

Supplemental Digital Content is available in the text
Keywords: coagulation, meta-analysis, polymorphism, trial sequential analysis, venous thromboembolism
Abstract
Background:
Recently, several studies showed that the polymorphisms in the coagulation-related genes might be associated with venous thromboembolism (VTE); however, the results were still controversial. We performed a meta-analysis with trial sequential analysis to investigate the associations between the endothelial cell-activated protein C receptor (EPCR) rs9574, F11 rs2289252, F11 rs2036914, FGG rs2066865, FGG rs1049636, CYP4V2 rs13146272, SERPINC1 rs2227589, and GP6 rs1613662 polymorphisms with the risk of VTE.
Methods:
We searched both the common English-language databases and the Chinese literature databases. Two authors selected studies according to inclusion and exclusion criteria. Crude odds ratios with 95% confidence intervals (CI) were calculated to estimate the strength of this association. Between-study heterogeneity was assessed with the chi-square-based Q test and the I2 statistic.
Results:
Overall, a total of 20 studies were included. The meta-analysis revealed that the F11 rs2289252, F11 rs2036914, FGG rs2066865, and CYP4V2 rs13146272 polymorphisms were closely related to the development of VTE in the white race under the best genetic models after multiple testing adjustments. The EPCR rs9574, FGG rs1049636, SERPINC1 rs2227589, and GP6 rs1613662 polymorphisms might be potential candidates in the pathogenesis of VTE, but trial sequential analyses and sensitivity analyses indicated that the evidences were limited. Larger scale studies were demanded to avoid false-positive outcomes.
Conclusions:
Finally, our study demonstrated the important role of rs2289252, rs2036914, rs2066865, and rs13146272 polymorphisms in the development of VTE in the white race. Rs9574, rs1049636, rs2227589 and rs1613662 polymorphisms might be risk factors of VTE. However, more studies involving diverse races are needed to probe the ethnic difference and the underlying mechanisms of significant associations.
1. Introduction
Venous thromboembolism (VTE) is a major public health concern. VTE associated with hospitalization is an important reason of disability-adjusted life years (DALYs) lost in both developed and developing countries.[1,2] As a multifactorial disease, VTE involves interactions between clinical risk factors and acquired or inherited predispositions to thrombosis.[3] In recent years, many lines of evidence have indicated that thrombotic risk may be in part genetically determined and genetic influence is potentially profound on account of its life-long presence.[4] The protein C (PC) pathway is a part of the natural anticoagulation system. Endothelial cell-activated PC receptor (EPCR), a transmembrane protein widely expressed on the endothelium of large vessels, can bind circulating PC, which further enhances the activity of the PC pathway via the thrombin–thrombomodulin complex by 5- to 20-fold.[5] Several single nucleotide polymorphisms (SNPs) have been explored in the EPCR gene so far. Thereinto, EPCR 4678G/C (rs9574), the tag SNP of haplotype 1 found in the 3’untranslated region (3’UTR), is one of the most studied SNPs recently.[6] Coagulation factors are various protein components that participate in the blood-clotting process. Factor XI (FXI) is a key component of the intrinsic coagulation pathway, which serves as a cofactor for coagulation factor IX.[7] Meijers and colleagues[8] have reported that high level of FXI is a risk factor for VTE. Furthermore, pharmacologic inhibition of FXI has showed an apparent opposite protection against blood clot in different animal models and human studies.[9–12] Two common F11 SNPs, rs2289252 and rs2036914, were found to be associated with VTE in 2 large well-designed population-based (PB) studies: the Leiden Thrombophilia Study (LETS) and the Multiple Environmental and Genetic Assessment of Risk Factors for Venous Thrombosis (MEGA study).[13] It is worth noting that 3 additional SNPs (CYP4V2 rs13146272, SERPINC1 rs2227589, and GP6 rs1613662) were also identified to be associated with VTE in LETS and MEGA studies. Fibrinogen, the precursor of fibrin, is an essential component of the fibrinolytic system. Structurally, it comprises 3 polypeptide chains (Aα, Bβ, and γ), encoded by 3 separate genes, fibrinogen alpha (FGA), fibrinogen beta (FGB), and fibrinogen gamma (FGG).[14] Two haplotype-tagging SNPs of the FGG gene including rs2066865 and rs1049636 have recently been proposed as potential risk factors for VTE.[15]
It is well known that normal hemostasis relies on a successful balance among the anticoagulation system, the coagulation system, and the fibrinolytic system. EPCR, F11, FGG, CYP4V2, SERPINC1, and GP6 play vital roles in these 3 systems. Several studies have investigated the associations between polymorphisms in these 6 genes with the risk of VTE. However, the results remain unclear or even contradictory. To some degree, it is possibly because of small sample size of a single study or insufficient statistical power to probe the latent relation. Hence, we performed a meta-analysis with trial sequential analysis (TSA) to obtain a more precise estimation of the relationships between these 8 polymorphisms and the risk of VTE.
2. Materials and methods
2.1. Ethnic statement
Ethical approval is not necessary for the present meta-analysis and TSA.
2.2. Search strategy
We systematically collected all of the eligible literatures from 01/01/2000 to 01/05/2016 by searching both the common English-language databases (PubMed, Web of Science, and EMBASE) and the Chinese literature databases (CNKI [http://www.cnki.net] and WanFang [http://www. wanfangdata.com.cn ]). The following search phrases were used: (EPCR or PROCR or FXI or F11 or FGG or CYP4V2 or SERPINC1 or GP6; polymorphism or mutation or variant; and VTE. Additional studies were identified by hand, searching the references in original articles and review articles.
2.3. Inclusion and exclusion criteria
The included studies were required to meet the following criteria: evaluating the EPCR rs9574, F11 rs2289252, F11 rs2036914, FGG rs2066865, FGG rs1049636, CYP4V2 rs13146272, SERPINC1 rs2227589, and GP6 rs1613662 polymorphisms with the risk of VTE; using a case–control design; supplying enough data for estimating an odds ratio (OR) with a 95% CI. The major reasons for exclusion of studies were as follows: not involving VTE; reviews, animal studies, comments, or duplicate publications; not designed as a case–control or cohort study; not providing the source of cases and controls and other essential information.
2.4. Data extraction
The information was carefully extracted from all eligible literatures independently by 2 investigators (Liu K and Jiang J) according to the inclusion criteria listed previously. For a conflicting evaluation, a third reviewer (Zou JJ) was consulted and a consensus was reached by discussion. The following variables were collected from each literature: the first author's name, the year of publication, country of origin, ethnicity, genotyping method, study object, source of controls, age, gender, numbers of genotyped cases and controls, and Hardy–Weinberg equilibrium (HWE) in the controls. The ethnicities mainly consisted of the white, yellow, and black races. The different sources of controls were categorized as PB controls and hospital-based (HB) controls. The genotyping methods were divided into polymerase chain reaction-restriction fragment length polymorphism, Taqman, allele-specific PCR, sequencing, and so on. The quality of the studies was assessed using the Newcastle–Ottawa scale (NOS).[16] Studies with scores ≥6 were considered to be of high quality.
2.5. Statistical analysis
Crude odds ratios (ORs) with their corresponding 95% CIs were used to assess the strength of associations between 8 SNPs and VTE risk. OR1, OR2, and OR3 regarding each polymorphism were calculated for genotypes MM versus WW, WM versus WW, and MM versus WM, respectively. The comparison between OR1, OR2, and OR3, and the P-values were used to determine the best genetic model.[17] Besides the best genetic model, several other possible genetic models were also assessed: allele contrast (M vs W), homozygote model (M/M vs W/W), heterozygote model (W/M vs W/W), dominant model (W/M + M/M vs W/W), and recessive model (M/M vs W/M + W/W). To assess the heterogeneity across included studies, the chi-square-based Q test and the I2 statistic were performed. P <0.10 or I2 >50% indicated evidence of heterogeneity.[18] The pooled OR was estimated in both the fixed-effects model (the Mantel–Haenszel method)[19] and the random-effects model (the DerSimonian and Laird methods).[20] The fixed-effects model would be used when the studies were found to be homogeneous. Otherwise, the random-effects model would be adopted. Galbraith plot was performed to further investigate the possible sources of heterogeneity. Stratified analyses were conducted by ethnicity, sample size, source of controls, and study objects. Sensitivity analyses were performed to assess the stability of the results. Publication bias was estimated using Begg's funnel plot and Egger's linear regression test. To adjust the values for multiple comparisons, we applied the Benjamini–Hochberg methods, which control for false discovery rate (FDR).[21] In addition, departure from HWE in the controls was tested by the chi-square test for goodness of fit, and a P <0.05 was considered as a significant disequilibrium. The meta-analysis was conducted using the Stata software (version 12.1; StataCorp LP, College Station, TX; Supplementary file), and the P value was 2-sided.
2.6. Trial sequential analysis
In order to examine the reliability and conclusiveness of the available evidence, we used a novel statistical analysis software called TSA (The Copenhagen Trial Unit, Center for Clinical Intervention Research, Denmark). We calculated the heterogeneity corrected optimal information size (HOIS) by considering an overall type-I error of 5%, a type-II error of 20%, and a relative risk reduction (RRR) assumption of 10%. A continuity correction of 0.5 was also applied in zero-event trials. TSA plotted a 2-sided graph where red straight dash lines indicate conventional P = 0.05 statistical boundaries, the blue line shows the cumulative Z-score of the meta-analysis, and the inward sloping red lines represent the truncated trial sequential monitoring boundaries.
3. Results
3.1. Characteristics of studies
Through the literature search and selection based on the inclusion criteria, a total of 20 papers were included in the meta-analysis (Fig. 1).[13,15,22–39] All the 20 papers were of high quality. Among the eligible studies, 2 included subjects of yellow race from China[22,39] and 3 of black race.[15,29,32] Most of the studies used peripheral blood samples for DNA extraction, and the polymerase chain reaction method or TaqMan method was often utilized for genotyping. All studies recruited age- and sex-matched subjects as controls, apart from 2 including prothrombin G20210A or FV Leiden carriers as research objects [37,42] and 2 were not mentioned.[30,35] All controls were PB in addition to 5 case-control studies including HB controls.[22,28,29,37,38] The distribution of genotypes in all of the controls was consistent with HWE, except for 1 article.[34] The main characteristics for all eligible studies are listed in Supplementary Table S1. All results were based on best genetic models.
Figure 1.
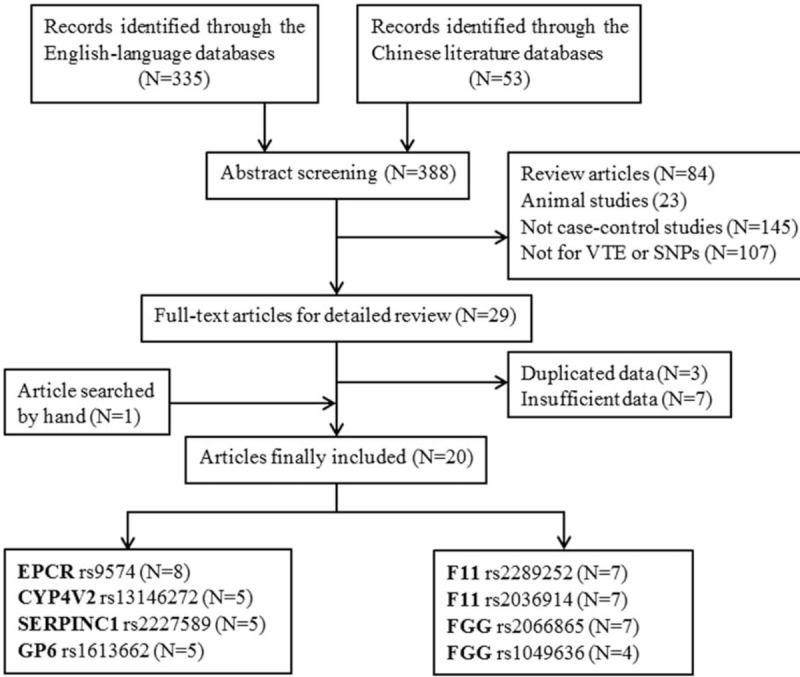
Studies identified with criteria for inclusion and exclusion.
3.2. EPCR rs9574
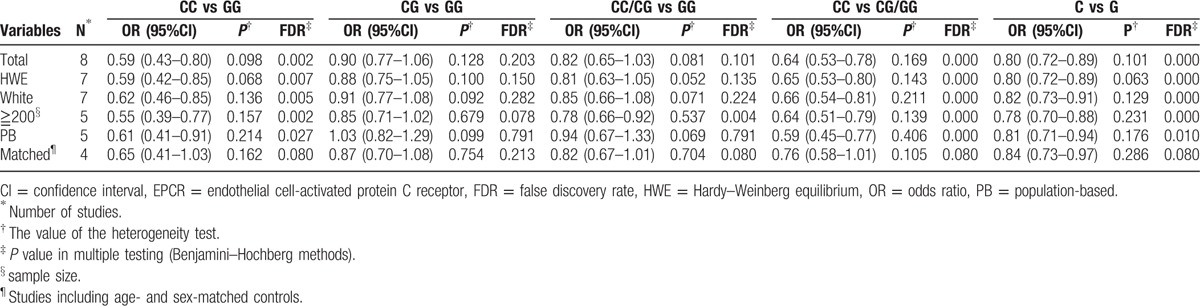
Table 1 lists the main results of the meta-analysis of the association between the EPCR 4678G/C polymorphism and VTE risk. After multiple testing adjustment, a significantly decreased risk of VTE was associated with the 4678G/C polymorphism in the recessive model (OR = 0.64, 95% CI = 0.53–0.78, FDR <0.001). The positive association did not change when we excluded 1 study with control inconsistent with HWE. In the subgroup analysis by ethnicity, source of controls, and sample size (≧200), we found that the EPCR 4678G/C polymorphism was significantly associated with VTE risk in the white race and in the studies with PB controls and relatively large sample size. However, when we merely included the studies which declared the controls were age- and sex-matched, the association was turned out to be meaningless. The heterogeneity across all of the studies was nonsignificant in the best genetic model.
Table 1.
Main results for the EPCR rs9574 mutation in the meta-analysis.
3.3. F11 rs2289252, F11 rs2036914, FGG rs2066865, and FGG rs1049636
As shown in Table 2, our meta-analysis showed that the F11 rs2289252 TT genotype and the F11 rs2036914 CC genotype were risk factors in the susceptibility of VTE for the codominant models. Stratification by ethnicity indicated that the associations were still significant in the white race. When restricting the analysis to the studies with larger sample size or PB controls, the statistically significant associations were still observed. Heterogeneity analysis verified that the heterogeneity across researches was mild.
Table 2.
Main results for the mutations in F11 and FGG genes in the meta-analysis.
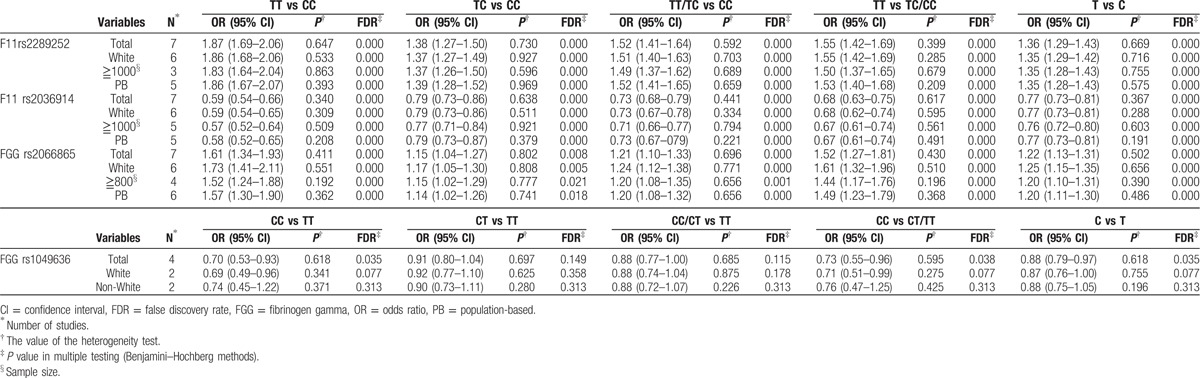
With regard to the FGG rs2066865 polymorphism, we found that individuals carrying C allele were more likely to suffer from VTE than those with T allele in the codominant model (Fig. 2). The results of stratification analyses according to ethnicity, sample size, and source of controls were similar to the overall results. As for the FGG rs1049636 polymorphism, our results suggested that the polymorphism was significantly related to VTE especially in the white race rather than the non-white race in the recessive model. Furthermore, the heterogeneity analysis stated that all the studies of the 2 polymorphisms had favorable homogeneity.
Figure 2.
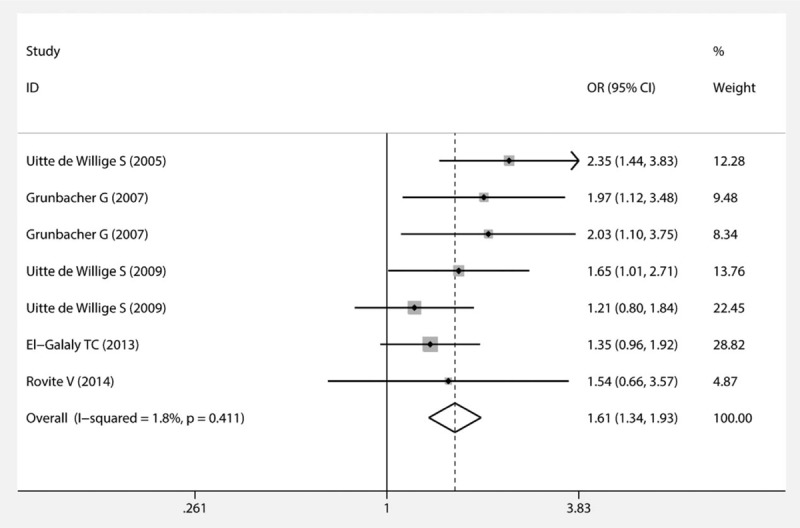
Forest plot of the association between the FGG rs2066865 polymorphism and VTE (codominant model). FGG = fibrinogen gamma, VTE = venous thromboembolism.
3.4. CYP4V2 rs13146272, SERPINC1 rs2227589, and GP6 rs1613662
In terms of the CYP4V2 rs13146272 polymorphism, the overall OR with its 95% CI exhibited a statistically significant association between this polymorphism and a reduced risk of VTE for the codominant model, particularly in the white race and in the studies with PB controls and relatively large sample size. No obvious heterogeneity was observed in the overall and subgroup analyses. Meta-analysis of the SERPINC1 rs2227589 polymorphism showed an elevated risk between this polymorphism and VTE susceptibility with low between-study heterogeneity under the overdominant model after the multiple testing adjustment. Likewise, the significant association did not change in the subgroup analyses based on the white race, sample size, and source of controls. With respect to the GP6 rs1613662 polymorphism, we detected that the A allele was a risk factor in the occurrence of VTE in the dominant model. Similarly, an obvious relationship was identified in subgroup analyses in light of ethnicity, sample size, and source of controls. Meanwhile, the heterogeneity across all of the studies was not prominent. Detailed results are presented in Table 3 and Supplementary Fig. S1.
Table 3.
Main results for the mutations in CYP4V2, SERPINC1, and GP6 genes in the meta-analysis.
3.5. Sensitivity analysis and publication bias
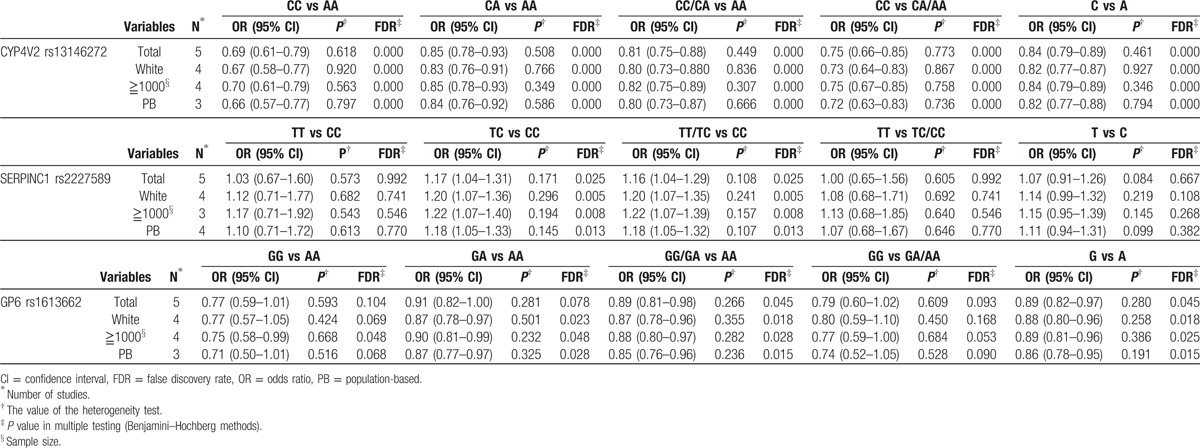
In the sensitivity analysis, the influence of each study on the pooled OR was assessed by repeating the meta-analysis while omitting each study, 1 at a time. To our surprise, the positive association between the SERPINC1 rs2227589 polymorphism and VTE risk turned out to be negative (Fig. 3) after removing the same study.[13] The sensitivity analyses of other polymorphisms certified that our results were reliable and robust.
Figure 3.
Sensitivity analysis for the SERPINC1 rs2227589 polymorphism under the overdominant model.
Begg's funnel plot and Egger's test were performed to evaluate the publication bias of the literatures (Supplementary Figs. S2 and S3). The shapes of the funnel plots seemed symmetrical for all of the polymorphisms. Then, the Egger's test was used to provide statistical evidence of funnel plot symmetry. Finally, the results only revealed that there was potential publication bias existed in the best genetic model for the SERPINC1 rs2227589 polymorphism (P = 0.023).
3.6. Trial sequential analysis
For the F11 rs2036914 and FGG rs2066865 polymorphisms, the number of patients reached the optimal information size and the blue cumulative Z curve crossed the red trial sequential monitoring boundary, which confirmed our positive results. As for the F11 rs2289252, CYP4V2 rs13146272, and GP6 rs1613662 polymorphisms, although the number of patients did not exceed the required information size, the blue cumulative Z curve crossed the red trial sequential monitoring boundary, which verified the reliability of our results. With respect to the EPCR rs9574, FGG rs1049636, and SERPINC1 rs2227589 polymorphisms, actually accrued number of participants did not meet the needs and the blue cumulative Z curve did not cross the trial sequential monitoring boundary. More studies are demanded for these 3 polymorphisms to get a solid conclusion. The results of TSA are shown in Fig. 4 and Supplementary Fig. S4.
Figure 4.
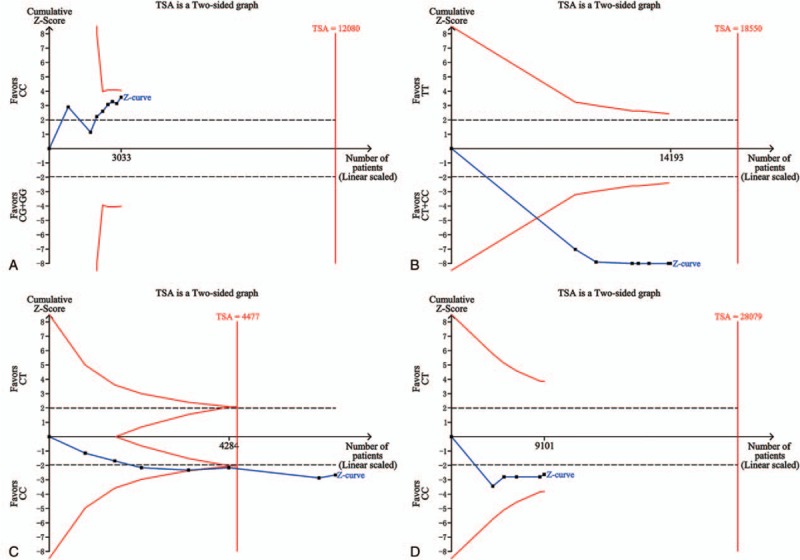
Trial sequential analyses of (A) the EPCR rs9574 polymorphism, (B) the F11 rs2289252 polymorphism, (C) the FGG rs2066865 polymorphism, and (D) the SERPINC1 rs2227589 polymorphism. The heterogeneity-corrected optimal information size was based on a relative risk reduction of 10%, an alpha of 5%, and a beta of 20%. The blue dash line represents the cumulative Z-score of the meta-analysis. The red straight lines represent the conventional P = 0.05 statistical boundaries. The inward sloping red lines represent the truncated trial sequential monitoring boundaries. EPCR = endothelial cell-activated protein C receptor, FGG = fibrinogen gamma.
4. Discussion
VTE is always regarded as a multifactorial disease with both established environmental and genetic risk factors.[40] The recognition that VTE is closely related to the genetic predisposition dates back to the 1960s. Jordan and Nandorff[41] were the first, in 1956, to report the familial tendency in thromboembolic disease. Two recognized and well-characterized genetic risk factors for VTE are the factor V Leiden R506Q and the prothrombin G20210A polymorphisms, which can increase the risk of DVT in carriers by 3- to 5-fold.[25,42] Other polymorphisms, such as SNPs in the EPCR, F11, FGG, CYP4V2, SERPINC1, and GP6 genes, have been reported to be related to VTE. Nevertheless, most conclusions were based on single case–control studies which might lead to false-positive or false-negative results. Pooling all available data using a genetic meta-analysis strategy can overcome the problem of insufficient sample size. A key benefit of this approach is the aggregation of information leading to a higher statistical power and more robust point estimate than is possible from the measure derived from any individual study.[43] The present meta-analysis and TSA demonstrated that the F11 rs2289252, F11 rs2036914, FGG rs2066865, and CYP4V2 rs13146272 polymorphisms played important roles in the development of VTE in the white race. The EPCR rs9574, FGG rs1049636, SERPINC1 rs2227589, and GP6 rs1613662 polymorphisms might be associated with the risk of VTE, but more studies involving different races were needed to support their associations.
4.1. The rs9574 polymorphism in EPCR
Medina et al[30] recruited 405 VTE patients and 401 unrelated controls and found that the EPCR rs9574 polymorphism might be involved in the development of VTE; however, Uitte de Willige et al[37] and Karabiyik et al[35] held an opposite viewpoint. They suggested no association between this polymorphism and VTE susceptibility. In our meta-analysis, pooled data indicated that there was a significant association between EPCR rs9574 and the risk of VTE in 3 genetic models after multiple testing adjustment. EPCR, a key component in the PC pathway, can bind to PC and also activated protein C with high affinity and promote PC activation by increasing the catalytic efficiency of the thrombin–thrombomodulin complex.[44] The possible explanation for the significant association is that the polymorphism may affect the activity of EPCR and further lead to a change of the PC pathway. In our opinion, the discrepancies of the results might partly attribute to different study designs, different study populations, SNP interaction, and the like. It is acknowledged that age and gender are risk factors for VTE.[45] In the subgroup analysis, no association between the rs9574 polymorphism and VTE risk was observed when we merely included the studies that declared the controls were age- and sex-matched, which suggested that study populations might interfere with the results. The study of Navarro et al[31] showed that the association between the rs9574 polymorphism and VTE did not reach statistical significance in carriers of the prothrombin G20210A polymorphism, which revealed that SNP interaction might play a role in different results. In addition, it was worth noting that the result of TSA reminded us to keep a cautious attitude toward the positive relationship. More studies were urgently needed to verify the association between the rs9574 polymorphism and VTE risk and further illustrate potential mechanisms.
4.2. Polymorphisms in F11 and CYP4V2
FXI, a unique coagulase, is shown as a zymogen in plasma, which contributes to the process of coagulation cascade amplification.[7] It is indicated that the thrombotic potential of FXI could be explained by both the ability of FXIa to promote the extrinsic pathway of thrombin generation via inactivation of tissue factor pathway inhibitor (TFPI) and by the feedback activation of FXI by thrombin to further amplify the extrinsic pathway.[46] In the past decade, 2 polymorphisms at the F11 locus, rs2289252 and rs2036914, were most studied and appeared to be associated with VTE. In our work, the ORs of the associations between these 2 polymorphisms and VTE were statistically significant under all of the genetic models in the overall and subgroup analyses. We speculated that polymorphisms in F11 might affect the activity and function of FXI, resulting in the alteration of the FXI level and further lead to the formation of VTE via above-mentioned mechanisms. Besides, another polymorphism in CYP4V2 (rs13146272) that is located close to the F11 gene was reported to be associated with VTE in a previously published genome-wide association study (GWAS) conducted by Bezemer et al.[13] Our meta-analysis indicated that available epidemiologic studies were consistent with the Bezemer's original report. Li et al[25] probed that the rs13146272 polymorphism was in linkage disequilibrium with rs2289252 and rs2036914 in the F11 gene. They supposed that the association of rs13146272 with VTE and FXI levels might be because of the presence of the risk allele for rs13146272 on a common haplotype that included the risk alleles for 2 polymorphisms in the F11 gene. Austin et al[29] and Rovite et al[24] further performed validation studies and supported this hypothesis. Therefore, it was not surprising that we found the rs13146272C allele to be a risk factor in VTE. Although final TSA affirmed our positive results, we advised that more large-scale experiments were still needed to support this conclusion in diverse races.
4.3. Polymorphisms in FGG, SERPINC1, and GP6
Two novel polymorphisms in the FGG gene (rs2066865 and rs1049636) were first proposed as novel risk factors for VTE in the Leiden Thrombophilia Study by Uitte de Willige et al.[23] They considered that polymorphisms in the FGG gene increased the risk for VTE by reducing fibrinogen γ’ levels and reducing ratios of fibrinogen γ’ to total fibrinogen. Then many researchers tried to verify the associations in different ethnicities. However, the results were quite different. Uitte de Willige et al[15] conducted a validation research in a second cohort in 2009 and demonstrated that rs2066865 and rs1049636 polymorphisms influenced VTE risk merely in the Caucasian population but not in the African–American population. Ko et al[39] declared that rs2066865 and rs1049636 polymorphisms were irrelevant to VTE in a Chinese population. In our meta-analysis, the results were accorded with the discrepancy in different race. When we conducted stratified analysis according to the race, the positive association tended to be insignificant in the non-white race. Even though the exact mechanism for the ethnic discrepancy was not well known, some reasons may account for it. One is that it may be because of the differences in the underlying genetic backgrounds, environmental factors, or lifestyles in different population studied. In addition, different minor allele frequency might be a reflection of natural selection stresses or a balance by other related functional polymorphisms.
Egeberg[47] was the first to show that the antithrombin (AT) deficiency was a risk factor for VTE in 1965. AT deficiency is owing to the polymorphisms in the SERPINC1 gene encoding AT.[48] SERPINC1 rs2227589 was the most studied polymorphism which was found to be related to VTE in LETS and MEGA studies conducted by Bezemer et al.[13] However, Austin et al[29] failed to observe any statistically significant association between the rs2227589 polymorphism and VTE in both the white race and the black race. Austin et al further performed a meta-analysis including 5 verification case–control studies (including the 3 reported by Tregouet et al[49]) and combined the OR values of 5 studies. The pooled overall OR was in line with the findings of Bezemer et al. But it was strange that only the result of the Bezemer et al's study was significant. The pooled overall OR became statistically insignificant when we removed the Bezemer et al's study and recombined the OR values. In our meta-analysis, Tregouet et al's study was not included as we did not obtain the original data, but we additionally included 2 recently published studies.[24,27] Sensitivity analysis revealed that Bezemer et al's study might influence the reliability and accuracy of our results. The significant association turned to be meaningless after we removed the MEGA2 study, which was similar to the results got by Austin et al. TSA also required us to include more studies to validate the actual relationship between the rs2227589 polymorphism and VTE. With respect to the GP6 rs1613662 polymorphism, the similar situation resembled to rs2227589 was happened. The significant association no longer existed on removing the MEGA2 study in both Austin et al's meta-analysis and this study. Although TSA showed that the results of the GP6 rs1613662 had statistical reliability, we should keep skeptical of the association and more studies are demanded to eliminate false-positive results.
4.4. Limitations of this study
Despite the overall robust statistical evidence generated through this analysis, some limitations should be addressed. First, our results were based on unadjusted estimates, whereas a more precise analysis should be performed if all individual raw data were available, which would allow for the adjustment by other confounders, including age, gender, activity condition, and lifestyle. Second, VTE is a multifactorial disease that results from complex interactions among many genetic and environmental factors. Hence, more detailed subgroup analyses and combined effects analyses of different SNPs are required. Third, the majority of original studies were from Caucasians and data involving other ethnicities were limited. Therefore, it was doubtful whether the obtained conclusions were generalizable to other populations. Finally, the sample size was still relatively small for some polymorphisms.
5. Conclusion and recommendations
In conclusion, our meta-analysis with TSA showed the important role of rs2289252, rs2036914, rs2066865, and rs13146272 polymorphisms in the development of VTE in the white race. Rs9574, rs1049636, rs2227589, and rs1613662 polymorphisms might be risk factors of VTE, but more evidences were demanded. We believed that the identification of VTE susceptible variants can provide new insight into its etiology. Well-established genetic markers surely would contribute to the early screening and prevention of VTE. Therefore, we recommend the following: first, it is unlikely that applying a single SNP to predict a polygenic disease. A combination of genetic markers and clinical risk factors should be advocated for the sake of accuracy. Also, we previously mentioned that only 2 studies investigated the yellow race and only 3 studies were involved in the black race. Larger scale studies and combined analyses are warranted to further confirm ethnic difference in effect of these polymorphisms on VTE risks. Last but not the least, more basic researches are demanded to clarify underlying biologic mechanisms that drive the positive associations between these polymorphisms and VTE.
Acknowledgments
The authors would like to thank editor and anonymous referees for their valuable and informative comments.
Supplementary Material
Footnotes
Abbreviations: CI = confidence interval, DALYs = disability adjusted life years, EPCR = endothelial cell-activated protein C receptor, FGG = fibrinogen gamma, FXI = factor XI, HB = hospital-based, HWE = Hardy–Weinberg equilibrium, OR = odds ratio, PB = population-based, PC = protein C, SNPs = single nucleotide polymorphisms, TSA = trial sequential analysis, VTE = venous thromboembolism.
JJ, KL, and JZY contributed equally to this work.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].ISTH Steering Committee for World Thrombosis Day. Thrombosis: a major contributor to global disease burden. Thromb Res 2014;134:931–8. [DOI] [PubMed] [Google Scholar]
- [2].Rosendaal FR, Raskob GE. On world thrombosis day. Lancet 2014;384:1653–4. [DOI] [PubMed] [Google Scholar]
- [3].Heit JA. Epidemiology of venous thromboembolism. Nat Rev Cardiol 2015;12:464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lane DA, Grant PJ. Role of hemostatic gene polymorphisms in venous and arterial thrombotic disease. Blood 2000;95:1517–32. [PubMed] [Google Scholar]
- [5].Esmon CT. The endothelial cell protein C receptor. Thromb Haemost 2000;83:639–43. [PubMed] [Google Scholar]
- [6].Navarro S, Bonet E, Estelles A, et al. The endothelial cell protein C receptor: its role in thrombosis. Thromb Res 2011;128:410–6. [DOI] [PubMed] [Google Scholar]
- [7].Gailani D. Advances and dilemmas in factor XI. Curr Opin Hematol 1994;1:347–53. [PubMed] [Google Scholar]
- [8].Meijers JC, Tekelenburg WL, Bouma BN, et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med 2000;342:696–701. [DOI] [PubMed] [Google Scholar]
- [9].Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood 2003;102:953–5. [DOI] [PubMed] [Google Scholar]
- [10].van Montfoort ML, Knaup VL, Marquart JA, et al. Two novel inhibitory anti-human factor XI antibodies prevent cessation of blood flow in a murine venous thrombosis model. Thromb Haemost 2013;110:1065–73. [DOI] [PubMed] [Google Scholar]
- [11].Buller HR, Bethune C, Bhanot S, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 2015;372:232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Girolami A, Peroni E, Girolami B, et al. Congenital factor XI and factor VII deficiencies assure an apparent opposite protection against arterial or venous thrombosis: an intriguing observation. Hematology 2016;21:486–9. [DOI] [PubMed] [Google Scholar]
- [13].Bezemer ID, Bare LA, Doggen CJ, et al. Gene variants associated with deep vein thrombosis. JAMA 2008;299:1306–14. [DOI] [PubMed] [Google Scholar]
- [14].Acharya SS, Dimichele DM. Rare inherited disorders of fibrinogen. Haemophilia 2008;14:1151–8. [DOI] [PubMed] [Google Scholar]
- [15].Uitte de Willige S, Pyle ME, Vos HL, et al. Fibrinogen gamma gene 3′-end polymorphisms and risk of venous thromboembolism in the African–American and Caucasian population. Thromb Haemost 2009;101:1078–84. [PubMed] [Google Scholar]
- [16].Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol 2010;25:603–5. [DOI] [PubMed] [Google Scholar]
- [17].Thakkinstian A, McElduff P, D’Este C, et al. A method for meta-analysis of molecular association studies. Stat Med 2005;24:1291–306. [DOI] [PubMed] [Google Scholar]
- [18].Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539–58. [DOI] [PubMed] [Google Scholar]
- [19].Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719–48. [PubMed] [Google Scholar]
- [20].DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clinic Trials 1986;7:177–88. [DOI] [PubMed] [Google Scholar]
- [21].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B 1995;57:289–300. [Google Scholar]
- [22].Su W, Lv M, Xu X, et al. Association of coagulation factors VIII/XI/XIII polymorphisms with coagulation factor activities and deep vein thrombosis after artificial joints replacement. Am J Ther 2016;23:e1547–53. [DOI] [PubMed] [Google Scholar]
- [23].Uitte de Willige S, de Visser MC, Houwing-Duistermaat JJ, et al. Genetic variation in the fibrinogen gamma gene increases the risk for deep venous thrombosis by reducing plasma fibrinogen gamma’ levels. Blood 2005;106:4176–83. [DOI] [PubMed] [Google Scholar]
- [24].Rovite V, Maurins U, Megnis K, et al. Association of F11 polymorphism rs2289252 with deep vein thrombosis and related phenotypes in population of Latvia. Thrombo Res 2014;134:659–63. [DOI] [PubMed] [Google Scholar]
- [25].Li Y, Bezemer ID, Rowland CM, et al. Genetic variants associated with deep vein thrombosis: the F11 locus. J Thromb Haemost 2009;7:1802–8. [DOI] [PubMed] [Google Scholar]
- [26].Soria JM, Morange PE, Vila J, et al. Multilocus genetic risk scores for venous thromboembolism risk assessment. J Am Heart Assoc 2014;3:e001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].El-Galaly TC, Severinsen MT, Overvad K, et al. Single nucleotide polymorphisms and the risk of venous thrombosis: results from a Danish case-cohort study. Brit J Haematol 2013;160:838–41. [DOI] [PubMed] [Google Scholar]
- [28].Delluc A, Gourhant L, Lacut K, et al. Association of common genetic variations and idiopathic venous thromboembolism. Results from EDITh, a hospital-based case-control study. Thromb Haemost 2010;103:1161–9. [DOI] [PubMed] [Google Scholar]
- [29].Austin H, De Staercke C, Lally C, et al. New gene variants associated with venous thrombosis: a replication study in White and Black Americans. J Thromb Haemost 2011;9:489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Medina P, Navarro S, Estelles A, et al. Contribution of polymorphisms in the endothelial protein C receptor gene to soluble endothelial protein C receptor and circulating activated protein C levels, and thrombotic risk. Thromb Haemost 2004;91:905–11. [DOI] [PubMed] [Google Scholar]
- [31].Navarro S, Medina P, Mira Y, et al. Haplotypes of the EPCR gene, prothrombin levels, and the risk of venous thrombosis in carriers of the prothrombin G20210A mutation. Haematologica 2008;93:885–91. [DOI] [PubMed] [Google Scholar]
- [32].Zoheir N, Eldanasouri N, Abdel-Aal AA, et al. Endothelial cell protein C receptor gene 6936A/G and 4678G/C polymorphisms as risk factors for deep venous thrombosis. Blood Coagul Fibrinolysis 2016;27:259–65. [DOI] [PubMed] [Google Scholar]
- [33].Anastasiou G, Politou M, Rallidis L, et al. Endothelial protein C receptor gene variants and risk of thrombosis. Clin Appl Thromb/Hemost 2016;22:199–204. [DOI] [PubMed] [Google Scholar]
- [34].Pecheniuk N, Elias DJ, Xu X, et al. Failure to validate association of gene polymorphisms in EPCR, PAR-1, FSAP and protein S Tokushima with venous thromboembolism among Californians of European ancestry. Thromb Haemost 2008;99:453–5. [DOI] [PubMed] [Google Scholar]
- [35].Karabiyik A, Yilmaz E, Egin Y, et al. The effects of endothelial protein C receptor gene polymorphisms on the plasma sEPCR level in venous thrombosis patients. Turk J Haematol 2012;29:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Medina P, Navarro S, Estelles A, et al. Influence of the 4600A/G and 4678G/C polymorphisms in the endothelial protein C receptor (EPCR) gene on the risk of venous thromboembolism in carriers of factor V Leiden. Thromb Haemost 2005;94:389–94. [DOI] [PubMed] [Google Scholar]
- [37].Uitte de Willige S, Van Marion V, Rosendaal FR, et al. Haplotypes of the EPCR gene, plasma sEPCR levels and the risk of deep venous thrombosis. J Thromb Haemost 2004;2:1305–10. [DOI] [PubMed] [Google Scholar]
- [38].Grunbacher G, Weger W, Marx-Neuhold E, et al. The fibrinogen gamma (FGG) 10034C>T polymorphism is associated with venous thrombosis. Thromb Res 2007;121:33–6. [DOI] [PubMed] [Google Scholar]
- [39].Ko YL, Hsu LA, Hsu TS, et al. Functional polymorphisms of FGA, encoding alpha fibrinogen, are associated with susceptibility to venous thromboembolism in a Taiwanese population. Hum Genet 2006;119:84–91. [DOI] [PubMed] [Google Scholar]
- [40].Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008;28:370–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jordan FL, Nandorff A. The familial tendency in thrombo-embolic disease. Acta Medica Scand 1956;156:267–75. [DOI] [PubMed] [Google Scholar]
- [42].Zoller B, Garcia de Frutos P, Hillarp A, et al. Thrombophilia as a multigenic disease. Haematologica 1999;84:59–70. [PubMed] [Google Scholar]
- [43].Walker E, Hernandez AV, Kattan MW. Meta-analysis: its strengths and limitations. Cleve Clin J Med 2008;75:431–9. [DOI] [PubMed] [Google Scholar]
- [44].Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, et al. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci USA 1996;93:10212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Park MS, Perkins SE, Spears GM, et al. Risk factors for venous thromboembolism after acute trauma: a population-based case-cohort study. Thromb Res 2016;144:40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res 2016;141Suppl 2:S8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diathesis Haemorrh 1965;13:516–30. [PubMed] [Google Scholar]
- [48].Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med 2001;344:1222–31. [DOI] [PubMed] [Google Scholar]
- [49].Tregouet DA, Heath S, Saut N, et al. Common susceptibility alleles are unlikely to contribute as strongly as the FV and ABO loci to VTE risk: results from a GWAS approach. Blood 2009;113:5298–303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.