

Abstract
Mutations in isocitrate dehydrogenase (IDH) are the most prevalent genetic abnormalities in lower grade gliomas. The presence of these mutations in glioma is prognostic for better clinical outcomes with longer patient survival. In the present study, we found that defects in oxidative metabolism and 2-HG production confer chemosensitization in IDH1-mutated glioma cells. In addition, temozolomide (TMZ) treatment induced greater DNA damage and apoptotic changes in mutant glioma cells. The PARP1-associated DNA repair pathway was extensively compromised in mutant cells due to decreased NAD+ availability. Targeting the PARP DNA repair pathway extensively sensitized IDH1-mutated glioma cells to TMZ. Our findings demonstrate a novel molecular mechanism that defines chemosensitivity in IDH-mutated gliomas. Targeting PARP-associated DNA repair may represent a novel therapeutic strategy for gliomas.
Introduction
Mutations in isocitrate dehydrogenase (IDH1/2) are common genetic abnormalities in grade II and III diffusive astrocytomas and oligodendrogliomas(1). In WHO grade II/III gliomas, IDH mutated tumors are highly prevalent comprising nearly 80% of all clinical cases(2). In glioma, IDH mutations cluster in an arginine residue at the center of the catalytic domain (IDH1 R132, IDH2 R172). Mutant IDH confers neomorphic enzymatic activity that, catalyzes α-ketoglutarate (α-KG) into 2-hydroxyglutarate (2-HG), an oncometabolite closely related to the deactivation of α-KG-dependent deoxygenases(3,4). For example, IDH1 mutant derived 2-HG promotes hypoxia signaling by perturbing the catalytic activity of prolyl hydroxylase, resulting in constitutive activation of hypoxia-inducible factor 1α (HIF-1α)(5). Additionally, 2-HG has also been found to affect collagen maturation and basement membrane function, which may facilitate cancer cell infiltration and promote glioma progression(6). Clinically, the occurrence of IDH mutations predicts longer survival and greater sensitivity to chemotherapy in low-grade gliomas and secondary glioblastomas(7,8). A phase III clinical trial has provided the direct link between IDH mutation and survival benefit from chemotherapy(9). Combined with O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation status, IDH mutations serve as an important prognostic marker for gliomas treated with radiation and chemotherapy(10). Although there has been increasing awareness of the correlation between IDH mutations and chemo-sensitivity, the molecular mechanism that determines the vulnerability that results from IDH mutations remains unanswered.
DNA repair is defined as a series of molecular changes that occur in response to compromised chromosomal integrity. DNA repair is integral to cancer therapies based on generating DNA damage in chromosomal DNA, such as radiation therapy and cytotoxic chemotherapies(11). The activation and expression level of DNA repair pathways largely determines the efficacy of chemotherapies and resulting clinical outcomes(12,13). Several studies shed light on the changes in DNA repair mechanism in IDH1-mutated cells, including RAD51 and ATM pathways(14,15). The distinct connection between DNA repair pathway and chemo-sensitivity in IDH-mutated cells, however, must be further elucidated.
In the present study, we established cell lines that stably expresses either the IDH1R132C or IDH1R132H pathogenic mutations. We demonstrate that our cell lines recapitulate the IDH1-mutant phenotypes found in glioma patients. The IDH1 mutations resulted in the metabolic reprogramming and cytotoxic effects via 2-HG production. In addition, cells with mutant IDH failed to form the poly (ADP-ribose) polymer (pADPR), and therefore were unable to maintain genomic integrity. Furthermore, targeting the PARP DNA repair mechanism remarkably potentiated the cytotoxic effects of chemotherapy. Taken together, our findings indicate a potential molecular mechanism of chemo-sensitization in IDH mutant gliomas and, suggests a novel therapeutic strategy for clinical therapies.
Materials and Methods
Cell Culture and Reagents
The U251 MG cell line was obtained from Sigma Aldrich in 2015. The U87 MG, HT1080 and LN-18 glioma cell lines were acquired from the American Type Culture Collection (ATCC) in 2015. Each cell line was tested and authenticated by their manufacturers. Cells were cultured in Dulbecco Modified Eagle Medium-Ham F-12 medium (DMEM/F-12, 1:1, Life Technologies) supplemented with the 10% fetal bovine serum (Cellgro) and 1% antibiotics (100 U/ml penicillin and 10 µg/ml streptomycin) at 37 °C in a humidified air with 5% CO2. Temozolomide (TMZ, Sigma) or DMSO (Fisher Scientific) was added to cell cultures. Dimethyl ester of 2-hydroxyglutaric acid was synthesized and kindly provided by Dr. Stephen Frye’s lab at UNC School of Pharmacy.
Lentivirus or retrovirus production and stable cell line generation
U87 and U251 stable cell lines ectopically expressing wild type IDH1, R132C or R132H variants, as well as HT1080 cells with stable IDH1 gene knockdown were created. For IDH1 knockdown, pLKO.1 plasmids carrying either a non-target or IDH1-specific short hairpin RNA (shRNA) sequences were purchased from Sigma (St. Louis, MO). The IDH1-specific shRNA sequence was: 5’- CCG GGC TGC TTG CAT TAA AGG TTT ACT CGA GTA AAC CTT TAA TGC AAG CAG CTT TTT-3’. The lentivirus was packaged using HEK 293T cells. Virus containing supernatant was added to cells and the infection was carried out for another 48 hr. Medium was changed to fresh culture medium supplemented with puromycin (1-2 µg/ml). IDH1 gene expression and knockdown was confirmed by Western blot analysis.
Cell Viability Analysis
The MTS cell proliferation (Promega) and CCK-8 assays (Dojindo) were used to determine the number of viable cells according to the manufacturer’s protocol. Cell viability was measured by optical absorbance on an Epoch plate reader (BioTek).
Flow Cytometry
Cells were collected and fixed in ice-cold 75% ethanol overnight, and centrifuged at 3,000 g for 10 min. Cell pellets were mixed with 200 µl ethanol and RNA was removed by RNase treatment. Cellular DNA content was investigated by propidium iodide (PI) staining. Stained cells were analyzed on FACSCanto II (BD).
Apoptosis Assay
Cell apoptosis was analyzed by Annexin V/7-AAD or Annexin V/PI kit (Thermo Fisher) according to the manufacturer’s protocol. Cells were collected and analyzed on FACS Canto II (BD) flow cytometer.
DNA fragmentation assay
Genomic DNA was isolated from cells using DNeasy Blood & Tissue Kit (QIAGEN). Fifty nanograms of DNA was resolved by electrophoresis on a 10% Novex TBE gel. The gel was probed with SYBR Safe DNA Gel stain and visualized on the Bio-Rad ChemiDoc Imaging System.
Immunoblotting
Total protein was isolated from cultured cells using RIPA lysis buffer supplemented with protease and phosphatase inhibitor cocktail (Thermo Fisher). Cell lysates were sonicated and centrifuged at 12,000 g for 25 minutes. Protein content was determined using the BCA protein assay (Thermo Scientific). Protein extracts (25 μg/lane, for all tested proteins) were separated on NuPAGE 4 - 12% Bis-Tris minigels (Life Technologies) and transferred to Immobilon-P polyvinylidene fluoride (PVDF) membranes (Millipore). Blots were probed with primary antibodies and visualized by chemiluminescence according to the manufacturer’s protocol (Thermo Fisher). The primary antibodies used include: All histone specific antibodies were used according to manufacturer’s recommendation (CST), endostatin (Abcam), IDH1 (CST), IDH1 R132H (Dianova), PARP1 (CST), pADPR (Abcam), β-actin (Sigma), and FLAG M2-HRP (Sigma).
Comet assay
The Comet assay was performed as previously described(16). Cells were resuspended in 0.75% agarose and placed on glass slides. Cells were lysed in 1.2 M NaCl, 100 mM EDTA, 0.1% sodium lauryl sarcosinate, and 0.26 M NaOH (pH > 13). Electrophoresis of the cells was performed at a voltage of 0.6 V/cm for 30 min. DNA were labelled with SYBR Safe DNA Gel stain and visualized under a Zeiss LSM710 confocal microscope.
Seahorse metabolic assay
Seahorse XF Cell Mito Stress assays were performed according to the manufacturer’s protocol. Twenty thousand cells were seeded in a poly-D-lysine coated 96-well plate. Cells were treated with oligomycin (1 µM), FCCP (2 µM), and antimycin/rotenone (0.5 µM) sequentially. The oxygen consumption rate was recorded and calculated for baseline respiration and maximal respiration.
Determining 2-hydroxyglutarate concentration.
Intracellular 2-HG concentration was determined by either nuclear magnetic resonance (NMR) or colorimetric assay (BioVision). For NMR analysis, culture media were removed and cells were scraped in ice-cold acetonitrile (Sigma) and mixed with 2/3 volume of cold water. The total extract was evaporated in a Speed-Vac. Prior to analysis, the dried cell extract pellet was suspended in water containing 0.1 mM 3-trimethylsilyl-propionic acid (TSP), vortexed, and filtered into 5 mm NMR tubes. Chenomx NMR Suite (version 6.01; Chenomx Inc.) was used for spectral processing, metabolite identification and quantification.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (Version 6.05, GraphPad Software, Inc.). Data were displayed as mean ± SEM. Statistical comparisons were conducted using One-way ANOVA or independent Student’s t-test.
Results
IDH1-mutated glioma cells are sensitive to TMZ
Emerging clinical observations indicate remarkably different clinical outcomes in glioma patients depending on the IDH mutation status of their cancer. IDH mutations appear to be associated with a better response to chemotherapies such as TMZ(9,10 ,17). To better understand the molecular basis of IDH-associated chemo-sensitivity, we established IDH1 mutant cells by stably expressing IDH1 mutants (IDH1R132C or IDH1R132H) in U87 MG or U251 MG cell lines. We found that IDH1 mutant cells were more vulnerable to TMZ treatment in vitro. Assessing cell viability under different doses of TMZ revealed that the dose-response curves were significantly shifted leftwards in mutant cells (p<0.01, Figure 1A). The IC50 of TMZ was reduced in IDH1R132C and IDH1R132H cells (for U251 cells, IC50 IDH1WT=1791 µM; IC50 IDH1R132C=715.9 µM; IC50 IDH1R132H= 772.6 µM), suggesting that a smaller dose of TMZ are sufficient to achieve cytotoxic effects in IDH mutant cells. TMZ-induced apoptosis was enhanced in IDH mutant cells, as Annexin-V labeled cells were more abundant in IDH1R132H cells (Figure 1B, Annexin-V+ cells IDH1WT=22.5%; Annexin-V+ cells IDH1R132H=34.2%). The enhanced chemo-sensitivity was further verified through propidium iodide (PI) staining followed by cell cycle analyses (Figure 1C). We demonstrated impaired cell cycle progression with G2/M arrest in both IDH1WT and IDH1R132H cells treated with TMZ, which is consistent with the finding that TMZ induces G2/M arrest in glioma cells(18). Notably, the population of cells in G2/M arrest was markedly increased in IDH1 mutant cells (for U251 cells, G2/M IDH1WT=26.6%; G2/M IDH1R132H=56.8%), suggesting a greater incidence of failed cell cycle progression in mutant cells during treatment. As a further validation of cellular apoptosis, we also investigated the dynamic change of TMZ-induced DNA fragmentation (Figure 1D). TMZ treatment resulted in the accumulation of small molecular weight of fragmented DNA over a 4-day time course. The amount of fragmented DNA was increased by 2.21-fold in IDH1R132H cells, indicating an increased tendency for apoptosis in IDH mutant cells in the presence of TMZ.
Figure 1. IDH1 mutant glioma cells are sensitive to TMZ.

A. TMZ dose-response curves of U87 MG and U251 MG cells with IDH1 mutation for 96 hr (n=3).
B. Annexin-V apoptosis analysis of IDH1 mutant cell with TMZ treatment.
C. Cell cycle analysis of IDH1 mutant cells with TMZ treatment (left panel). The overview of cell cycle progression with clear G2/M arrest (right panel).
D. DNA fragmentation test for IDH1 mutant cells treated with TMZ.
Enhanced DNA damage in IDH1 mutant glioma cells
The increase in G2/M arrest is a common phenomenon when cells fail to proceed to the DNA damage checkpoint, suggesting that the chemo-sensitivity of IDH1 mutant cells is linked to their vulnerability to DNA damage. To test this hypothesis, we evaluated TMZ-induced DNA damage in IDH1 mutant cells by measuring γH2A.X (Figure 2A). TMZ exposure resulted in a remarkable increase in γH2A.X expression in all cell lines. TMZ induced more robust expression of γH2A.X in IDH1R132C cells compared to wild type (0.89 AU and 1.84 AU for IDH1WT and IDH1R132C, respectively, by densitometry analysis). Interestingly, the baseline H2A.X phosphorylation in mutant cells was also elevated, indicating severe intrinsic dysfunction in DNA repair machinery (0.21 AU and 0.57 AU for IDH1WT and IDH1R132C, respectively). We confirmed DNA damage through in situ labeling of γH2A.X puncta in the nucleus (Figure 2B). Consistent with previous findings, the baseline DNA damage was also elevated in mutant cells (0.0036 AU, 0.018 AU and 0.032 AU for IDH1WT, IDH1R132C and IDH1R132H, respectively). To better understand the integrity of genomic DNA in IDH1 mutant cells, we analyzed the proportion of fragmented DNA through comet assay (Figure 2C). We confirmed the baseline DNA damage by increased fragmented DNA in IDH1R132C and IDH1R132H cells without any treatment, as shown by elongated comet tail under electric field. TMZ treatment resulted in significant DNA fragmentation, and the fragmented DNA is more abundant in IDH1R132C and IDH1R132H cells. Quantitative analysis confirmed more DNA damage occurred in IDH1 mutant cells (Figure 2D).
Figure 2. Enhanced DNA damage in IDH1 mutant glioma cells.

A. Western blotting for γH2A.X expression in IDH1 mutant cells. Protein expression level was determined by densitometry analysis.
B. Immunostaining for γH2A.X puncta in IDH1 mutant cells.
C. Comet assay demonstrates DNA fragmentation in IDH1 mutant cells.
D. Quantitative analysis showed enhancement in DNA fragmentation in IDH1-mutated cells.
Metabolic reprogramming results in chemo-sensitivity in IDH1-mutated cells
Mutations in IDH1 establishes neomorphic catalytic activity, which redirects carbon metabolites away from the Kreb cycle towards 2-HG production(19,20). Metabolic defects and reprogramming could be one of the causes of chemo-sensitivity. To test this, we studied the oxidative metabolism in IDH1 mutant cells through the Seahorse metabolic assay (Figure 3A). The decrease in oxygen consumption rate (OCR) in IDH1-mutated cells suggested significantly reduced oxidative metabolism. Quantitative analyses confirmed a significant decrease in both baseline and maximum capacity of oxidative metabolism in mutant cells (Figure 3B and C). Moreover, we found a significant increase in intracellular 2-HG in mutant cells (Figure 3D). The 2-HG production could be reversed by treatment with AGI-5198, a specific chemical inhibitor targeting the R132H variant of IDH1 enzyme(21). However, AGI-5198 did not result in obvious difference in TMZ-induced cytotoxicity in our assay (Figure S1). Taken together, our findings demonstrate that the capacity for oxidative metabolism is greatly limited in the presence of IDH1 mutation, suggesting a significant shift in energy metabolism in IDH1 mutant cells.
Figure 3. Metabolic reprogramming results in chemo-sensitivity in IDH1 mutant cells.

A. Seahorse XF Cell Mito Stress analysis showed alteration in oxidative metabolism in IDH1 mutant cells (n=4).
B. Quantification of baseline oxidative metabolism in IDH1 mutant cells (n=4, **p<0.01).
C. Quantification of maximum oxidative metabolism in IDH1 mutant cells (n=4, **p<0.01).
D. Quantification of D-2-HG in IDH1 mutant cells. AGI-5198 treatment (1 µM) was used to determine specific activity from IDH1 mutant enzyme (n=3, **p<0.01)
E. Cell viability assessment of U87 MG cells in the presence of TMZ and dimethyl-2-HG (n=3).
F. Cell viability measurement of HT-1080 cells with the presence of TMZ and dimethyl-α-KG (n=3, *p<0.05 as compared with DMSO group, #p<0.05 as compared to TMZ group).
G. Cell viability assessment of HT-1080 cells with stable expression of IDH1 shRNA (n=3, *p<0.05 as compared to 0 µM TMZ, #p<0.05, ##p<0.01 as compared to scramble RNA)
H. Quantitative PCR (upper panel) and western blot analysis (lower panel) for IDH1 expression in HT-1080 cells (n=3).
We then investigated the relationship between 2-HG production and cellular sensitivity to TMZ. To investigate whether 2-HG directly leads to chemo-sensitization for TMZ treatment, we analyzed cell viability in the presence of cell permeable dimethyl-2-HG by MTS assay (Figure 3E). We found that the 2-HG levels correlated with their vulnerability to chemotherapy, as the introduction of 2-HG potentiated the cytotoxic effect of TMZ. With 500 µM TMZ treatment, 2-HG reduced cell viability by 31.0% in U87 cells. Conversely, increasing oxidative metabolism by introducing α-KG reversed the cytotoxic effect of TMZ (Figure 3F). Ten millimolar α-KG resulted in a 99.1% increase in cell viability after TMZ treatment. To further test the role of IDH1 mutation in chemo-sensitivity, we investigated the cell viability of HT-1080, a colon cancer cell line with an intrinsic IDH1R132H variant, with TMZ treatment (Figure 3G to H). As expected, TMZ treatment resulted in decreased cell viability in a dose-dependent manner. The cytotoxic effect of TMZ was alleviated by reducing 2-HG production via shRNA targeting of IDH1. In sum, our findings suggested an important role of 2-HG centered metabolic reprogramming in chemo-sensitization in IDH1 mutant cells.
Dysfunction of PARP1 DNA repair pathway in IDH1 mutant cells
In addition to alterations in cellular metabolism, the remarkably increased susceptibility for acquiring DNA damage in mutant cells suggested that the chemo-sensitivity may also be mitigated by defunct DNA repair mechanisms, such as base excision repair (BER), nucleotide excision repair, mismatch repair and/or DNA recombination. A seminal study indicated that the metabolic defects in NAD+ productivity, prompted the vulnerability of IDH1 mutant cells(22). NAD+ acts as a coenzyme in many redox reactions by donating ADP-ribose moieties to ADP-ribosylation reactions. Importantly, NAD+ is an essential donor molecule for PARP-mediated single strand DNA repair. Insufficient NAD+ may disable PARP-associated DNA repair, which could be fatal for IDH1-mutated cells when confronting DNA damage. To test this hypothesis, we treated IDH1 mutant cells with TMZ, and olaparib (Ola), a PARP inhibitor(23). DNA damage was assessed by γH2A.X staining (Figure 4A). We found that TMZ-induced DNA damage was significantly enhanced when PARP-associated DNA repair pathway was inhibited. Quantitative analysis confirmed the remarkable enhancement in DNA damage (Figure 4B). Consistent with this, the comet assay demonstrated a significantly elongated tail formation in both IDH1WT and IDH1R132H cells demonstrating that the fragmented DNA is more abundant in mutant cells in either TMZ treatment or combination treatment involving TMZ and Ola (Figure 4C and D).
Figure 4. Dysfunction of the PARP1 DNA repair pathway in IDH1 mutant cells.

A. Immunostaining of γH2A.X in IDH1 mutant cells treated with TMZ and Ola (Bar=10 µm).
B. Quantification of γH2A.X puncta in IDH1 mutant cells treated with TMZ and Ola (n=3, *p<0.05, **p<0.01).
C. Comet assay measuring DNA fragmentation in IDH1 mutant cells treated with TMZ and Ola.
D. Quantification of DNA fragmentation in IDH1 mutant cells.
E. Western blot analysis of pADPR, PARP and β-actin expression in IDH1 mutant cells treated with TMZ and Ola.
F. Quantification of NAD in IDH1 mutant cells treated with TMZ (n=3, *p<0.05, **p<0.01).
To better understand whether PARP-associated DNA repair is a key factor that defines the difference in chemo-sensitivity in IDH1 mutant cells, we measured the quantity of ADP-ribose-conjugated PARP (pADPR) through immunoblotting (Figure 4E). The formation of pADPR is a post-translational modification on PARP, which serves as a key step for PARP-associated DNA repair(24). We found that TMZ treatment induced a 1.73-fold increase in pADPR in IDH1WT cells, suggesting a mobilization of the intrinsic DNA repair mechanisms during chemotherapy. Interestingly, the quantity of pADPR was found to be extensively reduced in untreated IDH1R132C and IDH1R132H cells (2.92 AU, 0.42 AU and 0.61 AU for IDH1WT, IDH1R132C and IDH1R132H, respectively). Moreover, TMZ treatment did not induce an obvious elevation in pADPR in mutant cells (5.05 AU, 0.01 AU and 0.82 AU for IDH1WT, IDH1R132C and IDH1R132H, respectively). As expected, Ola treatment depleted pADPR formation and completely abolished PARP-associated DNA repair, while the expression of PARP1 was found unchanged across all treatment groups. The significant difference in the pADPR/PARP ratio suggested that although PARP DNA repair machinery was intact, DNA damage was more likely to accumulate due to lack of NAD+ as substrate for BER repair. Finally, we confirmed this hypothesis by measuring changes in NAD+ in mutant cells (Figure 4F and S2). The total and nuclear NAD+ quantity was slightly lower in IDH1R132C and IDH1R132H cells without treatment. TMZ treatment reduced the quantity of NAD+, presumably a direct effect on pADPR formation and DNA repair. Interestingly, the NAD+ quantity was increased after washing out TMZ in the microenvironment, suggesting that active NAD+ synthesis provides substrate for BER DNA repair. The quantity of NAD+ was significantly reduced in mutant cells after washing off TMZ, suggesting that the NAD+ deficiency is the key factor causing a malfunction of BER repair (−32.6% and −25.7% in IDH1R132C and IDH1R132H, respectively).
Targeting the PARP DNA repair pathway enhanced cytotoxicity induced by chemotherapy
The significantly enhanced DNA damage in IDH1 mutant cells that occurs as a consequence of inhibiting the PARP-associated DNA repair pathway suggested that a combination therapy could be more effective than current chemotherapy options, potentially generating a better tumor-inhibiting effect but at a lower dosage of chemotherapy. To better understand the potentiation effect of Ola in IDH1 glioma, we performed cell viability assays with combination therapy involving TMZ and Ola. We found that Ola potently enhanced the cytotoxic effect of TMZ regardless of the IDH1 mutation status (Figure 5A), as the dose-response curves were shifted leftwards in all genotypes. For clarity, we also showed that while cell viability was impacted a TMZ dose of 1 mM, the addition of Ola resulted in remarkable potentiation of cell death at the same dose of TMZ (Figure S3). Consistent with the findings above, the IC50 of TMZ was significantly smaller for IDH1R132C and IDH1R132H cells (Table S1). Moreover, the potentiation of TMZ cytotoxicity was confirmed by Annexin V/PI apoptosis analysis (Figure 5B). Similar to previous findings, quantitative analysis showed that TMZ induced more apoptosis in IDH1 mutant cells. The population of apoptotic cells was significantly increased in both cell lines with the presence of Ola; however, IDH1 mutation predicted a more extensive cellular apoptosis (Figure 5C).
Figure 5. Targeting the PARP DNA repair pathway enhanced cytotoxicity induced by chemotherapy.

A. Dose-response curves for IDH1 mutant cells treated with TMZ and Ola (n=3).
B. Annexin-V apoptosis analysis demonstrated the potentiation effect of Ola for TMZ chemotherapy.
C. Quantification of apoptotic cells in IDH1 mutant cells treated with TMZ and/or Ola.
Discussion
In the present study, we confirmed that pathogenic IDH1 mutations result in metabolic reprogramming and compromised oxidative metabolism. Production of 2-HG by the mutant enzyme activity confers a vulnerability to TMZ treatment. Furthermore, the compromised oxidative metabolism and decreased NAD+ pool in IDH1-mutated cells impairs the PARP-dependent DNA repair mechanism, resulting in substantially elevated DNA damage and cell death. As a consequence of these molecular changes, IDH1-mutated cells are sensitized to therapies such as TMZ and Ola (Figure 6). TMZ treatment in tandem with DNA repair inhibitor is a promising therapeutic strategy for future gliomas therapies.
Figure 6. Impaired PARP1 DNA repair defines chemo-sensitivity.
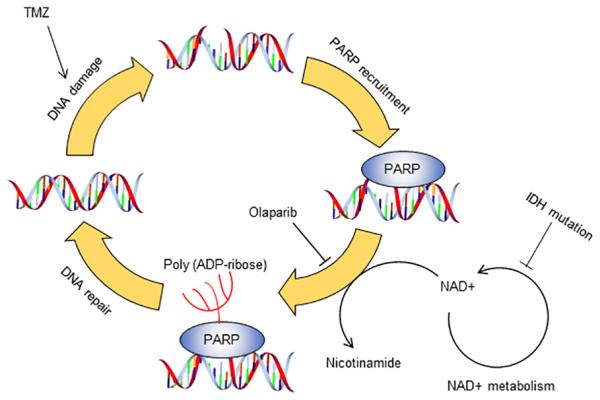
Chemotherapy induces damages in chromosomal DNA in cancer cells. PARP serves an important role in maintaining genomic integrity and counteract with chemo-agent. In tumors with IDH mutation, cells are less capable of maintaining DNA repair mechanism, as deficiency in NAD+ metabolism deprives substrates for the BER pathway. Targeting PARP by olaparib significantly potentiates the therapeutic effect of TMZ in IDH mutant cells.
IDH mutation and chemo-sensitivity
Emerging studies have demonstrated the increase in chemo-sensitivity in IDH mutant glioma(7). Although the prognostic value of an IDH mutation in glioma has been widely accepted, little is known about the unique molecular features that defines the chemo-sensitivity in IDH1 mutant cells. In the present study, we established cell line models by introducing IDH1 mutation in a glioma biologic background. Our cell lines recapitulate the molecular signatures of IDH1 mutant gliomas, such as expression of mutant enzyme, 2-HG production, as well as an increase in histone 3 methylation (Figure S4). Moreover, we confirmed that IDH1 mutant cells are more vulnerable to TMZ treatment, which replicated the clinically observed increased chemo-sensitivity in IDH1 mutant cells (Figure 1A). Additionally, analysis of apoptosis confirmed increased sensitivity to TMZ in IDH1 mutant cells (Figure 1B to D). These findings suggested that the IDH mutant cell line models can be valuable and reliable tools to understand the molecular and cellular responses to TMZ chemotherapy. We believe that our models are more suitable than several recently developed cell lines from individual patient with intrinsic IDH1 mutation. Because we introduced IDH1 mutants to cell lines with the same genetic background, as such the bias from genomic/transcriptomic variation is extensively reduced.
To validate the observed increase in chemo-sensitivity, we carefully measured DNA damage in IDH1 mutant cells by measuring γH2A.X as well as DNA fragmentation (Figure 2). TMZ treatment introduced significant DNA damage in our cell line models. Consistent with the changes in cell viability and fraction of apoptotic cells, we demonstrated greater DNA damage in IDH1- mutated cells compared with their wild type counterpart. Interestingly, we found an accumulation of DNA damage in untreated IDH1-mutated cells, suggesting elevated basal level of DNA damage resulted from the metabolic deficiencies. Presumably, the intrinsic DNA repair mechanism limits the baseline DNA damage to a certain extent, allowing the cells to survive and proliferate. In this case, mutant cells are more likely to suffer additional DNA damage with chemotherapy, when DNA repair mechanisms are compromised.
Metabolic defects in IDH-mutated cells confer cytotoxicity
Three distinct IDH enzymes, IDH1, IDH2 and IDH3, have been found in mammalian cells(25,26). All three enzymes catalyze the oxidative decarboxylation of isocitrate to α-KG. In contrast to IDH3, which is a heterotrimeric enzyme that uses NAD+ as a cofactor for it enzymatic reaction, IDH1 and 2 are homodimeric enzyme complexes and use NADP+ as cofactor to produce α-KG(26). Currently, only IDH1 and IDH2 mutations have been found in glioma cells. Neomorphic changes in IDH redirect carbon-based metabolites into unusable 2-HG(3,20,27). Metabolite deprivation leads to reprogramming of the Kreb’s cycle by altering glutamine utilization and pyruvate metabolism(27,28). The adjustment in cellular metabolism may limit physiologically important metabolic routes, such as ATP generation and macromolecule synthetic pathways, and therefore result in the increased vulnerability of IDH1 mutant cells. In the present study, we confirmed impaired oxidative metabolism (Figure 3A to C) as well as robust production of 2-HG (Figure 3D) in mutant cells. Moreover, introducing 2-HG to native U87 cells mimics the introduction of IDH1 mutant enzyme, as shown by increased cell death and chemo-sensitivity (Figure 3E). Furthermore, in HT-1080 cells, the TMZ chemo-sensitivity could be salvaged by enhancing cellular metabolism (Figure 3F) or reducing IDH1 biologic activity (Figure 3G to I). Taken together, our findings demonstrate reduced oxidative metabolism and 2-HG production lead to chemotherapy sensitization. Our finding also indicated that relieving the metabolic stress in IDH1-mutated cancer may prompt chemo-resistance by enhancement in cellular metabolism. Indeed, Molenaar et al has reported that targeting IDH1 mutation confer radioprotection(29). The contradictory therapeutic mechanisms between genotoxic agent and metabolic modulator should be considered in the future drug design and therapeutic strategies.
The importance of NAD+ in chemo-sensitivity in IDH1-mutated glioma
Mammalian cells develop DNA repair machinery that could be of great importance in maintaining genomic integrity. In cancer therapies, DNA repair pathways sometimes are taken advantaged to fix DNA damage sites introduced by chemotherapy. IDH1-mutated gliomas exhibit unique pattern in DNA repair enzymes. For example, ATRX/RAD54 is frequently mutated in IDH1-mutated glioma (30), whereas other DNA repair enzymes, such as ATM, CHEK1, CHEK2, and RAD52, are amplified in the genome of IDH1-mutated glioma (Figure S5). Several studies shed light on the changes in DNA repair mechanism in IDH1-mutated cells, including RAD51 and ATM pathways (14,15). In-depth study is encouraged to elucidate the unique pattern in DNA repair mechanisms in IDH1-mutated glioma, and therefore better guide drug discovery and cancer therapy. The question whether DNA repair mechanism is the key factor that determines in the chemo-sensitivity in IDH1-mutated cells remains unanswered. A seminal study by Tateishi et al. highlighted the essential role of NAD+ in IDH1-mutated glioma, suggesting that impairment of NAD+ synthesis could be one of the key metabolic defects in IDH mutant cells(22). In metabolic intact cells, the NAD concentrations in each of the subcellular compartment are steadily maintained (31). IDH1 mutant deplete NAD in all subcellular compartment due to inhibition of Naprt1 activity. Targeting the damaged NAD synthetic pathway by Nampt inhibitors FK866 or GMX1778 further limit the availability of NAD (22). In additional to the many important roles in cellular metabolism and redox balance, NAD+ plays a key role in BER pathway through PARP(24). Indeed, our results confirmed that PARP1 is essential for DNA repair during TMZ treatment, as chemical inhibition of PARP1 through Ola significantly potentiated TMZ induced DNA damage (Figure 4A-C). Interestingly, we observed a significant reduction in NAD+ in IDH1 mutant cells (Figure 4F), which confirmed that the limited DNA repair capacity in IDH1 mutant cells is caused by insufficient substrate for the biological function of PARP. Additionally, we observed pADPR, the intermediate molecule that represents active PARP DNA repair, is depleted in IDH1 mutant cells (Figure 4D). TMZ treatment led to an induction of pADPR formation in cells with wild type IDH1, but not the mutant cells, suggesting a significantly reduced DNA repair mechanism in IDH1 mutant cells. Interestingly, the amount of total PARP remained stable within all the treatments, suggesting the vulnerability based on functional ablation of DNA repair, but not changes in the level of expression. Notably, several recent findings showed PARP inhibition further damages energy metabolism, which may exert synergistic effect with genotoxic agents (32,33). In summary, our findings extend the current understanding of the molecular mechanisms that determine the vulnerability to NAD+ depletion in IDH1-mutated gliomas, through compromised PARP-associated DNA repair.
Combined treatment of TMZ and Ola is a promising therapeutic strategy for IDH-mutated gliomas
Radiation with daily TMZ followed by adjuvant TMZ is the current standard care for newly diagnosed glioblastoma. Compared to radiation therapy alone, additional TMZ chemotherapy extends median survival from 12.1 to 14.6 months(34,35). Despite the clear statistical improvement of this regimen compared to radiation alone, less than one third of the patients survive beyond 2 years. A phase III study in patients with grade II glioma provided the level I evidence that radiation therapy plus chemotherapy should be considered a new standard of care for patients who had subtotal resection and older than 40 years old (Mehta et al, 2014 RTOG9802). Although patients with low grade glioma survive longer following radiation plus chemotherapy compared with those who receive radiation only, the current standard care hasn’t prevented the lower grade tumor from going through higher grade transformation. Despite the fact that patients with IDH-mutated glioma have a better prognosis compared to those whose tumors lack this mutation a fatal clinical outcome is seen in nearly all patients with IDH-mutated lower grade glioma. Therefore, developing more effective therapies remain an important priority for patients with even lower grade gliomas.
As an orally available alkylating agent, TMZ has been widely used in lower grade glioma. Our study has provided the preclinical evidence that combined treatment with TMZ and Olaparib is more effective in against IDH-mutated glioma compared to the IDH wild type tumors. IDH-mutated tumor exhibited deficiency in NAD+ production and pAPDR production, whereas greatly limited the PARP DNA repair pathway (Figure 4E and F). The addition of Olaparib, an inhibitor of PARP, targeting a “selective vulnerability” of IDH-mutated tumors, further limiting DNA repair, leading to more DNA damage and cell death (Figure 5).
The synergistic effects between TMZ and Olaparib in patients with the IDH mutation provides the possibility to achieve better cytotoxic effect with less amount of alkylating agent, which leads to the reduced toxicities and improved patients’ quality of life(36,37). The findings from our study provide the foundation for a hypothesis driven clinical therapeutic study the clinical efficacy of the combined treatment of TMZ and Olaparib in IDH-mutated glioma; prospectively investigating IDH mutation status as a predictive marker for this combined therapy.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NCI, CCR. All authors received Intramural Research Grant from NIH, NCI, CCR.
Footnotes
All author claimed no conflict of interest in this study.
References
- 1.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458–68. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 3.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–5. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26:2038–49. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75:1560–6. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- 8.SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z, et al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012;103:269–73. doi: 10.1111/j.1349-7006.2011.02134.x. [DOI] [PubMed] [Google Scholar]
- 9.Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32:783–90. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minniti G, Scaringi C, Arcella A, Lanzetta G, Di Stefano D, Scarpino S, et al. IDH1 mutation and MGMT methylation status predict survival in patients with anaplastic astrocytoma treated with temozolomide-based chemoradiotherapy. J Neurooncol. 2014;118:377–83. doi: 10.1007/s11060-014-1443-0. [DOI] [PubMed] [Google Scholar]
- 11.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 12.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 13.Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–91. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 14.Ohba S, Mukherjee J, See WL, Pieper RO. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014;74:4836–44. doi: 10.1158/0008-5472.CAN-14-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell. 2016;30:337–48. doi: 10.1016/j.ccell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–9. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 17.van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010;16:1597–604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- 18.Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001;61:1957–63. [PubMed] [Google Scholar]
- 19.Borodovsky A, Seltzer MJ, Riggins GJ. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr Opin Oncol. 2012;24:83–9. doi: 10.1097/CCO.0b013e32834d816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74:3317–31. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–30. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell. 2015;28:773–84. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51:6581–91. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 24.Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci. 1995;20:405–11. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 25.Narahara K, Kimura S, Kikkawa K, Takahashi Y, Wakita Y, Kasai R, et al. Probable assignment of soluble isocitrate dehydrogenase (IDH1) to 2q33.3. Hum Genet. 1985;71:37–40. doi: 10.1007/BF00295665. [DOI] [PubMed] [Google Scholar]
- 26.Gabriel JL, Zervos PR, Plaut GW. Activity of purified NAD-specific isocitrate dehydrogenase at modulator and substrate concentrations approximating conditions in mitochondria. Metabolism. 1986;35:661–7. doi: 10.1016/0026-0495(86)90175-7. [DOI] [PubMed] [Google Scholar]
- 27.Izquierdo-Garcia JL, Viswanath P, Eriksson P, Cai L, Radoul M, Chaumeil MM, et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015;75:2999–3009. doi: 10.1158/0008-5472.CAN-15-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci U S A. 2014;111:14217–22. doi: 10.1073/pnas.1409653111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molenaar RJ, Botman D, Smits MA, Hira VV, van Lith SA, Stap J, et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 2015;75:4790–802. doi: 10.1158/0008-5472.CAN-14-3603. [DOI] [PubMed] [Google Scholar]
- 30.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–22. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, et al. Biosensor reveals multiple sources for mitochondrial NAD(+) Science. 2016;352:1474–7. doi: 10.1126/science.aad5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbi de Moura M, Feinstein T, et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8:1819–31. doi: 10.1016/j.celrep.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–14. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 35.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 36.Bailey ML, O'Neil NJ, van Pel DM, Solomon DA, Waldman T, Hieter P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol Cancer Ther. 2014;13:724–32. doi: 10.1158/1535-7163.MCT-13-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014;21:258–69. doi: 10.1038/cdd.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.