with the HCM phenotype cannot be excluded.
The p.Ala13Thr variant in MYL2, which is among the first set of causal genes identified for familial HCM 10, was shared by the two siblings who had inherited the variant from proband’s father. The diagnosis of HCM in proband’ father could not be established unambiguously due to the presence of severe aortic stenosis, which could explain the cardiac hypertrophy. The MYL2 variant was also present in a 7-year old boy that did not exhibit HCM, likely due to age-dependent penetrance. The MYL2 variant, however, is absent in proband’s mother, who had a definitive diagnosis of HCM. Therefore, MYL2 variant alone is insufficient to explain HCM in this family. Accordingly, the MYL2 variant, which was present in the siblings with severe HCM, might be a modifier of the phenotype.
The ALPK3 variant, shared by the affected trio and is considered pathogenic by some of the algorithms, alone is unlikely to be the cause of HCM in this family. It was detected in individual II-1 who was 66 years old at the time of last evaluation and did not show evidence of HCM. In addition, a 7-year boy also had inherited the ALPK3 variant but showed no phenotype, albeit the young age precludes from making a firm conclusion, due to age-dependent penetrance. Finally, only compound ALPK3 variants, but not a single variant, are implicated in cardiomyopathies 12. Therefore, the ALPK3 p.Ala13Thr variant is unlikely to be a high penetrance causal variant in HCM.
Despite identification of the pathogenic variants in genes implicated in hereditary cardiomyopathies, because of the small size of the family, the lack of a perfect co-segregation, and the presence of concomitant diseases, unambiguous ascertainment of the causal role of each of the three variants identified in affected members of this family cannot be established. Considering the adequate read coverage of the exons in the known HCM genes, it is unlikely that any of the known genes is responsible for HCM in this family. However, one cannot exclude the possibility of a causal gene, previously not implicated in hereditary cardiomyopathies, to be responsible for HCM in the present family.
In a genetic testing by exome sequencing, often a number of pathogenic variants are identified, which are not directly related to the phenotype of interest, and referred as the “incidental” findings. A notable incidental finding in the present family was the presence of the non-synonymous pathogenic variant p.Asp382Gly in the ADRB1 gene, encoding adrenergic receptor β1. This rare variant is present in the non-Finnish European population at a frequency of 0.00075. The variant was present in three affected individuals who exhibited sinus tachycardia at rest and hypersensitivity to β blockers. These observations are in accord with the known effects of adrenergic control of the heart rate and consistent with data showing that genetic variants in the ADRB1 gene influence heart rate rest and response to beta blockers 19, 20.
Thus, the data illustrate presence of multiple pathogenic variants in genes previously implicated in familial HCM, raising the possibility of an oligogenic etiology of HCM in small families and perhaps sporadic cases. Accordingly, we posit that a fraction of “missing causal genes” in conventionally single gene disorders, such as HCM, might be due to an oligogenic etiology 2. The data advocate for the hypothesis but do not prove it. To prove or reject this hypothesis, large-scale sequencing studies in hundreds of small families, trios, and sporadic cases would be necessary to assess enrichment of multiple pathogenic variants in HCM. Finally, the findings illustrate the difficulty in ascertaining causality in small families and sporadic cases with single gene disorders, even when the pathogenic variants reside in genes implicated in cardiomyopathies.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Hypertrophic cardiomyopathy (HCM) is a classic single gene disorder.
Genes encoding sarcomere proteins are the main causes HCM and are responsible for approximately 60% of HCM, typically in large families.
The remaining causal genes, referred to as the “missing causal genes”, typically in small families and sporadic cases, remains to be identified.
What New Information Does This Article Contribute?
We hypothesize that the difficulty in identifying the “missing causal genes” is in part due to an oligogenic etiology of HCM. Accordingly, multiple pathogenic variants that do not perfectly co-segregate with the phenotype and hence, conventionally are not considered the causal variants, are responsible for HCM in small families and sporadic cases.
In support of this hypothesis, we present a small family wherein the affected family members carry two or three pathogenic variants in TTN, MYL2, and ALPK3 genes, previously implicated as the causal genes in HCM.
The siblings carrying the three pathogenic variants show a severe phenotype, while members with two or one variant show a mild or an ambiguous phenotype.
Each human genome contains a large number of genetic variants (GVs), including several hundred rare and pathogenic variants. GVs exert a spectrum of effect sizes that vary from negligible to very large. GVs with the largest effect sizes are responsible for single gene disorders with Mendelian patterns of inheritance, such as familial HCM. Those with small effect sizes are typically responsible for the complex traits. We posit that a subset of HCM might be oligogenic caused by multiple pathogenic GVs with moderate to large effect sizes. In support of this hypothesis, we describe a family with members carry three pathogenic variants in TTN, MYL2, and ALPK3 genes, previously implicated as the causal genes in HCM. While the data are consistent with the hypothesis, they are insufficient to prove or reject the hypothesis. Large-scale multi-center whole exome or genome sequencing studies would be required to properly test a potential oligogenic etiology and identify the “missing causal genes” for HCM, which is conventionally considered a single gene disorder.
Acknowledgments
SOURCES OF FUNDING
This work was supported in part by grants from NIH, National Heart, Lung and Blood Institute (NHLBI, R01 HL088498, 1R01HL132401, and R34 HL105563), Leducq Foundation (14 CVD 03), Roderick MacDonald Foundation (13RDM005), TexGen Fund from Greater Houston Community Foundation, Texas Heart Institute Foundation, and George and Mary Josephine Hamman Foundation.
Nonstandard Abbreviations and Acronyms
- HCM
Hypertrophic cardiomyopathy
- WES
Whole Exome Sequencing
- TTN
Titin
- MYL2
Myosin light chain 2
- ALPK3
Alpha kinase 3
- MYH7
Myosin heavy chain 7
- MYBPC3
Myosin binding protein C
- GV
Genetic variant
- CMR
Cardiac magnetic resonance
- DCM
Dilated cardiomyopathy
- LVH
Left ventricular hypertrophy
- LVOT
Left ventricular outflow tract
- LVEF
Left ventricular ejection fraction
Footnotes
This manuscript was sent to Sumanth D. Prabhu, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
In January 2016, the average time from submission to first decision for all original research papers submitted to Circulation Research was 13.77 days.
DISCLOSURES
None.
References
- 1.Sen-Chowdhry S, Jacoby D, Moon JC, McKenna WJ. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nature reviews Cardiology. 2016;13:651–675. doi: 10.1038/nrcardio.2016.140. [DOI] [PubMed] [Google Scholar]
- 2.Marian AJ. The case of “missing causal genes” and the practice of medicine: A sherlock holmes approach of deductive reasoning. Circulation research. 2016;119:21–24. doi: 10.1161/CIRCRESAHA.116.308830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation C, MacArthur DG, Farrall M, Cook SA, Watkins H. Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genetics in medicine : official journal of the American College of Medical Genetics. 2016 doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marian AJ, Belmont J. Strategic approaches to unraveling genetic causes of cardiovascular diseases. Circulation research. 2011;108:1252–1269. doi: 10.1161/CIRCRESAHA.110.236067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J Med Genet. 2005;42:e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, Alders M, van Hagen JM. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the mybpc3 gene. J Med Genet. 2006;43:829–832. doi: 10.1136/jmg.2005.040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodatsu A, Konno T, Hayashi K, Funada A, Fujita T, Nagata Y, Fujino N, Kawashiri MA, Yamagishi M. Compound heterozygosity deteriorates phenotypes of hypertrophic cardiomyopathy with founder mybpc3 mutation: Evidence from patients and zebrafish models. American journal of physiology. Heart and circulatory physiology. 2014;307:H1594–1604. doi: 10.1152/ajpheart.00637.2013. [DOI] [PubMed] [Google Scholar]
- 8.Maron BJ, Maron MS, Semsarian C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: A potential link to sudden death in the absence of conventional risk factors. Heart rhythm : the official journal of the Heart Rhythm Society. 2012;9:57–63. doi: 10.1016/j.hrthm.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. Journal of the American College of Cardiology. 2010;55:1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- 10.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nature genetics. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 11.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. The New England journal of medicine. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almomani R, Verhagen JM, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, van der Zwaag PA, Frohn-Mulder IM, Bertoli-Avella AM, Boven LG, van Slegtenhorst MA, van der Smagt JJ, van IWF, Timmer B, van Stuijvenberg M, Verdijk RM, Saffitz JE, du Plessis FA, Michels M, Hofstra RM, Sinke RJ, van Tintelen JP, Wessels MW, Jongbloed JD, van de Laar IM. Biallelic truncating mutations in alpk3 cause severe pediatric cardiomyopathy. Journal of the American College of Cardiology. 2016;67:515–525. doi: 10.1016/j.jacc.2015.10.093. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, Consortium WGS. Wilkie AO, McVean G, Lunter G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nature genetics. 2014;46:912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM. Molecular findings among patients referred for clinical whole-exome sequencing. Jama. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bainbridge MN, Armstrong GN, Gramatges MM, Bertuch AA, Jhangiani SN, Doddapaneni H, Lewis L, Tombrello J, Tsavachidis S, Liu Y, Jalali A, Plon SE, Lau CC, Parsons DW, Claus EB, Barnholtz-Sloan J, Il’yasova D, Schildkraut J, Ali-Osman F, Sadetzki S, Johansen C, Houlston RS, Jenkins RB, Lachance D, Olson SH, Bernstein JL, Merrell RT, Wrensch MR, Walsh KM, Davis FG, Lai R, Shete S, Aldape K, Amos CI, Thompson PA, Muzny DM, Gibbs RA, Melin BS, Bondy ML, Gliogene C. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2015;107:384. doi: 10.1093/jnci/dju384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O’Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O’Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Science translational medicine. 2015;7:270ra276. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranade K, Jorgenson E, Sheu WH, Pei D, Hsiung CA, Chiang FT, Chen YD, Pratt R, Olshen RA, Curb D, Cox DR, Botstein D, Risch N. A polymorphism in the beta1 adrenergic receptor is associated with resting heart rate. American journal of human genetics. 2002;70:935–942. doi: 10.1086/339621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, Weber SA, Greene SM, Hodne D, Nelson B, Morrison J, Domanski MJ, Wagoner LE, Abraham WT, Anderson JL, Carlquist JF, Krause-Steinrauf HJ, Lazzeroni LC, Port JD, Lavori PW, Bristow MR. A polymorphism within a conserved beta(1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11288–11293. doi: 10.1073/pnas.0509937103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.