

Abstract
Although docetaxel (DOX) is the standard of care for advanced prostate cancer (PCa), most patients develop resistance to DOX. Therefore, elucidating the mechanism that underlies resistance to DOX is critical to enhance therapeutic intervention. Mining cDNA microarray from the PC-3 PCa cell line and its DOX-resistant derivative (PC3-TxR) revealed decreased latexin (LXN) expression in the resistant cells. LXN expression was inversely correlated with taxane resistance in a panel of PCa cell lines. LXN knockdown conferred DOX resistance to PCa cells in vitro and in vivo; whereas LXN overexpression reduced DOX resistance in several PCa cell lines. A mouse model of PCa demonstrated that PCa cells developed resistance to DOX in the bone microenvironment, but not the soft tissue microenvironment. This was associated with decreased LXN expression in PCa cells in the bone microenvironment compared to the soft tissue microenvironment. It was identified that bone stromal cells decreased LXN expression through methylation and induced chemoresistance in PCa in vitro. These findings reveal that a subset of PCa develops DOX resistance through loss of LXN expression associated with methylation and that the bone microenvironment promotes this drug resistance phenotype.
Keywords: taxane, chemoresistance, PCa, bone microenvironment, docetaxel
Introduction
Prostate cancer (PCa) is the most commonly diagnosed malignant cancer in Western men and the second leading cause of cancer-related deaths in Western societies (1). Patients with advanced tumors commonly receive hormonal deprivation therapy (i.e. anti-androgen, luteinizing hormone); however, the response to this treatment is not durable, leading to the development of castration resistant PCa (CRPC) (2). Chemotherapy with docetaxel (DOX), a taxane based antimitotic agent, has proven efficient in a subset of cases and is currently used as the standard chemotherapy for patients with CRPC (3–5). The efficacy of DOX treatment has been further emphasized due to results reported from the ChemoHormonal Therapy Versus Androgen Ablation Randomized Trial for Extensive Disease (CHAARTED), which demonstrated that addition of six cycles of DOX to standard hormonal deprivation therapy increased survival of men with metastatic disease relative to treatment with hormonal deprivation therapy alone (6). These findings were further supported by the Systemic Therapy in Advanced or Metastatic PCa: Evaluation of Drug Efficacy (STAMPEDE) trial (7).
In spite of its efficacy, resistance to DOX remains a significant clinical problem. Approximately 50% of patients do not respond to initial therapy (8), and eventually all patients who initially benefit from this treatment develop drug resistance within 24 months of initial exposure (9). Even though there are multiple PCa-specific pathways that have been identified to contribute to DOX resistance, many have not resulted in any clinically targetable treatments indicating that defining these mechanisms of DOX resistance could lead to significant therapeutic gains (10).
In order to determine mechanisms that contribute to docetaxel resistance in PCa, we performed a previous study in which we derived DOX-resistant Du145 (Du145-TxR) and PC-3 cells (PC-3-TxR) (11). While we determined that development of DOX resistance in the Du145 was associated with upregulation of the MDR1 gene (p-glycoprotein), which is a well-known chemoresistance factor, the DOX-resistant PC-3 cells did not have upregulated MDR1 expression and thus achieved chemoresistance through an unknown mechanism (11). The goal of the current study was to delineate the mechanisms through which the PC-3 cells gained DOX resistance.
Materials and Methods
Cell Culture
PC-3-TxR cells were generated and maintained as described previously (11). DU145, LNCaP, PC-3; and BPH-1 (benign prostatic hyperplasia-1) cell lines were maintained in RPMI with 10% FBS (Sigma) and 1× penicillin-streptomycin (Life Technologies). RWPE-1 cell line was cultured in K-SFM supplemented with recombinant human Epidermal Growth Factor (rhEGF) and BPE (Gibco cat no. 17005-042). The C4-2B cell line, which is an LNCaP subline, (kindly provided by Dr. Leland Chung, Cedars Sinai, Hollywood, CA) was maintained in T medium [80% DMEM (Life Technologies, Inc.), 20% F12 (Invitrogen), 100 units/liter penicillin G, 100 Ag/mL streptomycin, 5μg/mL insulin, 13.6 pg/mL triiodothyronine, 5μg/mL transferrin, 0.25μg/mL biotin, and 25 μg/mL adenine] supplemented with 10% FBS. The MC3T3-E1 (clone MC-4) cell line, a murine pre-osteoblast cell line, (kindly provided by Dr. Renny Franceschi, University of Michigan, Ann Arbor, MI) and ST-2 cell line, a mouse bone marrow stromal cell line, (obtained from RIKEN Cell Bank, Ibaraki, Japan) were maintained in α-MEM containing 10% FBS and 1% penicillin-streptomycin (Life Technologies, Inc.). RAW cells are a murine pre-osteoclast cell line (ATCC). PC-3 cells infected with the pLazarus retroviral construct expressing luciferase were selected for stable transductants in G418 (12). All cultures were maintained at 37°C, 5% CO2, and 100% humidity. The majority of cell line identities were authenticated using short tandem repeat profiling.
Toxicity assay
Cells were cultured in 96-well plates for overnight and then cells were treated with DOX (Cell Signaling Technology) for 48hr. 10μl cell proliferation reagent WST-1 (Clontech, MK400) was added into 100ul medium and incubated at 37°C and 5% CO2 for 4hr. Absorbance was then measured at 440 nm with a plate reader (Multi-Mode Microplate Reader, SpectraMax M5, Molecular Devices MDS Analytical Technologies).
Lentiviral transduction
PC-3 cells were plated in fresh medium in 96-well plates to achieve a 70% confluency upon transduction. Transduction conditions were performed according to the manufacturer’s guide (Sigma-Aldrich). Briefly, hexadimethrine bromide was added to a final concentration of 8 mg/ml then 10 μl of human LXN hPGK-Puro-CMV-tGFP Lentiviral Transduction Particles (Sigma-Aldrich, TRCN0000073472) or the control (pLKO.1-puro-CMV-TurboGFP™ Positive Control Transduction Particles, Sigma-Aldrich, SHC003V) were add. After 48h, the cells were treated with10ug/ml puromycin for selection of transduced cells.
Transfection
Transfection was performed in 6-well plates with FuGENE 6 transfection reagent (Promega, USA, E2691) according to the manufacturer’s instructions. 100 μl of DMEM serum-free medium containing 2μg of vector DNA: LXN (Myc-DDK-tagged)-Human LXN (LXN) (Origene, RC202769) or pCMV6-Entry (C-terminal Myc and DDK Tagged) (Origene, PS100001) and 3 μl of FuGENE reagent was incubated for 15 minutes at room temperature before the mixture was added in 6 cm-well cell cultures (50% confluent) in the presence of 10% fetal bovine serum. After transfection for 48h, the cells were treated with 50–200ug/ml G418 for selection of transfected cells.
Western Blot
Cell were lysed in a buffer containing Tris.HCl, PH 8.0, 137 mM NaCl, 1% NP-40, 10% glycerol, 1 mM Na3VO4, 1 mM PMSF. Equal amounts of protein were resolved on a 10% or 12.5% Ready Gel J (Bio-Rad, Hercules, CA), transferred onto a PVDF membrane (Invitrogen), and blocked with 5% non-fat dry milk at room temperature. Membranes were probed overnight with primary antibodies against LXN (mouse monoclonal antibody, clone 1E10, Origene, TA503779) and β-actin (mouse monoclonal anti-β-actin antibody, Sigma-Aldrich, A5441) followed by HRP-conjugated secondary antibodies. An ECL system was used to detect chemiluminescent signals (SuperSignal West Pico Chemiluminescent Substrate; Pierce).
Real-Time PCR
RNA was extracted and purified by RNeasy Mini-Kit (Qiagen, Valencia, CA, USA), and reverse transcribed using the SuperScript III Reverse Transcriptase Kit (Invitrogen). Quantitative real time PCR was performed in triplicate using SYBR Green qPCR MasterMix (Qiagen) in a 10 μl reaction volume on a Roche LightCycler 480 (Roche, Indianapolis, IN, USA). LXN primer (RT2 qPCR Primer Assay for Human LXN (NM_020169, PPH11668A-200)) and HPRT-1 primers (RT2 qPCR Primer Assay for Human HPRT1 (NM_000194), PPH01018C-200) were purchased from SABiosciences (Qiagen). The primer β-actin sequence was as follows: sense, 5′-GATGAGTTGGCATGGCTTT-3′, antisense 5′-CACCTTCACCGTTCCAGTTT-3′. The RT-PCR conditions were 30 sec at 95 °C, followed by 45 cycles at 95 °C for 10 sec and 60 °C for 20 sec. Melting curve analysis was performed to verify the purity of the products and to exclude undesired primer dimers. All analyses were performed in triplicate in three independent experiments. The relative amount of target gene mRNA was normalized to that of the control (β-actin or HPRT-1).
In vitro bioluminescence measurement of cells
PC-3-luc cells (1.5× 104/ml) were mixed with PC-3 (1.5× 104/ml), MC3T3 (1.5× 104/ml) or ST-2 (1× 104/ml) cells and plated into white, clear bottom 96-well plates (Costar, Corning, New York) at a concentration of 2.25 × 104/ml 200μl per well in RPMI medium with 10% FBS. After 48 hours, the cells were treated with 16nM DOX and incubated for 48 hours. Then 20μl luciferin (40 mg/ml, Regis Technologies, Inc.) was added to each well and incubated at 37°C and 5% CO2 for 2 minutes. Luminescence was then measured at integration 1000 ms with a plate reader (Multi-Mode Microplate Reader, SpectraMax M5, Molecular Devices MDS Analytical Technologies).
5-aza-2′-deoxycytidine Treatment
Cells were plated with 2 uM 5-aza-2′-deoxycytidine (Sigma-Aldrich; St. Louis, MO) and incubated for 4 days. The medium and the drug were replaced every 24 hours and cells were harvested for RNA extraction 4 days after treatment.
Animal Studies
All animal studies were performed in an AALAC-approved facility, with approval of the University of Michigan University Committee on Use and Care of Animals (UCUCA). Twelve mice per group were used for all in vivo experiments.
Subcutaneous in vivo model for evaluation of modulating LXN expression on sensitivity to DOX
Male nude mice aged 6–8 weeks were injected subcutaneously with PC-3 cells (1x106 in 100μl) expressing either shGFP or shLXN in RPMI 1640 medium. The mice were treated weekly with vehicle or 5mg/kg DOX by intraperitoneal injection once the tumors reached 100 mm3. Tumor volumes were measured weekly using calipers. The mice were euthanized after 4 weeks treatment.
In vivo model to compare sensitivity to DOX in soft tissue versus bone
For subcutaneous injection, single cell suspensions (1×106cells) of PC-3-luc cells in RPMI1640 were injected in the flank at 100μl/site using a 27-G3/8-inch needle under anesthesia with 2.5% isofluorane/air. Subcutaneous tumor growth was monitored by either caliper measurement or BLI weekly. For intratibial injection, mice were anesthetized with 2.5% isofluorane/air, and both legs were cleaned with betadine and 70% ethanol. The knee was flexed, and a 27-G3/8-inch needle was inserted into the proximal end of right tibia followed by injection of 20μl single-cell suspensions of PC-3-luc cells (5×105 cells). After 3 weeks, the mice were treated weekly with vehicle or 5mg/kg DOX by intraperitoneal injection. Tumor development in bone was evaluated weekly using BLI and radiography. For BLI, mice were injected intraperitoneally with 100μl luciferin (40 mg/ml in PBS), anesthetized with 1.5% isoflurane and imaged 15 minutes post-luciferin injection on the IVIS BLI system (Caliper Life Sciences) as previously described (13). Signal intensity was quantified as the sum of all detected photons within the region of interest during a 1-minute luminescent integration time.
Statistical Analyses
All in vitro experiments were performed at least three times. Numerical data are expressed as mean ± SD. Statistical analysis was performed by analysis of one-way ANOVA and/or student’s t-test for independent analysis. The value p <0.05 was considered statistically significant.
Results
LXN expression is reduced in PC-3-TxR cell line
We previously established a paclitaxel- and DOX-resistant PC-3 PCa cell line, PC-3-TxR, by incubating cells in increasing concentrations of paclitaxel (11). For purpose of the current study, we confirmed that DOX resistance was maintained in the PC-3-TxR cells compared to the PC-3 cells. PC-3-TxR had an increased IC50 (approximately 45 nm) compared to that of PC-3 (approximately 8 nM) (Fig. 1A). To determine candidate genes that contribute to DOX resistance in PC-3 cells, we examined our previously reported differential gene expression analysis between the PC-3 parental and PC-3-TxR cells (11). This led to identification of 3 genes that had the greatest magnitude of change between the PC-3 and PC-3-TxR cells. Namely, lxn, runx3 and plac8. To validate the gene array finding, mRNA differential expression of the three genes between PC-3 and PC-3-TxR was quantified using qPCR analysis. LXN showed the greatest differential mRNA expression compared to the other genes (Fig. 1B and Sup. Fig. 1). Specifically, LXN mRNA was observed to be significantly down regulated in the PC-3-TxR cells compared to PC-3 cells (Fig. 1B). This observation was further validated at the protein level using western blot analysis (Fig. 1C). Based on the decreased LXN expression and the previous reports that LXN has been shown to inhibit tumor growth (14–16) we further explored the potential role of LXN in the chemoresistance of PCa.
Figure 1. LXN expression is decreased in PC-3 resistant cells.
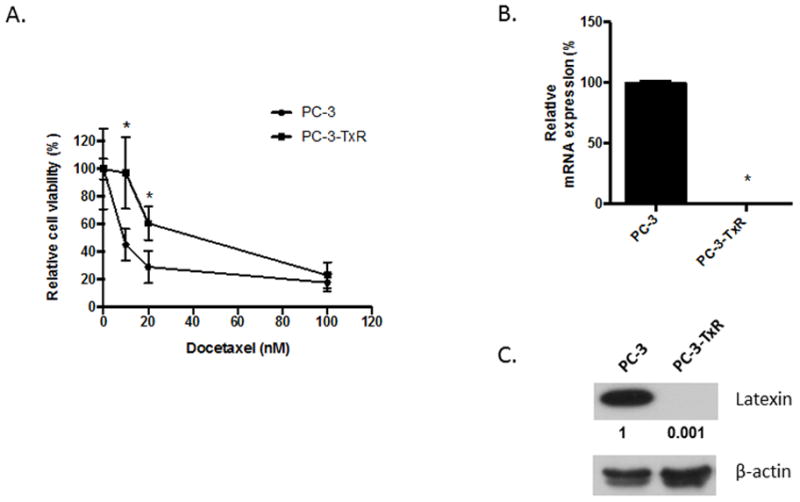
(A) PC-3 and PC-3-TxR cells were plated in 24-well plates overnight and cells were treated with 0–100 nM DOX. After 48 hours, cells were trypsinized and counted with a hemocytometer. *P<0.05 versus PC-3 parental cells (t test). (B) Total RNA from PC-3 parental and PC-3-TxR cells was subjected to real-time PCR for LXN (n = 3 per group); *P<0.0001 versus PC-3 parental cells (t test). (C) Protein from PC-3 parental and PC-3-TxR cells were subjected to Western blotting. Bands were measured using densitometry and values first normalized to respective β-actin bands and then reported below each gel as relative to PC-3.
LXN expression is correlated to cell resistance
To explore the relationship between LXN expression and the development of taxane resistance in PCa cell lines, we quantified LXN expression in additional PCa cell lines and non-neoplastic prostate cell lines. PCa cell lines LNCaP and C4-2B had lower LXN expression than non-neoplastic prostate cell lines, BPH-1 and RWPE-1 (Fig. 2A). Additionally, the expression of LXN was lower in the highly metastatic C4-2B cell line compared to its parental low-metastatic LNCaP cell line. To evaluate if there was an association with DOX resistance and endogenous LXN expression in the PCa cell lines, we evaluated their DOX resistance capability in vitro. C4-2B cells, which had undetectable levels of LXN as opposed to strong expression in the LNCaP cells, was more resistant to DOX than LNCaP (Fig. 2B). These findings suggest that LXN expression can modulate chemoresistance in multiple PCa cells.
Figure 2. LXN expression is correlated to docetaxel resistance in PCa cell lines.
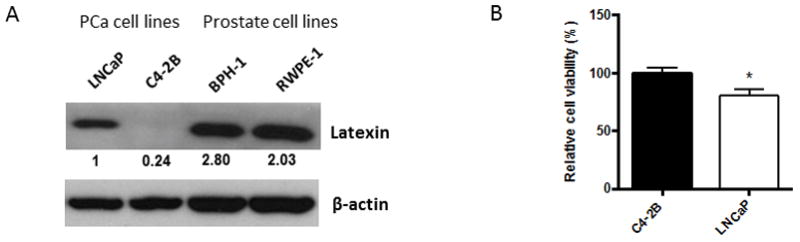
(A) Protein from PCa cell lines, LNCaP and C4-2B and non-cancer prostate cell lines, BPH-1 and RWPE-1 were subjected to western blotting. Bands were measured using densitometry and values first normalized to respective β-actin bands and then reported below each gel as relative to LNCaP. (B) C4-2B and LNCaP cells were cultured in 96-well plates overnight and cells were then treated with 64 nM docetaxel (DOX) for 48hr at which point 10μl cell proliferation reagent WST-1 was added into 100μl of medium and incubated for 4hr. Cell viability was obtained by measuring the absorbance of each well. *P=0.002 versus C4-2B (t test).
Down-regulation of LXN expression increases chemoresistance in PCa cells
To determine if down-regulation of LXN increases chemoresistance in PCa cells, PC-3 cells were stably transduced with shRNA directed against LXN (shLXN). Both shRNA constructs decreased LXN mRNA expression by approximately 50% and protein expression by over 90% compared to that of control non-targeting shRNA (shGFP) (Fig. 3A). To determine if loss of LXN impacted sensitivity to DOX, PC-3 shGFP cells and PC-3-shLXN cells were then subjected to a DOX toxicity assay. Downregulation of LXN increased viability of cells exposed to DOX compared to control shRNA (Fig. 3B). To determine if these in vitro findings extended to in vivo activity, PC-3 shGFP or PC-3 shLXN cells were injected subcutaneously into nude mice and once tumors were approximately 100mm3, the mice were treated weekly with vehicle or 5mg/kg DOX intraperitoneal injections for 4 weeks. Knockdown of LXN alone had not impact on tumor growth compared to shGFP-control tumors (Fig. 3C). DOX reduced tumor growth of shGFP-control cells by approximately 75%; whereas, DOX reduced tumor growth of shLXN cells by only 30%. These results demonstrate that reduction of LXN expression promotes resistance to DOX in PC-3 cells in vivo.
Figure 3. Down-regulation of LXN increases the resistance to docetaxel in PC-3 resistant cells.
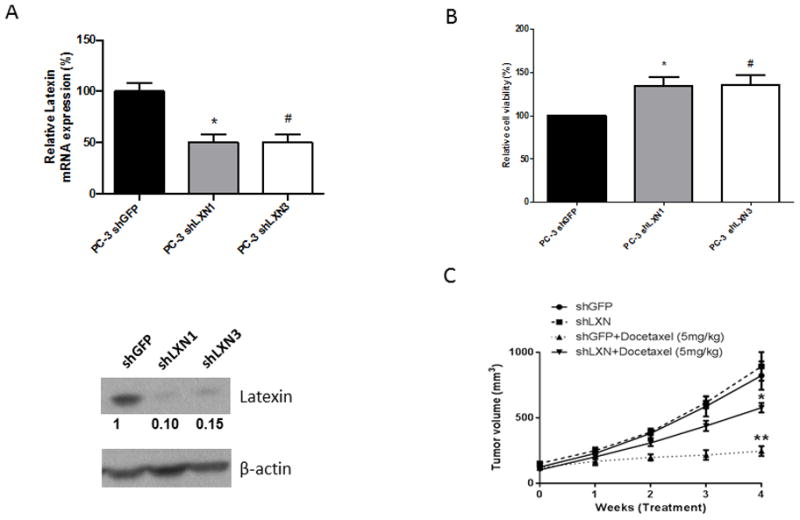
(A) Total RNA and protein from PC-3-shGFP, PC-3-shLXN1 and PC-3-shLXN3 cells were subjected to real-time PCR for LXN (n = 3 per group) and western blotting. Bands were measured using densitometry and values first normalized to respective β-actin bands and then reported below each gel as relative to PC-3 shGFP. *, P=0.0018 PC-3-shLXN1 versus PC-3-shGFP by t test. #, P=0.0018 PC-3-shLXN3 versus PC-3-shGFP by t test. (B) PC-3-shGFP, PC-3-shLXN1 and PC-3-shLXN3 were cultured in 96-well plates overnight and cells were then treated with 20nM docetaxel (DOX) for 48hr at which point 10μl cell proliferation reagent WST-1 was added into 100μl medium and incubated for 2hr. Cell viability was obtained by measuring the absorbance of each well. *, P=0.007 PC-3-shLXN1 versus PC-3-shGFP (t test). #P=0.007 PC-3-shLXN1 versus PC-3-shGFP (t test). (C) Male nude mice aged 6–8 weeks (n=12/group) were injected subcutaneously with PC-3 cells (1x106 in 100μl) expressing either shGFP or shLXN in RPMI 1640 medium. Tumors were allowed to reach approximately 100 mm3 at which time mice were treated weekly with vehicle or 5mg/kg DOX by intraperitoneal injection. Tumor volumes were measured weekly using calipers. The mice were euthanized after 4 weeks of treatment.
Up-regulation of LXN expression decreases chemoresistance in PC-3 cells
Although we have demonstrated that down-regulation of LXN confers DOX chemoresistance, it is unknown of LXN itself is able to confer sensitivity to DOX. To evaluate this possibility, PC-3-TxR cells were stably transfected with LXN human cDNA or empty vector control and confirmed LXN overexpression (Fig. 4A). The cells were then subjected to a toxicity assay with DOX for 48 hours. LXN overexpression in PC-3-TxR cells increased sensitivity to DOX by approximately 20% (Fig. 4A). To determine if this extended to other cell lines, we established additional stable LXN-overexpressing cell lines (or empty vector controls) using LNCaP, C4-2B and DU145 cells and confirmed their LXN expression (Fig. 4B–D). The cells were then subjected to a toxicity assay with DOX over 48 hours. Similar to the PC-3-TxR cell line, overexpression of LXN conferred sensitivity to DOX (between 15 to 30% for the various cell lines). Taken together, these results suggest that the up-regulation of LXN is sufficient to confer sensitivity to docetaxel in PCa cells.
Figure 4. Up-regulation of LXN decreases chemoresistance in PCa cells.
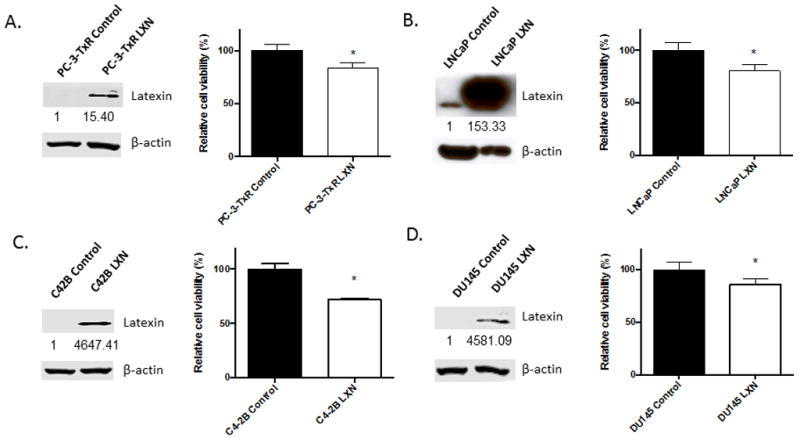
(A) A stable PC-3-TxR LXN overexpressing cell line (PC-3-TxR-LXN) was established by transfecting PC-3-TxR cells with LXN-human vector and selecting with G418. PC-3-TxR-LXN and PC-3-TxR control empty vector cells were subjected to Western blotting (Bands were measured using densitometry and values first normalized to respective β-actin bands and then reported below each gel as relative to PC-3-TxR Control) and cultured in 96-well plates for overnight and then cells were treated with 300nM docetaxel (DOX) for 48hr followed by addition of 10μl cell proliferation reagent WST-1 which was added into 100μl medium and incubated for 2hr. Cell viability was obtained by measuring the absorbance of each well. *P=0.0015 PC-3-TxR-LXN versus PC-3-TxR cells (t test). (B)–(D) Stable LNCaP, C4-2B or DU145 LXN-overexpressing cell lines were established as in (A). The LXN-overexpressing cell lines and their parental control empty vector cells were subjected to Western blotting (Bands were measured using densitometry and values first normalized to respective β-actin bands and then reported below each gel as relative to each control cell line) and cultured in 96-well plates overnight and then cells were treated with 20nM, 20nM or 10nM DOX for 48hr. 10μl cell proliferation reagent WST-1 was added into 100μl medium and incubated at 37°C and 5% CO2 for 2hr. Cell viability was obtained by measuring the absorbance of each well. (B) *P=0.0021 LNCaP-LXN versus LNCaP control cells (t test); (C) *P=0.0001 C4-2B-LXN versus C4-2B control cells (t test); (D) *P=0.0072 DU145-LXN versus DU145 control cells (t test).
Bone stromal cells confer chemoresistance on PCa cells which is associated with methylation-induced reduction of LXN expression
Bone is the most common site of PCa metastases. LXN has been shown to negatively regulate hematopoietic stem cell (HSC) self-renewal, possibly by influencing the interactions between HSC and the osteoblast niche (17,18). Recently several reports demonstrate a link between PCa cells and the HSC osteoblast niche, which may also promote protection chemotherapy treatments (19–21). To determine if the bone microenvironment conferred a protective effect against chemotherapy, we injected mice either subcutaneously or intratibially with PC-3-luciferase-expressing (PC-3-luc) cells. After three weeks of tumor growth, DOX (5 mg/kg weekly) administration was initiated for 4 weeks. While DOX diminished tumor growth by approximately 55% in the subcutaneous tumors, it had no significant impact on the intratibial tumors (Fig. 5A and Sup. Fig. 2). These findings suggest that the bone microenvironment confers a chemoresistance phenotype on the PCa cells. Accordingly, we then tested to see if LXN expression was altered in tumors localized within bone versus soft tissue. CB17/SCID mice were injected with PC-3 cells into either the tibia or subcutaneously and the tumors were allowed to become established for 2 weeks at which time they were harvested for qPCR analysis. Tumors growing in the bone marrow microenvironment had approximately a 46% decrease in LXN mRNA expression compared to those growing subcutaneously (Fig. 5B).
Figure 5. The bone microenvironment confers a protective effect against docetaxel.
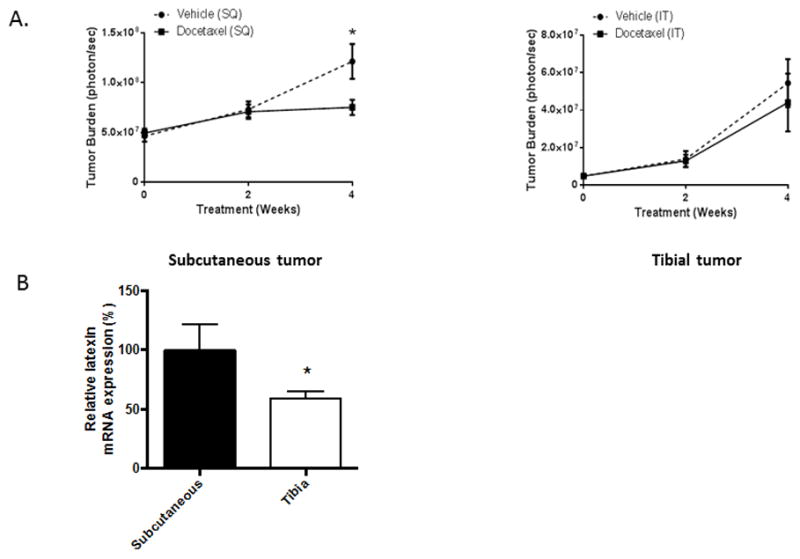
(A) Mice (n= 12/group) were injected either subcutaneously (SQ) with a single cell suspension of PC-3-luc cells (1×106cells) or intratibially with PC-3-luc cells (5×105 cells) in RPMI1640. After 3 weeks of tumor development, the mice were treated weekly with vehicle or 5mg/kg DOX by intraperitoneal injection was performed for 4 weeks. Tumor burden measurement using bioluminescence BLI was started at the time of DOX initiation and measured every two weeks. *P<0.05 PC-3 (SQ) vehicle versus PC-3 (SQ) DOX by (t test). (B) Mice were injected with PC-3 cells as in subcutaneously or intratibially as in (A) and tumors developed for 3 weeks and were then harvested. Total RNA was isolated and subjected to real-time PCR using human specific primers for HPRT and LXN. *P=0.034 for intratibial versus subcutaneous tumors (t test).
The in vivo results indicated that the bone microenvironment may alter chemosensitivity through down-regulating the expression of LXN. Therefore, to determine if bone marrow stromal cells could alter the expression of LXN we grew PC-3 cells alone or in co-culture with the mouse bone stromal cell lines MC3T3 or ST-2. LXN mRNA expression was analyzed with qPCR using human specific LXN and HPRT primers to ensure were detecting the impact on LXN in the PCa cells (which are human) and not the stromal cells (which are murine) in the co-cultures. To confirm the PCR primer/probes’ species specificity, we subjected total RNA from human PCa cells alone and compared to the murine stromal cells alone. While we observed LXN mRNA expression in the PC-3 cells, there was no expression in the murine stromal cells (Fig. 6A; PC-3-luc+PC-3 vs. MC3T3 only and ST2 only) confirming that any LXN mRNA observed was from the human PCa cells. Co-culture of PC-3 cells with MC3T3 or ST-2 cells reduced LXN expression by over 50% (Fig. 6A). To extend this finding to another cell line, we repeated the experiment with LNCaP PCa cells. Similar to the PC-3 cells, co-culture of MC3T3 or ST-2 cells with LNCaP cells, diminished latexin expression in LNCaP cells (Sup. Fig. 3A). These results suggest that bone stromal cells can decrease LXN expression in PCa cells.
Figure 6. Bone stromal cells decrease LXN expression and induce chemoresistance through methylation.
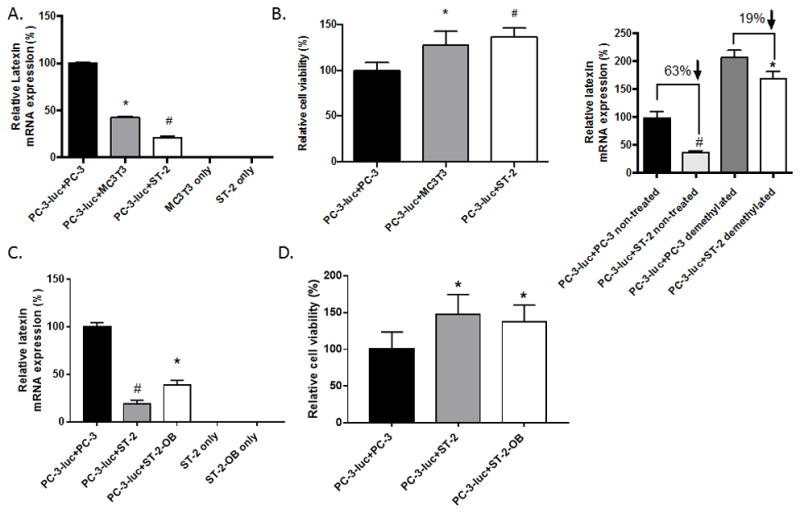
PC-3-luc cells (1.5× 104/ml) were mixed with PC-3 (1.5× 104/ml), MC3T3 (1.5× 104/ml) or ST-2 (1× 104/ml). (A) The cell mixtures were plated into 6-well plates. After 48 hours, cells were then collected and total mRNA subjected to real-time PCR for LXN and HPRT-1 using human-specific primers. LXN expression was normalized to HPRT-1. Note there is no detectable human LXN expression in MC3T3 or ST-2. *P=0.0004 PC-3-luc+MC3T3 cells versus PC-3-luc+PC-3 cells (t test). #P=0.0002 PC-3-luc+ST-2 cells versus PC-3-luc+PC-3 cells (t test). (B) The cell mixtures were plated into white, clear bottom 96-well plates. After 48 hours, the cells were treated with 16nM docetaxel (DOX) for 48 hours and then 20μl luciferin (40 mg/ml) was added to each well and the cells were incubated at 37°C and 5% CO2 for 2 minutes. Cell viability was obtained by measuring the luminescence at integration 1000 ms with a plate reader. *P=0.0194 PC-3-luc+MC3T3 cells versus PC-3-luc+PC-3 cells (t test). #P=0.0015 PC-3-luc+ST-2 cells versus PC-3-luc+PC-3 cells (t test). (C) ST-2 cells were grown in osteoblast differentiating medium then either undifferentiated or osteoblast-differentiated ST-2 cells were cocultured with PC-3 cells for testing LXN expression as in A. *P=0.0178 PC-3-luc+ST-2 versus PC-3-luc+PC-3 cells (t test). #P=0.008 PC-luc + ST-2 versus PC-3-luc+PC-3. (D) PC-3 cells were cocultured with ST-2 osteoblast-differentiated cells or ST-2 parental cells as in C and tested for DOX chemoresistance as in B. *P<0.05 versus PC-3-luc+PC-3 cells (t test). (E) PC-3-luc cells were mixed with PC-3 or ST-2 and plated into 6-well plates as in (A). PC-3-luc cells co-cultured with PC-3 or ST-2 for 48 hours and then either not treated or treated with 5-aza-2′-deoxycytidine. After a 4-day treatment period, cells were collected and total RNA was subjected to real-time PCR for LXN and HPRT-1 using human-specific primers. LXN expression was normalized to HPRT-1. *P<0.01 PC-3-luc+ST-2 non-treated versus PC-3-luc+PC-3 non-treated (t test); #P<0.05 PC-3-luc+ST-2 demethylated versus PC-3-luc+PC-3 demethylated (t test).
To determine if bone stromal cells confer chemoresistance, as observed in the PCa tumors growing in the bone microenvironment, we co-cultured the PC-3 cells alone or with the bone stromal cell lines and treated them with DOX. To determine the impact on PCa cell growth as opposed to an impact on stromal cells, the PC-3 cells were luciferase-labeled (PC-3-luc) allowing us to clearly identify only the PCa cells in the co-cultures. Additionally, to account for the addition of stromal cells to the PC-3-luc culture, we added PC-3 cells that do not express luciferase at similar numbers as the stromal cells to the PC-3 only cultures. After 48 hours of co-culture, we added DOX and then after an additional 48 hours, quantified the cell viability of PC-3-luciferase-expressing cells. Co-culture of PC-3-luc cells with bone stromal cells increased the PC-3 cells resistance to DOX compared to PC-3 cells alone (Fig. 6B). Similar results were found when LNCaP cells were cocultured with bone stromal cells (Sup. Fig. 3B). Taken together, these results suggest that cells within the bone microenvironment reduce latexin expression and confer taxane resistance in PCa cells.
In order to determine if these results extended into additional bone cells within the bone microenvironment, we first evaluated the effect of differentiating the ST-2 bone stromal cells into mature osteoblast phenotype on their ability to modulate LXN expression. Both native ST-2 cells and osteoblast-differentiated ST-2 cells decreased LXN expression in PC-3 cells (Fig. 6C). Similar to the effect of MC3T3 and native ST-2 cells, the osteoblastic-differentiated ST-2 cells promoted resistance to DOX (Fig. 6D). In contrast, coculture of the PC-3 cells with the osteoclast differentiated RAW cell line had no impact on LXN expression or DOX sensitivity in the PC-3 cells (Sup. Fig. 4). Taken together, these findings demonstrate that bone microenvironment can alter taxane sensitivity in PCa cells, and specifically suggests that the stromal and osteoblastic components of the bone microenvironment decreases LXN expression within tumors cells, allowing for the development of taxane resistance in PCa cells.
LXN has been reported to be hypermethylated in several cancers such as gastric cancer, lymphoma and malignant melanoma (14–16). To explore if hypermethylation of the LXN promoter might contribute to the downregulation of LXN expression induced by bone stromal cells, we treated PC-3-luc cells, which have been co-cultured with ST-2 cells or with similar amounts of PC-3 cells as control for 2 days, and treated with 5-aza-2′-deoxycytidine, a DNA demethylating reagent, for 4 days. For the control group which didn’t get the demethylation treatment, the LXN expression in PC-3-luc being co-cultured with ST-2 cells decreased by 63% compared to that in PC-3-luc cells grown with PC-3 cells; whereas, in the 5-aza-2′-deoxycytidine-treated group, the LXN expression in PC-3-luc being co-cultured with ST-2 cells decreased by only 19% compare to that in PC-3-luc co-cultured with PC-3 cells (Fig. 6E). Thus, presence of a demethylation drug decreased the ability of the stromal cells to decrease LXN expression by 44%. These results indicate that bone stromal cells inhibit LXN expression, in part, through inducing methylation in the PCa cells.
Discussion
In the current study, we identified downregulation of LXN expression in a DOX-resistant PCa cell line. We further demonstrated that LXN expression inversely regulates DOX-chemoresistance in PCa cells. Finally, we identified that the bone microenvironment confers a DOX-resistant phenotype in PCa cells through its ability to down-regulate LXN expression, in part through promoting methylation in the PCa cells.
Development of resistance to taxanes is a well-recognized phenomenon in cancer therapy. A variety of mechanisms have been identified including mutations in tubulin binding regions and changes to the expression and function of apoptosis and cell cycle checkpoint proteins (e.g., Bcl-2 and p53) (22–24). In addition, upregulation and activation of EGFR and genes encoding P-glycoprotein such as ABCG2 have shown to lead to chemoresistance in several tumor types (25–27). The current study provides a novel mechanism that contributes towards the multiple pathways through which PCa cells develop chemoresistance.
In our prior study, we created taxane resistant Du145 and PC-3 cancer cells through longitudinal culture in increasing levels of paclitaxel (11). While increased MDR expression was observed in the Du145 cells, accounting for its chemoresistance, it was not found in the PC-3TxR cells indicating they achieved taxane resistance through another mechanism. The current study identifies LXN as a mediator of chemoresistance and suggesting it has potential as a novel biomarker and therapeutic target.
LXN was originally isolated and described as a marker of lateral neocortex neurons in developing rat brains (28–30). In our study, we observed that LXN was significantly reduced in PC-3 cells that were made taxane-resistant through longitudinal growth in increasing concentrations of paclitaxel. These were consistent with the possibility that loss of LXN in PC-3 cells increases their chemoresistance.
LXN is a carboxypeptidase inhibitor with specificity for carboxypeptidase A4 in humans (31). LXN mediates various functions including modulation of sensory perception (32), pain transmission (33,34) and regulation of inflammatory responses (35). LXN has homology to two other proteins/genes, tazarotene-induced gene 1 (TIG1) and cystatin C, both of which have been reported to be tumor suppressors (36,37). LXN expression has been suggested to be a tumor suppressor itself as it has been shown to inhibit tumor cell growth in gastric cancer (14), leukemia (15), melanoma (16), PCa (38), hepatocellular carcinoma (39) and breast cancer (40). In addition to being a potential tumor suppressor, LXN negatively regulates hematopoietic stem cell self-renewal in mice, resulting in an inverse relationship between LXN expression and HSC population size (17,18,41). In our study, we did not observe that downregulation of LXN expression promoted tumor growth. In contrast, we identified a novel function (i.e., chemosensitivity) for LXN in the context of cancer biology. Specifically, we observed that decreasing LXN expression increased resistance to DOX and that overexpressing LXN promoted sensitivity to DOX. These results demonstrate that in the proper cellular context, LXN can regulate chemoresistance. However, LXN may only modulate chemoresistance in a subset of PCas as in the Du145-TxR cells the key chemoresistance factor was MDR-1 and LXN expression was not decreased (11).
As altered LXN expression can contribute to the development of taxane resistance in PCa, it was important to understand what leads to a decrease in LXN expression in PCa cells. It has been previously demonstrated that hypermethylation of CpG islands was found to be highly correlated with the transcriptional silencing of LXN gene (14–16). Furthermore, consistent with its role as tumor suppressor, the LXN gene has been shown to be silenced by methylation in several different cancers (14–16,38–40). Our findings that demethylation treatment of the PCa cells restored LXN expression are consistent with these previous reports suggesting that methylation of the LXN gene contributes to its downregulation in the chemoresistant PCa cells. However, based on the current experiments, we cannot rule out that demethylation indirectly impacted LXN gene expression trough modulating some alternative factor within the cell.
The skeleton is the most common site for PCa metastases (42). Once localized to the bone, the cancer is virtually incurable as it is highly resistant to chemotherapy or the chemotherapy fails within a short period of time (43). This suggests that there is a link between tumor localization to bone and the development of chemoresistance. Our observation that PCa tumors localized within the bone are less responsive to DOX treatment than tumors localized subcutaneously suggests that the bone microenvironment provides a protective effect against DOX. Furthermore, that LXN expression was decreased to a greater extent in bone versus soft tissue sites supports the concept that bone mediated its chemoprotective effect, in part, through downregulationg LXN expression in the PCa cells. This possibility was further supported by the observation that bone stromal cells downregulated LXN expression, through promoting methylation, in the PCa cells. Our finding that RAW cells did not reduce LXN expression or increase resistance to DOX; whereas stromal-derived cells, including osteoblast-differentiated cells, suggests that the effect is specific to stromal-derived cells as opposed to the osteoclastic cells. Our studies do not rule out additional mechanisms that could also contribute to the chemoprotective nature of the bone microenvironment, such as the possibility of decreased taxane bioavailability.
In summary, we have identified that LXN expression inversely modulates taxane chemoresistance in the PC-3 PCa cell line. Inhibition of LXN expression in the bone microenvironment promotes chemoresistance, in part through a methylation-dependent mechanism. These findings suggest that evaluating for LXN expression may serve as a biormarker to identify a subset of PCa patients for which targeting LXN-modulated pathways may provide a therapeutic effect to restore sensitivity to taxanes.
Supplementary Material
Implications.
This study suggests that the latexin pathway should be further explored as a viable target for preventing or reversing taxane resistance in PCa.
Acknowledgments
This work was supported in part by NIH grant P01 CA093900.
Financial support: To ET Keller: NIH grant P01 CA093900
Footnotes
Conflict of Interest: All authors have no conflict of interest with the manuscript contents
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6(2):76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amaral TM, Macedo D, Fernandes I, Costa L. Castration-resistant PCa: mechanisms, targets, and treatment. PCa. 2012;2012:327253. doi: 10.1155/2012/327253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced PCa. N Engl J Med. 2004;351(15):1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 5.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory PCa. N Engl J Med. 2004;351(15):1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 6.Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive PCa. The New England journal of medicine. 2015;373(8):737–46. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in PCa (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. The Lancet. 2016;387(10024):1163–77. doi: 10.1016/S0140-6736(15)01037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrylak DP. The treatment of hormone-refractory PCa: docetaxel and beyond. Rev Urol. 2006;8(Suppl 2):S48–55. [PMC free article] [PubMed] [Google Scholar]
- 9.Magadoux L, Isambert N, Plenchette S, Jeannin JF, Laurens V. Emerging targets to monitor and overcome docetaxel resistance in castration resistant PCa (review) Int J Oncol. 2014;45(3):919–28. doi: 10.3892/ijo.2014.2517. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong CM, Gao AC. Drug resistance in castration resistant PCa: resistance mechanisms and emerging treatment strategies. Am J Clin Exp Urol. 2015;3(2):64–76. [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda M, Mizokami A, Mamiya K, Li YQ, Zhang J, Keller ET, et al. The establishment of two paclitaxel-resistant PCa cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate. 2007;67(9):955–67. doi: 10.1002/pros.20581. [DOI] [PubMed] [Google Scholar]
- 12.Comstock KE, Hall CL, Daignault S, Mandlebaum SA, Yu C, Keller ET. A bioluminescent orthotopic mouse model of human osteosarcoma that allows sensitive and rapid evaluation of new therapeutic agents In vivo. In Vivo. 2009;23(5):661–8. [PubMed] [Google Scholar]
- 13.Dai J, Lu Y, Yu C, Keller JM, Mizokami A, Zhang J, et al. Reversal of chemotherapy-induced leukopenia using granulocyte macrophage colony-stimulating factor promotes bone metastasis that can be blocked with osteoclast inhibitors. Cancer Res. 2010;70(12):5014–23. doi: 10.1158/0008-5472.CAN-10-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Basang Z, Ding H, Lu Z, Ning T, Wei H, et al. Latexin expression is downregulated in human gastric carcinomas and exhibits tumor suppressor potential. BMC Cancer. 2011;11:121. doi: 10.1186/1471-2407-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Howard D, Rector K, Swiderski C, Brandon J, Schook L, et al. Latexin is down-regulated in hematopoietic malignancies and restoration of expression inhibits lymphoma growth. PLoS One. 2012;7(9):e44979. doi: 10.1371/journal.pone.0044979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muthusamy V, Premi S, Soper C, Platt J, Bosenberg M. The hematopoietic stem cell regulatory gene latexin has tumor-suppressive properties in malignant melanoma. J Invest Dermatol. 2013;133(7):1827–33. doi: 10.1038/jid.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Y, Jansen M, Aronow B, Geiger H, Van Zant G. The quantitative trait gene latexin influences the size of the hematopoietic stem cell population in mice. Nat Genet. 2007;39(2):178–88. doi: 10.1038/ng1938. [DOI] [PubMed] [Google Scholar]
- 18.Mitsunaga K, Kikuchi J, Wada T, Furukawa Y. Latexin regulates the abundance of multiple cellular proteins in hematopoietic stem cells. J Cell Physiol. 2012;227(3):1138–47. doi: 10.1002/jcp.22834. [DOI] [PubMed] [Google Scholar]
- 19.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human PCa metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298–312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, et al. GAS6/AXL axis regulates PCa invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12(2):116–27. doi: 10.1593/neo.91384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang N, Docherty FE, Brown HK, Reeves KJ, Fowles AC, Ottewell PD, et al. PCa cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: evidence from in vivo models. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2014;29(12):2688–96. doi: 10.1002/jbmr.2300. [DOI] [PubMed] [Google Scholar]
- 22.Barbuti AM, Chen ZS. Paclitaxel Through the Ages of Anticancer Therapy: Exploring Its Role in Chemoresistance and Radiation Therapy. Cancers (Basel) 2015;7(4):2360–71. doi: 10.3390/cancers7040897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hari M, Loganzo F, Annable T, Tan X, Musto S, Morilla DB, et al. Paclitaxel-resistant cells have a mutation in the paclitaxel-binding region of beta-tubulin (Asp26Glu) and less stable microtubules. Mol Cancer Ther. 2006;5(2):270–8. doi: 10.1158/1535-7163.MCT-05-0190. [DOI] [PubMed] [Google Scholar]
- 24.Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV. Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr Cancer Drug Targets. 2003;3(1):1–19. doi: 10.2174/1568009033333754. [DOI] [PubMed] [Google Scholar]
- 25.Kim KA, Cha YJ, Lee HM, Joo HJ, Park JY. Effects of the ABCG2 and ABCB1 drug transporter polymorphisms on the pharmacokinetics of bicalutamide in humans. Clin Chim Acta. 2015;438:7–11. doi: 10.1016/j.cca.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Volk EL, Farley KM, Wu Y, Li F, Robey RW, Schneider E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002;62(17):5035–40. [PubMed] [Google Scholar]
- 27.Zhang W, Meng Y, Liu N, Wen XF, Yang T. Insights into Chemoresistance of PCa. Int J Biol Sci. 2015;11(10):1160–70. doi: 10.7150/ijbs.11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arimatsu Y. Latexin: a molecular marker for regional specification in the neocortex. Neurosci Res. 1994;20(2):131–5. doi: 10.1016/0168-0102(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 29.Hatanaka Y, Uratani Y, Takiguchi-Hayashi K, Omori A, Sato K, Miyamoto M, et al. Intracortical regionality represented by specific transcription for a novel protein, latexin. Eur J Neurosci. 1994;6(6):973–82. doi: 10.1111/j.1460-9568.1994.tb00592.x. [DOI] [PubMed] [Google Scholar]
- 30.Arimatsu Y, Miyamoto M, Nihonmatsu I, Hirata K, Uratani Y, Hatanaka Y, et al. Early regional specification for a molecular neuronal phenotype in the rat neocortex. Proc Natl Acad Sci U S A. 1992;89(19):8879–83. doi: 10.1073/pnas.89.19.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Yu L, Gao J, Fu Q, Zhang J, Zhang P, et al. Cloning, tissue expression pattern and genomic organization of latexin, a human homologue of rat carboxypeptidase A inhibitor. Mol Biol Rep. 2000;27(4):241–6. doi: 10.1023/a:1010971219806. [DOI] [PubMed] [Google Scholar]
- 32.Bai WZ, Ishida M, Arimatsu Y. Chemically defined feedback connections from infragranular layers of sensory association cortices in the rat. Neuroscience. 2004;123(1):257–67. doi: 10.1016/j.neuroscience.2003.08.056. [DOI] [PubMed] [Google Scholar]
- 33.Jin M, Ishida M, Katoh-Fukui Y, Tsuchiya R, Higashinakagawa T, Ikegami S, et al. Reduced pain sensitivity in mice lacking latexin, an inhibitor of metallocarboxypeptidases. Brain Res. 2006;1075(1):117–21. doi: 10.1016/j.brainres.2005.12.099. [DOI] [PubMed] [Google Scholar]
- 34.Takiguchi-Hayashi K, Sato M, Sugo N, Ishida M, Sato K, Uratani Y, et al. Latexin expression in smaller diameter primary sensory neurons in the rat. Brain Res. 1998;801(1–2):9–20. doi: 10.1016/s0006-8993(98)00496-x. [DOI] [PubMed] [Google Scholar]
- 35.Aagaard A, Listwan P, Cowieson N, Huber T, Ravasi T, Wells CA, et al. An inflammatory role for the mammalian carboxypeptidase inhibitor latexin: relationship to cystatins and the tumor suppressor TIG1. Structure. 2005;13(2):309–17. doi: 10.1016/j.str.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Wegiel B, Jiborn T, Abrahamson M, Helczynski L, Otterbein L, Persson JL, et al. Cystatin C is downregulated in PCa and modulates invasion of PCa cells via MAPK/Erk and androgen receptor pathways. PLoS One. 2009;4(11):e7953. doi: 10.1371/journal.pone.0007953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahab ZJ, Hall MD, Me Sung Y, Dakshanamurthy S, Ji Y, Kumar D, et al. Tumor suppressor RARRES1 interacts with cytoplasmic carboxypeptidase AGBL2 to regulate the alpha-tubulin tyrosination cycle. Cancer Res. 2011;71(4):1219–28. doi: 10.1158/0008-5472.CAN-10-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oldridge EE, Walker HF, Stower MJ, Simms MS, Mann VM, Collins AT, et al. Retinoic acid represses invasion and stem cell phenotype by induction of the metastasis suppressors RARRES1 and LXN. Oncogenesis. 2013;2:e45. doi: 10.1038/oncsis.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ni QF, Tian Y, Kong LL, Lu YT, Ding WZ, Kong LB. Latexin exhibits tumor suppressor potential in hepatocellular carcinoma. Oncol Rep. 2014;31(3):1364–72. doi: 10.3892/or.2014.2966. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Ren Y, Pang D, Liu C. Clinical implications of AGBL2 expression and its inhibitor latexin in breast cancer. World J Surg Oncol. 2014;12:142. doi: 10.1186/1477-7819-12-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Zant G, Liang Y. Natural genetic diversity as a means to uncover stem cell regulatory pathways. Ann N Y Acad Sci. 2009;1176:170–7. doi: 10.1111/j.1749-6632.2009.04567.x. [DOI] [PubMed] [Google Scholar]
- 42.Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of PCa: an autopsy study of 1,589 patients. Hum Pathol. 2000;31(5):578–83. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 43.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nature reviews Cancer. 2011;11(6):411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.