

Abstract
Proteins change their charge state through protonation and redox reactions as well as through binding charged ligands. The free energy of these reactions are dominated by solvation and electrostatic energies and modulated by protein conformational relaxation in response to the ionization state changes. Although computational methods for calculating these interactions can provide very powerful tools for predicting protein charge states, they include several critical approximations of which users should be aware. This chapter discusses the strengths, weaknesses, and approximations of popular computational methods for predicting charge states and understanding their underlying electrostatic interactions. The goal of this chapter is to inform users about applications and potential caveats of these methods as well as outline directions for future theoretical and computational research.
Keywords: Dielectric constant, Electrostatics, Monte Carlo, Molecular Simulation, pKa, Poisson Boltzmann
Introduction
Methods that use continuum electrostatics have been developed to calculate the energies of protein charge states as they change through processes such as residue protonation, redox chemistry, or ion binding. While only a subset of amino acids are titratable, they play key roles in protein function (Bartlett, Porter, Borkakoti & Thornton, 2002). The model pKa of isolated amino acid in aqueous solution (Richarz & Wüthrich, 1975), can be used to calculate the probability that an isolated residue is charged at a given pH. Aspartate (Asp), glutamate (Glu), arginine (Arg), and lysine (Lys) comprise approximately 25% of average proteins and their pKa,sol values favor their ionization at physiological pHs (Kim, Mao & Gunner, 2005). The termini of amino acid chains also have model pKa values that cause them to frequently be ionized at physiological pH. Isolated histidine (His) has a pKa value near 7, which makes it easy to titrate at physiological pH values. It not surprising that His is highly enriched in active sites (Holliday, Almonacid, Mitchell & Thornton, 2007). Cysteine (Cys) (Go & Jones, 2013) and tyrosine (Tyr) (Styring, Sjoholm & Mamedov, 2012) are acids with higher model pKa values and are therefore less frequently ionized; however, these residues can play important functional roles as proton donors and as redox active sites. The residues in the active sites of proteins are often made of clusters of residues with linked protonation equilibria, leading to “non-ideal” titration curves (Ondrechen, Clifton & Ringe, 2001). A remarkable number of the mutations that lead to cancer involve protonatable residues (Webb, Chimenti, Jacobson & Barber, 2011). Although nucleic acids (Wong & Pollack, 2010) and phospholipid membranes (Argudo, Bethel, Marcoline & Grabe, 2016) also have titratable groups and strong electrostatic interactions, this chapter will focus on proteins.
The ionization states of small molecules are important to their functions as substrates, cofactors, and control factors. Many protein ligands are charged with ionization states that can change during the binding process or enzymatic reactions (Schindler, Bornmann, Pellicena, Miller, Clarkson & Kuriyan, 2000; Dissanayake, Swails, Harris, Roitberg & York, 2015; Lee, Miller & Brooks, 2016). Cofactors such as NAD or FAD have charged groups such as phosphates that do not participate in reactions but must be bound to the protein for enzyme catalysis. Metal ions are often used by proteins to enhance stability such as in Zn fingers and as participants in redox reactions (Williams, 1997). Biological processes often occur at salt concentrations of 150 mM (or higher (Bowers & Wiegel, 2011)) such that all biomolecules are surrounded by a bath of small ions. The resulting ion cloud interacts with the protein in several ways, including salt-specific protein binding, electrostatic screening, and changing the thermodynamic activity of the protein in solution (Record, Anderson & Lohman, 1978; Grochowski & Trylska, 2008)
Charged groups have very favorable interactions with water that strongly influence their behavior in aqueous solutions (Warshel & Russell, 1984; Ren et al., 2012). They are often found on the on protein exterior surfaces maximizing their interaction with water while ensuring protein solubility and influencing interactions with other biomolecules. Supercharged proteins with total charges in excess of ±30 e are now used in protein design to prevent aggregation (Lawrence, Phillips & Liu, 2007). However, an important minority of charges are found within proteins, where they play functional roles.
Protein interiors are not simple hydrophobic environments and thus can tolerate internal charges through favorable electrostatic interactions (Spassov, Ladenstein & Karshikoff, 1997; Kim et al., 2005). Interaction with other buried charges can form stabilizing ion pairs. Additionally, polar and polarizable groups are also present in protein interiors: the amide backbone dipole moment is larger than water’s (Gunner, Saleh, Cross, ud-Doula & Wise, 2000), many amino acid side chains that are polar or polarizable, and many crystal structures show water molecules and ions in protein interiors (Nayal & Di Cera, 1994, 1996; Makarov, Pettitt & Feig, 2002) One of the goals of the electrostatic calculation methods described in this chapter is to quantitatively understand the nature of these electrostatic interactions.
This review will describe current methods for computing the charge states of residues and ligands as a function of pH, Eh, or solution salt concentrations. The key reactions are thus:
| (1a) |
| (1b) |
| (1c) |
where the species include acids (A), bases (B), ligands (L), oxidized species (O), and reduced species (R). Redox reactions are characterized by the number of electrons (n) and hydrogens (m) transferred. Ligand binding may also be accompanied by changes in protonation of the protein or ligand (Lee et al., 2016). Computational methods for modeling these reactions attempt to predict how the energetics of proton (pKa), electron (Em), or ligand (Ka) change as a function of environment (e.g., protein interior vs. solution). A wide range of experimental data are available for testing the predicted pKa (Stanton & Houk, 2008; Gosink, Hogan, Pulsipher & Baker, 2014), Em (Reedy & Gibney, 2004) and Kd (Gilson, Liu, Baitaluk, Nicola, Hwang & Chong, 2016) values. The goal of matching specific numerical values creates a high bar for testing these calculation methodologies.
The pKa value is the solution pH where the activities of A− and AH (or B and BH+) are equal. However, proton/electron/ion binding affinities depend on the pH-dependent ionization states of other protein residues, thus making the proton affinity and thus the in situ pKa pH-dependent. Therefore, a single pKa is often insufficient for characterizing the behavior of a titratable residue. Titration curves, describing the charge state as a function of pH can provide valuable information about the energetics influencing protein charge regulation (H. Webb et al., 2011). In addition, as reactions often occur far from the pKa of the reactant, it is often important to determine the proton affinity at physiological pH (Goyal, Lu, Yang, Gunner & Cui, 2013). Insight into protein electrostatics is ideally obtained through the (favorable) comparison of experimental and calculated titration curves together with the microscopic information (e.g., electrostatic potentials, hydrogen bonding networks, etc.) obtained from computational methods (Nielsen, Gunner & Garcia-Moreno, 2011).
The calculation of the free energy of a group of charges in a protein or other macromolecule by continuum methods has been reviewed extensively (Gunner & Alexov, 2000; Bashford, 2004; Garcia-Moreno & Fitch, 2004; Warshel, Sharma, Kato & Parson, 2006; Alexov et al., 2011). Thus, rather than providing a detailed methods review, we will highlight the information and choices that guide continuum electrostatics calculation and discuss emerging strategies for improving these methods.
Biomolecular structure and flexibility
Structures are a required starting point for contemporary continuum electrostatic calculations (Berman et al., 2000). Charge state calculations are sensitive to structural details so care should be taken to access the structural quality using tools now found associated with all structures in the PDB database. However, rigid, single structures are inadequate for accurate charge state calculations due to the importance of changes in flexibility and conformation that occur upon introduction of new charges into a protein.
One of the key choices in charge state modeling involves the degrees of freedom (DOFs) included in the model. In the simplest case of rigid molecules, the only DOFs are the protonation or redox states or the ligand binding state. Because the protein will move in response to changes in the protonation, redox, or binding states, sampling DOFs for multiple structural “conformers” available to protein or ligand allows more a more “physical” analysis of the process. Thus, continuum electrostatics simulations balance implicit DOFs which, as described below, are approximated by the dielectric constant of the protein (εsolute) and explicit degrees of freedom.
A protein microstate is a defined choice for each element that has any DOF. Each microstate has an associated energy that is used to generate the thermodynamic averages for titration curves, pKas, Ems, binding probabilities, etc. The energy Gα of a microstate α can be written as a sum of contributions, which is implicitly summed over each group with DOF:
| (2) |
where mα μα is the free energy of mα bound species with chemical potential μα, is the non-electrostatic molecular mechanical energy, is the electrostatic energy, the Uα terms depend explicitly on the state of the other residues in the microstate, is the polar solvation energy, and is the nonpolar solvation energy. The chemical potential μα varies for the quantity of interest; for protonation, it can be written as:
| (3) |
where kBT ≈ 2.5 kJ/mol (0.43 pKa units) is the thermal energy at room temperature and log(10) ≈ 2.3. Most energies represent energy differences between the microstate in the protein interior and a reference state in solution. The quantity pKα,α is the model value for the residue in solution: the positive form of the expression is used for acidic sites, and the negative form is used for basic sites. Given this reference value, the other terms in the equation represent an effective shift to the model pKα,α to account for the influence of the protein environment.
The probability of a microstate β is given by the ensemble average
| (4) |
If n groups each sample 2 protonation states, then there are 2n microstates. If residue i has mi protonation or steric conformers there are ∏i mi microstates where the product runs over all residues with DOF. The high dimensionality of this sum makes it impractical to evaluate for most protein systems. Instead of direct evaluation, ρβ is often calculated through limited conformational sampling; e.g., via Monte Carlo (MC) simulations (Song, Mao & Gunner, 2009; Polydorides & Simonson, 2013). Conformational degrees of freedom can range from sampling side chain rotameric states to relatively inexpensive optimization of steric clashes (Song, 2011) and simple enumeration of different tautomeric forms for the hydrogen position on protonated side chains.
Allowing only dipolar groups to reorient and sample multiple rotameric and tautomer states has significant advantages (Nielsen & Vriend, 2001). Modifying these positions, remodels the hydrogen bond network in response to charge changes, which can provide a significant energetic stabilization of titration events. Note that this form of limited sampling can require ad hoc entropy corrections to compensate for larger numbers of neutral state tautomers or conformers (Song et al., 2009).
One significant approximation in conformational sampling involves the treatment of the intramolecular interactions, which use force fields with only self and pairwise energetics to greatly improve the efficiency when evaluating microstate energies: the Uα terms include only pairwise additive energetics between two groups and are independent of the state of any third group. Evaluation of all pairwise interactions yields an energy matrix of dimension m2 for m conformers. This pairwise decomposition is possible for less accurate non-polarizable force fields and algorithms that sample proton positions and side chain rotameric states. However, more recent polarizable force fields and larger collective protein motions such as backbone displacements generally cannot be represented in this pairwise form. However, while most methods that utilize Monte Carlo sampling make this approximation, it should be recognized that it misses motions that are likely to be important (Richman, Majumdar & Garcia-Moreno, 2014).
Monte Carlo methods can be used to incorporate side chain conformer sampling on a rigid protein backbone (Rabenstein, Ullmann & Knapp, 1998; Song et al., 2009; Polydorides & Simonson, 2013). Such sampling attempts to explicitly evaluate the ensemble average described above and thus incorporates a significant amount of side chain response to charge state changes. However, adding conformational degrees of freedom requires new approximations. In particular, the shape of the protein can change when sampling different side chain or backbone conformations. Calculation costs can increase dramatically if the shape is each microstate is calculated explicitly. To maintain the cost for calculating the interaction energies of O(m2) often relies on all conformers being present when the continuum electrostatic pairwise interactions between conformers are determined. This can exaggerate the low dielectric space of the protein. Some early approaches scaled the electrostatic interactions by an empirical screening function (Georgescu, Alexov & Gunner, 2002). Newer methods correct for dielectric boundary errors due to excess conformers by using information obtained from a small number of calculations with an exact boundary (Song et al., 2009).
Molecular dynamics calculations can also be used to provide conformations in different protonation states. Given a particular titratable site, two sets of simulations are performed: one with the charged state of the site and a second with the neutral state. For sufficiently small energetic differences between the two protonation states (i.e., when linear response theory is valid), these ensembles will substantially overlap and the titration probability can be calculated via simple ensemble averages (Sham, Chu & Warshel, 1997). Molecular dynamics-based linear response approaches have two key limitations. The first is the computational expense of running O(2n) molecular dynamics simulations to sample the n distinct neutral and ionized charge states. Rational choices can help pick consequential protonation states to sample (Witham et al., 2011; Meyer & Knapp, 2015). The second limitation is the underlying linear response assumption requiring the energy difference between neutral and ionized states is small – which is often not true for the important titration events in protein systems (Di Russo, Marti & Roitberg, 2014).
Molecular dynamics (MD) simulations can also be performed in open constant-pH ensembles. Unlike the fixed charge state simulations described above, constant-pH methods allow charged sites to exchange protons with the surrounding solution based on the pH of the bulk media, the model pKa of the site, and the energetics of the conformational ensemble. One class of methods performs MD for 10s of fs followed by a continuum electrostatics pKa calculation as described above to modify the protonation states in the MD trajectory (Baptista, Martel & Peterson, 1997; Swails, York & Roitberg, 2014; Lee, Miller, Damjanovic & Brooks, 2015). Alternatively, protonation states can be changed continuously via λ dynamics (Khandogin & Brooks, 2005; Goh, Hulbert, Zhou & Brooks, 2014). The primary hurdle to adoption of such continuous-pH MD methods is the difficulty of reaching convergence of the simulations. The use of pH replica exchange has led to significant improvements, but these methods are still not routine for the study of large proteins.
Solvent models or: How I learned to stop worrying and love the dielectric coefficient
Most methods use continuum models of solvation behavior to incorporate the effects of solvent on charging energetics because of the computation effort associated with explicit descriptions of water molecules. The simplest continuum model for electrostatics represents the solvent as a dielectric material, usually with a dielectric coefficient of approximately 80 to represent water. The Poisson equation (Nicholls & Honig, 1991; Baker, 2004) describes polar solvation (electrostatic) energies within this dielectric approximation. This equation is can be combined with a nonpolar solvation term that describes the non-electrostatic contributions from the solvent when conformers with significantly different surface exposure are sampled (Song et al., 2009). For calculations of ligand affinity and for ligand partition coefficients, continuum models generally include a shape-related contribution, to describe the work associated with inserting the uncharged solute into water, and a Lennard-Jones-like term to describe the weak solute-solvent dispersive interactions (Lee et al., 2016). Although small, such weak dispersive forces play an important role in protein solvation and in titration state calculations (Levy, Zhang, Gallicchio & Felts, 2003; Wagoner & Baker, 2004; Song et al., 2009). Popular models for the cavity term generally contain a term that scales as the area of the molecule times the surface tension of the solution and often also include a term that multiples solution (hard sphere) pressure with the volume of the solute (Wagoner & Baker, 2006).
Use of the Poisson equation – or other related continuum models – assumes that all polarization in the system (molecule and solvent) is linear, local, and time-independent. Linear response implies that, no matter how large the electric field, the system will polarize in a proportional manner. However, given the finite density and polarizability of water and molecular solutes, this assumption is clearly violated at high charge densities and field strengths, such as found near nucleic acids (Lipfert, Doniach, Das & Herschlag, 2014). Local response implies that system polarization always occurs in the same location as an applied field. However, given the non-zero size and hydrogen bonding structure of water and most biomolecular species, this assumption is nearly always violated in biologically relevant systems (Mobley, Barber, Fennell & Dill, 2008; Xie, Jiang, Brune & Scott, 2012). Finally, the static response assumes no time dependence for molecular polarization. However, even in bulk solvent, this time-independence is violated, with the optical dielectric constant of water of 2, which increases to the static value of 80 on the picosecond-nanosecond timescale (Fernandez, Mulev, Goodwin & Sengers, 1995; Zasetsky, 2011).
Given that nearly all of the assumptions of continuum electrostatics are violated in biologically relevant systems, the reader is probably wondering “why bother?” The answer lies in the power of heuristics. Although there are many arguments about its accuracy at microscopic levels (for example, (Schutz & Warshel, 2001; Kukic et al., 2013; Simonson, 2013)), the continuum model of water with a dielectric coefficient of 78–80 has proven remarkably useful for a wide range of applications. Likewise, while the ab initio derivation of solute dielectric constants is likely an exercise in futility, several heuristics have been useful in extending the applicability of continuum electrostatics to real biomolecular systems. These heuristics are described below; however, it is essential that the users of these continuum electrostatics heuristics are aware that they are using imperfect surrogates for complicated molecular phenomena. In particular, continuum electrostatics calculations should always be benchmarked for accuracy against real experimental data.
Simple – but imperfect – heuristics can be used to guide the selection of a molecular dielectric coefficient value (εsolute). These are presented in Figure 1 and comprise three basic regimes:
An εsolute of 2 represents the electronic polarization that will be found in any condensed matter system (Landau, Lifshit︠s︡ & Pitaevskiĭ, 1984)]. This interpretation has an important implication for continuum electrostatics calculations: εsolute ≥ 2 should be used for all calculations with non-polarizable force fields (Leontyev & Stuchebrukhov, 2009).
An εsolute of 4 has been ascribed to dried proteins and can be interpreted to include a very constrained polarization response of the protein dipoles (Gilson & Honig, 1986). This interpretation has an important implication for continuum electrostatics: an εsolute ≥ 4 should be used for all calculations that do not allow backbone rearrangement; e.g., through molecular dynamics or Monte Carlo configuration sampling.
Larger values of εsolute allow more of the dipolar rearrangement of the backbone and side chains to be treated in an averaged manner with a single, compact parameter. Values of 4 < εsolute < 12 have been successfully used to predict protein-protein binding and are often attributed to limited side chain rearrangement. Values of εsolute above 12 are associated with larger scale backbone rearrangement and water penetration. Early continuum electrostatics attempts to model pKa values in proteins showed that εsolute = 20 gave the best predictive power for calculations using a single dielectric constant and a single conformation (Antosiewicz, McCammon & Gilson, 1994).
Figure 1.
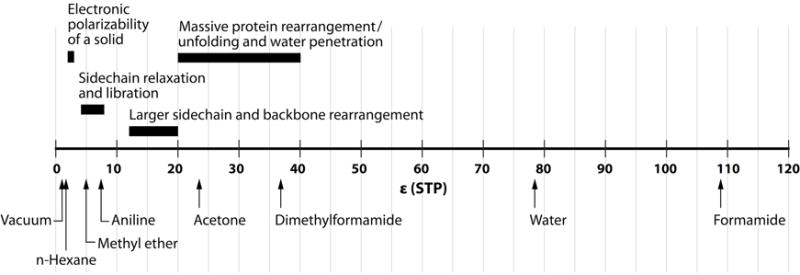
A summary of dielectric constants of model compounds (bottom) and their application to protein continuum electrostatics modeling (top).
Not all continuum electrostatics treatments use a constant dielectric coefficient for the solute interior; some models use larger dielectric values for regions of the protein with greater responses to charge changes. Alexov has varied the dielectric constant based on the atomic packing density (Li, Li, Zhang & Alexov, 2013). While such variation does not explicitly take into account the chemical nature of the side chains, it does provide a mechanism for modeling internal degrees of freedom through the dielectric coefficient. Because these calculations avoid explicit conformational sampling, they offer the possibility of improved dielectric descriptions with the efficiency of standard continuum electrostatic methods.
Most continuum electrostatics software packages will identify interior cavities large enough to accommodate a water molecule – and many will assign these interior cavities a bulk dielectric value of εsolute = 80. However, the high-dielectric treatment of internal cavities comes with a few important caveats. First, it is difficult to provide a physical justification for a single water molecule having the dielectric behavior of the bulk solvent. Second, this procedure is sensitive to small conformational changes that may cause regions to switch between εsolute and εsolvent. To address this issue, Knapp has explored the effects of modeling the cavities with higher detail using a finer grid, which can accept smaller or less spherical wet regions, which improves the fit to benchmark pKas (Meyer, Kieseritzky & Knapp, 2011). Other methods make use of Gaussian dielectric boundaries in the calculation of the Poisson-Boltzmann equation, which also raises the effective internal dielectric constant (Word & Nicholls, 2011; Li, Li & Alexov, 2014).
As an alternative to high-dielectric models of internal cavities, continuum electrostatics software such as MCCE can include explicit water molecules within the protein (Song, Mao & Gunner, 2003). The included waters require explicit sampling. They must be optimized for each charge state and the number of waters may change with the charge state. Waters are often found in clusters so this optimization must be performed for multiple water molecules simultaneously. As a result, the inclusion of explicit water molecules can substantially increase the computational expense of the charge state calculation. The pKas obtained with implicit or explicit waters in the cavities have been found to agree surprisingly well in limited testing.
The various modifications of the methods described above all improve the fit to known data essentially by increasing the effective interior dielectric constant. The electrostatic energy of a charge depends on the atomic charge distribution, the radius and the interior and exterior dielectric constant. Thus, the effective interior dielectric constant can be raised by increasing εsolute directly, or by smoothing the dielectric surface, or by enhancing cavities in the interior. The effects of changing these parameters have been explored separately. Without a better sense of exactly how the various parameters interact the search through parameter space remains Balkanized with different laboratories exploring their favorite parameters. However, it should be noted that all of these changes do lead to significant improvement in the correspondence between experimental and calculated values.
Modeling ion-solute interactions
Ions are arguably more difficult to model than solvent. The simplest – and most widely used – model of ion behavior is based on Debye-Hückel descriptions of aqueous ions as a diffuse “cloud” that non-specifically screens electrostatic behavior in solution. The only major determinants of ion behavior in Debye-Hückel-like models are the ion concentration and charge valencies. However, this treatment has extreme limitations in describing realistic protein-ion interactions that often include specific ion binding to protein sites as well as strong dependence on ion species, even for ions with the same charge. To address these issues, some researchers have begun to use models that combine implicit solvent descriptions with explicit simulation of the ions (often via Monte Carlo sampling) (Sharp, Friedman, Misra, Hecht & Honig, 1995; Chen, Marucho, Baker & Pappu, 2009; Song & Gunner, 2009). Nevertheless, many charge state calculations still use Poisson-Boltzmann (PB) methods, which combine a Poisson treatment of the solvent with the Boltzmann Debye-Hückel-like ion description.
Force field and parameter choices
The microstate energy calculations described above require several different types of parameters to describe molecular mechanics interactions, solvent characteristics, as well as atomic size and charge. The molecular mechanics energies, atomic charges, and solute-solvent Lennard-Jones interactions are often specified by standard molecular simulation force fields such as AMOEBA (Schnieders, Baker, Ren & Ponder, 2007; Shi et al., 2013), AMBER (Pearlman et al., 1995) or CHARMM (Brooks et al., 2009). These force fields can also be used to specify the solute-solvent boundary through atomic radii; however, custom parameter sets such as PARSE (Tannor et al., 1994) or ZAP (Word & Nicholls, 2011) are generally preferred because they have been optimized to reproduce solvation energies. In addition to atomic radii, the user must specify the algorithm used to determine the shape of the solute-solvent interface. A variety of choices are available for these shape algorithms ranging from simple unions of spheres (Lee & Richards, 1971) or Gaussians (Grant, Pickup, Sykes, Kitchen & Nicholls, 2007) to heuristic molecular-accessible surfaces (Connolly, 1983) to thermodynamically defined self-consistent solute-solvent interface definitions (Cheng, Dzubiella, McCammon & Li, 2007; Chen, Baker & Wei, 2010, 2011). Additionally, the user must choose a function to define the ion-accessible regions around the protein; however, this interface is commonly chosen as an ion-accessible union of spheres with radii equal to the atomic radii plus a nominal ionic radius of 0.2 nm. It is important to note that the optimal choices for radii, charges, and surface definitions are strongly correlated; i.e., the radii are often optimized for a specific surface definition (Dong & Zhou, 2002). These many choices of parameters are then presented to a program that will solve the Poisson-Boltzmann equation to provide the solvation energy of individual conformers (within the environment of the protein) and the pairwise interactions between all pairs of conformers. For example, the programs DelPhi (Li, Li, Zhang & Alexov, 2012) or APBS (Baker, Sept, Joseph, Holst & McCammon, 2001) have been employed within programs such as MCCE (Song et al., 2009), and Karlsberg (Meyer et al., 2011), DelPhi pKa (Wang, Li & Alexov, 2015), and PDB2PKA (Dolinsky et al., 2007; Olsson, Sondergaard, Rostkowski & Jensen, 2011) to calculate the equilibrium protonation, redox and ligand binding states as a function of the appropriate chemical potential.
Titration state prediction methods must be benchmarked against datasets of in situ pKas, Ems, or Kds to determine their accuracy. There are approximately ≈350 wild-type residues with known pKas that are used extensively for such benchmarking (Song et al., 2009). These include a large number surface residues where the protein does not significantly influence the proton affinity. A subset of 100 residues has been selected to yield better range of pKas for training and testing (Stanton & Houk, 2008). The “null model” for charge state prediction assigns the model amino acid pKa value to all residues in the protein, regardless of their location or interactions. When the null model is used with the 100-residue subset, the RMSD between predicted and experimental pKa values is ≈1 pH unit. This sets a challenging metric for evaluating the performance of more sophisticated titration prediction methods. For example, the RMSD using modern Monte Carlo methods with continuum electrostatics force field, an εsolute between 4 and 8 and addition of conformational sampling or modification of the dielectric boundary and distribution can be between 0.9 and 1.1 (Song et al., 2009; Polydorides & Simonson, 2013; Wang et al., 2015). However, informatics-based methods such as PROPKA3 can do much better while sacrifice the underlying physical interactions for knowledge-based potentials (Olsson, 2011).
The Garcia-Moreno lab has placed >100 mutated residues into the core of Staphylococcal nuclease (Isom, Castaneda, Cannon, Velu & Garcia-Moreno, 2010; Isom, Castaneda, Cannon & Garcia-Moreno, 2011; Richman, Majumdar & Garcia-Moreno, 2015). These residues formed the basis of the only blind challenge; i.e., where pKas were calculated without the experimental value being known (Nielsen et al., 2011). A meta-analysis (Gosink et al., 2014) of the blind predictions found that the RMSD for the null model is ≈3.5, indicating that the pKas for these residues were very shifted from the model values due to their burial in the protein. Empirical methods such as PROPKA3 (Olsson, 2011) did significantly better than the null model, methods with added conformational sampling did slightly better than the null model, while methods without added sampling did worse. Papers submitted after the pKas were revealed were able to obtain RMSDs <2 for this challenging dataset, as different modifications were explored once the errors were known. Particular improvement was found for methods that increased the response of the protein; e.g.: by using more explicit sampling via continuous-pH molecular dynamics (Wallace et al., 2011), by adding ensembles of structures obtained with MD (Witham et al., 2011), Rosetta (Song, 2011), through increased side-chain conformation sampling (Gunner, Zhu & Klein, 2011), by increasing the effective εsolute to implicitly model more internal water (Meyer et al., 2011), or by using a smoother dielectric boundary (Word & Nicholls, 2011). The errors for calculations with rigid backbones were smaller when crystal structures of the mutants were used rather then when the mutation was made in silico. Ensemble models which aggregated all of the predictions using Bayesian model averaging gave the best overall results (Gosink et al., 2014).
Conclusions
The goal of this chapter was to present an overview of computational methods for predicting charge states of proteins with an emphasis on the issues that arise when applying continuum electrostatic methods to these applications. Given the many choices that must be made when applying these computational methods, one of the most important issues for this field is the availability of well-curated experimental data sets for testing computational predictions. The pKa Cooperative is a collaborative activity focused on assembling such data sets, performing blind predictions, and discussing the results as well as how to improve computational predictions (http://pkacoop.org/). All of the methods described above can be tuned to provide reasonable agreement with experimental data in a postdiction setting. However, only a few methods perform with acceptable accuracy (~1 pKa unit error) in blind challenge predictions. Among these, constant-pH molecular dynamics methods generated the best predictions – at significantly increased computational expense and the risk of poor convergence of the molecular dynamics simulations. Thus, computational methods continue to evolve to make the calculation of the energy of charges in protein faster and more accurate while providing increased physical insight into the forces at work. The current methods, despite their limitations, provide guidance as to the proton affinities of sites in proteins as well as the atomic interactions that affect a specific charge in a specific site and thus can be invaluable in getting more understanding of protein structure/function relationships.
Acknowledgments
MRG gratefully acknowledges the support of Grant MCB1519640 from NSF, as well as National Institute on Minority Health and Health Disparities Grant 8G12MD7603-28 for infrastructure. NAB gratefully acknowledges support from NIH grants R01GM069702 and R01GM099450.
References
- Alexov E, Mehler EL, Baker N, Baptista AM, Huang Y, Milletti F, Word JM. Progress in the prediction of pKa values in proteins. Proteins. 2011;79:3260–3275. doi: 10.1002/prot.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antosiewicz J, McCammon JA, Gilson MK. Prediction of pH-dependent properties in proteins. J Mol Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- Argudo D, Bethel NP, Marcoline FV, Grabe M. Continuum descriptions of membranes and their interaction with proteins: towards chemically accurate models. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbamem.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA. Poisson-Boltzmann methods for biomolecular electrostatics. Methods Enzymol. 2004;383:94–118. doi: 10.1016/S0076-6879(04)83005-2. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista AM, Martel PJ, Peterson SB. Simulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titration. Proteins: Struct Funct Genet. 1997;27:523–544. [PubMed] [Google Scholar]
- Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–211. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- Bashford D. Macroscopic electrostatic models for protonation states in proteins. Front Biosci. 2004;9:1082–1099. doi: 10.2741/1187. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Bourne PE. The protein data bank. Nucl Ac Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers KJ, Wiegel J. Temperature and pH optima of extremely halophilic archaea: a mini-review. Extremophiles. 2011;15:119–128. doi: 10.1007/s00792-010-0347-y. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AA, Marucho M, Baker NA, Pappu RV. Simulations of RNA interactions with monovalent ions. Methods Enzymol. 2009;469:411–432. doi: 10.1016/S0076-6879(09)69020-0. [DOI] [PubMed] [Google Scholar]
- Chen Z, Baker NA, Wei GW. Differential geometry based solvation model I: Eulerian formulation. J Comput Phys. 2010;229:8231–8258. doi: 10.1016/j.jcp.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Baker NA, Wei GW. Differential geometry based solvation model II: Lagrangian formulation. J Math Biol. 2011;63:1139–1200. doi: 10.1007/s00285-011-0402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LT, Dzubiella J, McCammon JA, Li B. Application of the level-set method to the implicit solvation of nonpolar molecules. J Chem Phys. 2007;127:084503. doi: 10.1063/1.2757169. [DOI] [PubMed] [Google Scholar]
- Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- Di Russo NV, Marti MA, Roitberg AE. Underlying thermodynamics of pH-dependent allostery. J Phys Chem B. 2014;118:12818–12826. doi: 10.1021/jp507971v. [DOI] [PubMed] [Google Scholar]
- Dissanayake T, Swails JM, Harris ME, Roitberg AE, York DM. Interpretation of pH-activity profiles for acid-base catalysis from molecular simulations. Biochemistry. 2015;54:1307–1313. doi: 10.1021/bi5012833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Zhou HX. Electrostatic contributions to T4 lysozyme stability: solvent-exposed charges versus semi-buried salt bridges. Biophys J. 2002;83:1341–1347. doi: 10.1016/S0006-3495(02)73904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Mulev Y, Goodwin A, Sengers J. A database for the static dielectric constant of water and steam. J Phys Chem Ref Data. 1995;24:33. [Google Scholar]
- Garcia-Moreno EB, Fitch CA. Structural interpretation of pH and salt-dependent processes in proteins with computational methods. Methods Enzymol. 2004;380:20–51. doi: 10.1016/S0076-6879(04)80002-8. [DOI] [PubMed] [Google Scholar]
- Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson MK, Honig BH. The dielectric constant of a folded protein. Biopolymers. 1986;25:2097–2119. doi: 10.1002/bip.360251106. [DOI] [PubMed] [Google Scholar]
- Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM, Jones DP. The redox proteome. J Biol Chem. 2013;288:26512–26520. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh GB, Hulbert BS, Zhou H, Brooks CL., 3rd Constant pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins. 2014;82:1319–1331. doi: 10.1002/prot.24499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosink LJ, Hogan EA, Pulsipher TC, Baker NA. Bayesian model aggregation for ensemble-based estimates of protein pKa values. Proteins. 2014;82:354–363. doi: 10.1002/prot.24390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal P, Lu J, Yang S, Gunner MR, Cui Q. Changing hydration level in an internal cavity modulates the proton affinity of a key glutamate in cytochrome c oxidase. Proc Natl Acad Sci U S A. 2013;110:18886–18891. doi: 10.1073/pnas.1313908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant JA, Pickup BT, Sykes MJ, Kitchen CA, Nicholls A. The Gaussian Generalized Born model: application to small molecules. Phys Chem Chem Phys. 2007;9:4913–4922. doi: 10.1039/b707574j. [DOI] [PubMed] [Google Scholar]
- Grochowski P, Trylska J. Continuum molecular electrostatics, salt effects, and counterion binding–a review of the Poisson-Boltzmann theory and its modifications. Biopolymers. 2008;89:93–113. doi: 10.1002/bip.20877. [DOI] [PubMed] [Google Scholar]
- Gunner MR, Alexov E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim Biophys Acta. 2000;1458:63–87. doi: 10.1016/s0005-2728(00)00060-8. [DOI] [PubMed] [Google Scholar]
- Gunner MR, Saleh MA, Cross E, ud-Doula A, Wise M. Backbone dipoles generate positive potentials in all proteins: origins and implications of the effect. Biophys J. 2000;78:1126–1144. doi: 10.1016/S0006-3495(00)76671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunner MR, Zhu X, Klein MC. MCCE analysis of the pKas of introduced buried acids and bases in staphylococcal nuclease. Proteins. 2011;79:3306–3319. doi: 10.1002/prot.23124. [DOI] [PubMed] [Google Scholar]
- Holliday GL, Almonacid DE, Mitchell JB, Thornton JM. The chemistry of protein catalysis. J Mol Biol. 2007;372:1261–1277. doi: 10.1016/j.jmb.2007.07.034. [DOI] [PubMed] [Google Scholar]
- Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno B. Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci U S A. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Castaneda CA, Cannon BR, Velu PD, Garcia-Moreno EB. Charges in the hydrophobic interior of proteins. Proc Natl Acad Sci U S A. 2010;107:16096–16100. doi: 10.1073/pnas.1004213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandogin J, Brooks CL., 3rd Constant pH molecular dynamics with proton tautomerism. Biophys J. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mao J, Gunner MR. Are acidic and basic groups in buried proteins predicted to be ionized? J Mol Biol. 2005;348:1283–1298. doi: 10.1016/j.jmb.2005.03.051. [DOI] [PubMed] [Google Scholar]
- Kukic P, Farrell D, McIntosh LP, Garcia-Moreno EB, Jensen KS, Toleikis Z, Nielsen JE. Protein dielectric constants determined from NMR chemical shift perturbations. J Am Chem Soc. 2013;135:16968–16976. doi: 10.1021/ja406995j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau LD, Lifshit︠s︡ EM, Pitaevskiĭ LP. Electrodynamics of continuous media. 2nd. Oxford Oxfordshire; New York: Pergamon; 1984. [Google Scholar]
- Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Richards FM. The interpretations of protein structures: Estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Lee J, Miller BT, Brooks BR. Computational scheme for pH-dependent binding free energy calculation with explicit solvent. Protein Sci. 2016;25:231–243. doi: 10.1002/pro.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Miller BT, Damjanovic A, Brooks BR. Enhancing Constant-pH Simulation in Explicit Solvent with a Two-Dimensional Replica Exchange Method. J Chem Theory Comput. 2015;11:2560–2574. doi: 10.1021/ct501101f. [DOI] [PubMed] [Google Scholar]
- Leontyev IV, Stuchebrukhov AA. Electronic continuum model for molecular dynamics simulations. J Chem Phys. 2009;130:085102. doi: 10.1063/1.3060164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RM, Zhang LY, Gallicchio E, Felts AK. On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. J Am Chem Soc. 2003;125:9523–9530. doi: 10.1021/ja029833a. [DOI] [PubMed] [Google Scholar]
- Li C, Li L, Zhang J, Alexov E. Highly efficient and exact method for parallelization of grid-based algorithms and its implementation in DelPhi. J Comput Chem. 2012;33:1960–1966. doi: 10.1002/jcc.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li C, Alexov E. On the modeling of polar component of solvation energy using smooth Gaussian-based dielectric function. Journal of theoretical & computational chemistry. 2014;13 doi: 10.1142/S0219633614400021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li C, Zhang Z, Alexov E. On the dielectric “constant” of proteins: smooth dielectric function for macromolecular modeling and Its implementation in DelPhi. J Chem Theory Comput. 2013;9:2126–2136. doi: 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Doniach S, Das R, Herschlag D. Understanding nucleic acid-ion interactions. Annu Rev Biochem. 2014;83:813–841. doi: 10.1146/annurev-biochem-060409-092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V, Pettitt BM, Feig M. Solvation and hydration of proteins and nucleic acids: a theoretical view of simulation and experiment. Acc Chem Res. 2002;35:376–384. doi: 10.1021/ar0100273. [DOI] [PubMed] [Google Scholar]
- Meyer T, Kieseritzky G, Knapp EW. Electrostatic pKa computations in proteins: role of internal cavities. Proteins. 2011;79:3320–3332. doi: 10.1002/prot.23092. [DOI] [PubMed] [Google Scholar]
- Meyer T, Knapp EW. pKa values in proteins determined by electrostatics applied to molecular dynamics trajectories. J Chem Theory Comput. 2015;11:2827–2840. doi: 10.1021/acs.jctc.5b00123. [DOI] [PubMed] [Google Scholar]
- Mobley DL, Barber AE, 2nd, Fennell CJ, Dill KA. Charge asymmetries in hydration of polar solutes. J Phys Chem B. 2008;112:2405–2414. doi: 10.1021/jp709958f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal M, Di Cera E. Predicting Ca2+-binding sites in proteins. Proc Natl Acad Sci U S A. 1994;91:817–821. doi: 10.1073/pnas.91.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256:228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Honig B. A rapid finite difference algorithm utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J Comp Chem. 1991;12:435–445. [Google Scholar]
- Nielsen JE, Gunner MR, Garcia-Moreno BE. The pKa Cooperative: a collaborative effort to advance structure-based calculations of pKa values and electrostatic effects in proteins. Proteins. 2011;79:3249–3259. doi: 10.1002/prot.23194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JE, Vriend G. Optimizing the hydrogen-bond network in Poisson-Boltzmann equation-based pKa calculations. Proteins: Struct Funct Genet. 2001;43:403–412. doi: 10.1002/prot.1053. [DOI] [PubMed] [Google Scholar]
- Olsson MH. Protein electrostatics and pKa blind predictions; contribution from empirical predictions of internal ionizable residues. Proteins. 2011;79:3333–3345. doi: 10.1002/prot.23113. [DOI] [PubMed] [Google Scholar]
- Olsson MH, Sondergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Ondrechen M, Clifton J, Ringe D. THEMATICS: a simple computational predictor of enzyme function from structure. Proc Natl Acad Sci USA. 2001;98:12473–12478. doi: 10.1073/pnas.211436698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman DA, Case DA, Caldwell JW, Ross WS, III, T EC, DeBolt S, Kollman P. AMBER a package of computer programs for applying molecular mechanics normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Computer Physics Communications. 1995;91:1–41. [Google Scholar]
- Polydorides S, Simonson T. Monte Carlo simulations of proteins at constant pH with generalized Born solvent, flexible sidechains, and an effective dielectric boundary. J Comput Chem. 2013;34:2742–2756. doi: 10.1002/jcc.23450. [DOI] [PubMed] [Google Scholar]
- Rabenstein B, Ullmann GM, Knapp EW. Calculation of protonation patterns in proteins with structural relaxation and molecular ensembles-application to the photosynthetic reaction center. Eur Biophys J. 1998;27:626–637. [Google Scholar]
- Record MT, Anderson CF, Lohman TM. Thermodynamic analysis of ion effects on binding and conformational equilibria of proteins and nucleic-acids - roles of ion association or release, screening, and ion effects on water activity Q. Rev Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- Ren P, Chun J, Thomas DG, Schnieders MJ, Marucho M, Zhang J, Baker NA. Biomolecular electrostatics and solvation: a computational perspective. Q Rev Biophys. 2012;45:427–491. doi: 10.1017/S003358351200011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richarz R, Wüthrich K. Carbon-13 NMR chemical shifts of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers. 1975;17:2133–2141. [Google Scholar]
- Richman DE, Majumdar A, Garcia-Moreno EB. pH dependence of conformational fluctuations of the protein backbone. Proteins. 2014;82:3132–3143. doi: 10.1002/prot.24673. [DOI] [PubMed] [Google Scholar]
- Richman DE, Majumdar A, Garcia-Moreno EB. Conformational reorganization coupled to the ionization of internal lys residues in proteins. Biochemistry. 2015;54:5888–5897. doi: 10.1021/acs.biochem.5b00522. [DOI] [PubMed] [Google Scholar]
- Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- Schnieders MJ, Baker NA, Ren P, Ponder JW. Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum. J Chem Phys. 2007;126:124114. doi: 10.1063/1.2714528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz CN, Warshel A. What are the “dielectric constants” of proteins and how to validate electrostatic models? Proteins: Struct Funct Genet. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- Sham YY, Chu ZT, Warshel A. Consistent calculations of pKa’s of ionizable residues in proteins: Semi-microscopic and microscopic approaches. J Phys Chem. 1997;101:4458–4472. [Google Scholar]
- Sharp KA, Friedman RA, Misra V, Hecht J, Honig B. Salt effects on polyelectrolyte-ligand binding: comparison of Poisson-Boltzmann, and limiting law/counterion binding models. Biopolymers. 1995;36:245–262. doi: 10.1002/bip.360360211. [DOI] [PubMed] [Google Scholar]
- Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P. The polarizable atomic multipole-based AMOEBA force feld for proteins. J Chem Theory Comput. 2013;9:4046–4063. doi: 10.1021/ct4003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson T. What Is the dielectric constant of a protein when Its backbone Is fixed? J Chem Theory Comput. 2013;9:4603–4608. doi: 10.1021/ct400398e. [DOI] [PubMed] [Google Scholar]
- Song Y. Exploring conformational changes coupled to ionization states using a hybrid Rosetta-MCCE protocol. Proteins. 2011 doi: 10.1002/prot.23146. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- Song Y, Gunner MR. Using Multi-Conformation Continuum Electrostatics to compare chloride binding motifs in a-amylase, human serum albumin, and Omp32. J Mol Biol. 2009;387:840–856. doi: 10.1016/j.jmb.2009.01.038. [DOI] [PubMed] [Google Scholar]
- Song Y, Mao J, Gunner MR. Calculation of proton transfers in bacteriorhodopsin bR and M intermediates. Biochemistry. 2003;42:9875–9888. doi: 10.1021/bi034482d. [DOI] [PubMed] [Google Scholar]
- Song YF, Mao JJ, Gunner MR. MCCE2: Improving Protein pKa Calculations with Extensive Side Chain Rotamer Sampling. Journal of Computational Chemistry. 2009;30:2231–2247. doi: 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov VZ, Ladenstein R, Karshikoff AD. Optimization of the electrostatic interactions between ionized groups and peptide dipoles in proteins. Protein Sci. 1997;6:1190–1196. doi: 10.1002/pro.5560060607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton CL, Houk KN. Benchmarking pKa prediction methods for residues in proteins. J Chem Theory Comput. 2008;4:951–966. doi: 10.1021/ct8000014. [DOI] [PubMed] [Google Scholar]
- Styring S, Sjoholm J, Mamedov F. Two tyrosines that changed the world: Interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Biochim Biophys Acta. 2012;1817:76–87. doi: 10.1016/j.bbabio.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Swails JM, York DM, Roitberg AE. Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J Chem Theory Comput. 2014;10:1341–1352. doi: 10.1021/ct401042b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannor DJ, Marten B, Murphy R, Friesner RA, Sitkoff D, Nicholls A, Goddard WA., III Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J Am Chem Soc. 1994;116:11875–11882. [Google Scholar]
- Wagoner J, Baker NA. Solvation forces on biomolecular structures: a comparison of explicit solvent and Poisson-Boltzmann models. J Comput Chem. 2004;25:1623–1629. doi: 10.1002/jcc.20089. [DOI] [PubMed] [Google Scholar]
- Wagoner JA, Baker NA. Assessing implicit models for nonpolar mean solvation forces: the importance of dispersion and volume terms. Proc Natl Acad Sci U S A. 2006;103:8331–8336. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Wang Y, Shi C, Pastoor KJ, Nguyen BL, Xia K, Shen JK. Toward accurate prediction of pKa values for internal protein residues: The importance of conformational relaxation and desolvation energy. Proteins. 2011 doi: 10.1002/prot.23080. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- Wang L, Li L, Alexov E. pKa predictions for proteins, RNAs, and DNAs with the Gaussian dielectric function using DelPhi pKa. Proteins. 2015;83:2186–2197. doi: 10.1002/prot.24935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A, Russell ST. Calculations of electrostatic interactions in biological systems and in solutions. Q Rev Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochim Biophys Acta. 2006;1764:1646–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- Webb H, Tynan-Connolly BM, Lee GM, Farrell D, O’Meara F, Sondergaard CR, Nielsen JE. Remeasuring HEWL pKa values by NMR spectroscopy: Methods, analysis, accuracy, and implications for theoretical pKa calculations. Proteins. 2011;79:685–702. doi: 10.1002/prot.22886. [DOI] [PubMed] [Google Scholar]
- Williams RJ. The natural selection of the chemical elements. Cell Mol Life Sci. 1997;53:816–829. doi: 10.1007/s000180050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witham S, Talley K, Wang L, Zhang Z, Sarkar S, Gao D, Alexov E. Developing hybrid approaches to predict pKa values of ionizable groups. Proteins. 2011;79:3389–3399. doi: 10.1002/prot.23097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong GC, Pollack L. Electrostatics of strongly charged biological polymers: ion-mediated interactions and self-organization in nucleic acids and proteins. Annual review of physical chemistry. 2010;61:171–189. doi: 10.1146/annurev.physchem.58.032806.104436. [DOI] [PubMed] [Google Scholar]
- Word JM, Nicholls A. Application of the Gaussian dielectric boundary in Zap to the prediction of protein pKa values. Proteins. 2011;79:3400–3409. doi: 10.1002/prot.23079. [DOI] [PubMed] [Google Scholar]
- Xie DX, Jiang Y, Brune P, Scott LR. A Fast Solver for a Nonlocal Dielectric Continuum Model. Siam J Sci Comput. 2012;34:B107–B126. [Google Scholar]
- Zasetsky AY. Dielectric relaxation in liquid water: two fractions or two dynamics? Phys Rev Lett. 2011;107:117601. doi: 10.1103/PhysRevLett.107.117601. [DOI] [PubMed] [Google Scholar]