

Abstract
Tamoxifen is a common adjuvant treatment for ERα-positive breast cancer patients, however acquired resistance abrogates the efficacy of this therapeutic approach. We recently demonstrated that G protein-coupled estrogen receptor 1 (GPER1) mediates tamoxifen action in breast cancer cells by inducing insulin-like growth factor binding protein-1 (IGFBP-1) to inhibit IGF-1-dependent signaling. To determine if dysregulation of IGFBP-1 induction is associated with tamoxifen resistance, IGFBP-1 transcription was measured in tamoxifen-resistant MCF-7 cells (TamR) after tamoxifen (Tam) treatment. IGFBP-1 transcription was not stimulated in Tam-treated TamR cells while decreased expression of FoxO1, a known modulator of IGFBP-1 was observed. Exogenous expression of FoxO1 rescued the ability of Tam to induce IGFBP-1 transcription in TamR cells. Since decreased IGF-1R expression is observed in tamoxifen-resistant cells, the requirement for IGF-1R expression on Tam-stimulated IGFBP-1 transcription was investigated. In TamR and SK-BR-3 cells, both characterized by low IGF-1R levels, exogenous IGF-1R expression increased FoxO1 levels and IGFBP-1 expression while IGF-1R knockdown in MCF-7 cells decreased Tam-stimulated IGFBP-1 transcription. Interestingly, both 17β-estradiol (E2)-stimulated ERα phosphorylation and progesterone receptor (PR) expression were altered in TamR; PR is a transcription factor known to modulate FoxO1 transcription. Additionally, IGF-1R knockdown decreased FoxO1 protein levels in MCF-7 cells. Furthermore, IGF-1R or FoxO1 knockdown inhibited the ability of Tam to induce IGFBP-1 transcription and Tam sensitivity in MCF-7 cells. These data provide a molecular mechanistic connection between IGF-1R expression and the FoxO1-mediated mechanism of tamoxifen action in breast cancer cells.
Keywords: IGF-1 receptor, FoxO1, IGFBP-1, tamoxifen resistance, breast cancer
Introduction
Breast cancer is one of the leading causes of cancer-related deaths in the world (1). Tamoxifen, a selective estrogen receptor modulator (SERM), is a commonly prescribed drug for estrogen receptor alpha (ERα)-positive breast cancer patients, however the therapeutic benefits of this drug are diminished by de novo and acquired resistance (2, 3). While ERα antagonism is a well-documented mechanism of tamoxifen action in breast cancer cells, tamoxifen also modulates breast cancer cells that do not express ERα (4–6). We previously demonstrated that G protein-coupled estrogen receptor (GPER1) is a critical component of tamoxifen action in breast cancer cells. After treatment with 4-hydroxytamoxifen (Tam), the active metabolite of tamoxifen, GPER1 mediates the inhibition of Insulin-like growth factor-1 (IGF-1)-dependent cell signaling by inducing the extracellular accumulation of IGF-binding proten-1 (IGFBP-1) (6).
IGF-1-dependent cell signaling is critical for growth of normal mammary tissue and contributes to breast carcinogenesis (7, 8). IGF-1 receptor (IGF-1R) is activated upon IGF-1 binding resulting in the stimulation of multiple downstream signal transduction pathways that enhance cell survival and induce cell proliferation (9–11). Cell signaling mediated by IGF-1R is regulated by IGFBPs, a family consisting of six members that modulate the bioavailability and binding capacity of IGFs to IGF-1R. IGFBP-1, for example, inhibits IGF-1-dependent signaling in several cell types including breast cancer cells (6, 12, 13). Loss of IGF-1R expression results in poor prognosis for postmenopausal breast cancer patients treated with tamoxifen (14) and loss IGF-1R expression is observed in tamoxifen-resistant breast cancer cells in multiple studies (15–17) suggesting that the expression of this receptor is necessary for tamoxifen action. These findings suggest that IGF-1R is an important component of tamoxifen action, however the molecular mechanisms of altered tamoxifen action in breast cancer cells with decreased IGF-1R expression has not been determined.
FoxO1 is a member of the Forkhead family of transcription factors that is stabilized and active in the absence of growth factors. When stabilized, FoxO1 translocates to the nucleus and induces the transcription of anti-proliferative and pro-apoptotic genes such as p21 and IGFBP-1 (18–21). FoxO1 phosphorylation by AKT and ERK kinases results in cytoplasmic localization and proteasome-dependent degradation thus decreasing FoxO1 protein levels (22–24). In breast cancer cells, decreased FoxO1 expression is observed relative to normal mammary epithelial cells (25), however the role of FoxO1 in tamoxifen-treated breast cancer cells has not been adequately characterized.
The goal of the current research was to characterize the molecular mechanisms of altered IGFBP-1 transcription in tamoxifen-resistant breast cancer cells. In Tam-resistant MCF-7 cells (TamR), IGFBP-1 transcription was not induced upon treatment with Tam. In these cells, FoxO1 expression was decreased suggesting that FoxO1 dysregulation contributes to the loss of Tam-induced IGFBP-1 transcription. Exogenous expression of FoxO1 rescued the ability of Tam to induce IGFBP-1 transcription in TamR cells and FoxO1 knockdown decreased Tam-induced IGFBP-1 transcription in MCF-7 cells. Since decreased IGF-1R expression is a characteristic of tamoxifen-resistant cells, the requirement for IGF-1R expression on Tam-stimulated IGFBP-1 transcription was determined. Exogenous IGF-1R expression in TamR and SK-BR-3 cells, both characterized by low IGF-1R levels increased FoxO1 protein levels and IGFBP-1 expression while IGF-1R knockdown in MCF-7 cells decreased Tam-stimulated IGFBP-1 transcription and decreased FoxO1 protein levels. IGF-1R knockdown in MCF-7 cells increased ERK1/2 phosphorylation; ERK1/2 has previously been shown to regulate FoxO1 protein levels. Progesterone receptor (PR) is a known inducer of FoxO1 expression, so we tested if E2-induced PR expression was altered in TamR cells. E2 treatment did not induce ERα phosphorylation or progesterone receptor (PR) expression in TamR cells, suggesting potential mechanisms for reducing FoxO1 protein levels in these cells. Finally, IGF-1R or FoxO1 knockdown resulted in decreased Tam sensitivity in MCF-7 cells. Collectively, these data provide a molecular mechanistic connection between IGF-1R expression and FoxO1-dependent mechanism of tamoxifen action in breast cancer cells.
Materials and Methods
Cell culture and treatment
MCF-7 and SK-BR-3 breast cancer cells validated using short tandem repeat (STR) profiling were obtained from ATCC, (Manassas, VA). Cells were cultured in our laboratory for less than 6 months in DMEM (Life Technologies, Carlsbad, CA) and DMEM/F12 (Life Technologies, Carlsbad, CA), respectively. Maintenance media were supplemented with 10% fetal bovine serum (Life Technologies, Carlsbad, CA). 48h prior to treatment, cells were washed with 1X PBS, and phenol red-free DMEM supplemented with 10% charcoal-stripped FBS (Life Technologies, Carlsbad, CA) was added to the cells. After 24h, cells were washed with 1X PBS, then serum-starved in phenol red-free DMEM overnight followed by treatment with the indicated dose of 4-hydroxytamoxifen (Tam, Fluka, St. Louis, MO), IGF-1 (I3769, Sigma, St. Louis, MO), 17β-Estradiol (E2, E2758, Sigma, St. Louis, MO), picropodophyllin (PPP, sc-204008, Santa Cruz Biotechnology, Dallas, TX), and Actinomycin D (15021, Cell Signaling Technology, Danvers, MA). Tam and E2 were dissolved in ethanol (vehicle), and the final concentration of ethanol in medium was 0.1%.
Establishment of tamoxifen-resistant MCF-7 cells (TamR)
TamR cells were generated by continuously culturing of MCF-7 cells in phenol-red free DMEM supplemented with 10% FBS containing 1 µM Tam as previously reported (26).
siRNA knockdown
IGF-1R siRNA (39584, Santa Cruz Biotechnology, Dallas, TX), FoxO1 siRNA (106652, Ambion, Austin, TX), or non-targeting, negative control siRNA (4390843, Ambion, Austin, TX) were performed using Lipofectamine 2000 reagent (Life Technology, Carlsbad, CA) for 6 h in serum-free Opti-MEM (Life Technology, Carlsbad, CA). After overnight recovery in maintenance media, the transfection was repeated followed by growing transfected cells in media containing 10% charcoal-striped FBS and analyzed for protein expression (immunoblot) after 48 h.
Exogenous gene expression
The IGF-1R expressing plasmid was a gift from Ronald Kahn (11212, Addgene, cambridge, MA) (27) and FoxO1 expressing plasmid was a gift from Kunlianf Guan (13507, Addgene, cambridge, MA) (28) were transfected using Lipofectamine 2000 reagent (Life Technology, Carlsbad, CA) for 6h in complete medium.
Total RNA isolation and quantitative PCR analysis
Total RNA was isolated with the PureLink RNA Mini Kit (Life Technologies, Carlsbad CA) with DNase-I following the manufacturer’s protocol. cDNA was synthesized from total RNA (1 µg) using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR (qPCR) reactions were performed using SYBR Green Master Mix and the 7300 Real-Time PCR system (Bio-Rad, Hercules, CA). RPL30 gene expression was used in all quantitative real-time PCR reaction as an internal reference gene to normalize relative changes in transcript levels. Primers for amplification of human IGFBP-1: forward 5′-CTA-TGA-TGG-CTC-GAA-GGC-TC-3′, reverse 5′-TTC-TTG-TTG-CAG-TTT-GGC-AG-3′ (29), human FoxO1: forward 5′-TAC-GAG-TGG-ATG-GTC-AAG-AGC-3', reverse 5'-TGA-ACT-TGC-TGT-GTA-GGG-ACA-3' (30) and RPL30: forward 5′-ACA-GCA-TGC-GGA-AAA-TAC-TAC-3′, reverse: 5′-AAA-GGA-AAA-TTT-TGC-AGG-TTT-3′ (31).
Immunoblot analysis
Whole-cell extracts were prepared in RIPA buffer containing protease and phosphatase inhibitor cocktails (87785, 78420, Thermo Scientific, Rockford, IL). Cell extract protein concentrations were determined using BCA assay (Thermo Scientific, Rockford, IL). 30 µg of whole-cell lysates were resolved using Bolt 4–12% Bis-Tris Plus gels (Life Technologies, Carlsbad, CA) and transferred to PVDF membrane. Blots were blocked in Tris-buffered saline-0.1% Tween 20 (1X TBST) containing 5% fat-free milk at room temperature for 1h then incubated with primary antibody overnight at 4°C using the following antibodies: IGFBP-1 (sc-13097, Santa Cruz Biotechnology, Dallas, TX); GPER1 (sc-48825-R, Santa Cruz Biotechnology, Dallas, TX); IGF-I receptor beta (3024, Cell Signaling Technology, Danvers, MA); p-AKT (S473) (4060, Cell Signaling Technology, Danvers, MA); AKT (4685, Cell Signaling Technology, Danvers, MA); p-ERK1/2 (Thr202/Tyr204) (9106, Cell Signaling Technology, Danvers, MA); ERK1/2 (9102, Cell Signaling Technology Danvers, MA); FoxO1 (2880, Cell Signaling Technology Danvers, MA); p-CREB (Ser133) (9198, Cell Signaling Technology, Danvers, MA); CREB (9197, Cell Signaling Technology Danvers, MA), ERα (8644, Cell Signaling Technology Danvers, MA), p-ERα (Ser118) (2511, Cell Signaling Technology Danvers, MA), p-ERα (Ser167) (5587, Cell Signaling Technology Danvers, MA), p21 (sc-397, Santa Cruz Biotechnology, Dallas, TX) and β-actin (sc-47778, Santa Cruz Biotechnology, Dallas, TX). After washing with 1X TBST, blots were incubated with anti-IgG horseradish peroxidase-conjugated secondary antibody (sc-81178, Santa Cruz Biotechnology, Dallas, TX) at room temperature for 1h. Prior to the addition of chemiluminescence reagent (34076, Thermo Scientific, Rockford, IL), blots were washed with 1X TBST. Each blot was stripped using restore plus western blot stripping buffer (46430, Thermo Scientific, Rockford, IL) when necessary to determine equivalent sample loading. Chemiluminescence was detected using Gel Doc™ XR ChemiDoc™ imaging system (BioRad, Hercules, CA) and quantitated using Quantity One® software (BioRad, Hercules, CA).
Cell viability assay
Cells were plated in 96-well plates at a density of 2500 cells per well in DMEM supplemented with fetal bovine serum. 12h prior to treatment, media were replaced with DMEM supplemented with charcoal-stripped serum (17), and cell viability was determined after five days using Alamar blue reagent (Life Technology, Carlsbad, CA) according to the manufacture’s procedure. The medium was replaced at day three.
Statistical analysis
All statistical analysis was performed using Kaleidagraph (Synergy Software, Reading, PA). Differences were considered significant if p < 0.05 using one-way ANOVA, with Tukey post hoc analysis, and the error bars are represented mean ± SEM.
Results
Tam-induced IGFBP-1 transcription and FoxO1 expression are dysregulated in TamR cells
We previously reported that treatment of breast cancer cells with the active metabolite of tamoxifen (Tam) inhibits IGF-1-stimulated breast cancer cell signaling and cell viability by inducing the extracellular accumulation of IGFBP-1 (6). Actinomycin D pretreatment blocked the accumulation of IGFBP-1 transcript in Tam-treated MCF-7 cells confirming that Tam stimulates IGFBP-1 transcription in MCF-7 cells (Supplementary Fig. S1). The ability of E2, also a GPER1 agonist (32), to stimulate IGFBP-1 transcription was determined, and as expected, IGFBP-1 transcription is stimulated by E2 in MCF-7 cells (Supplementary Fig. S2). To determine if IGFBP-1 transcription was altered during the development of tamoxifen resistance, Tam-resistant MCF-7 cells (TamR) were generated following a previously published protocol (26) (Fig. 1A). Whereas Tam treatment significantly increased IGFBP-1 transcription in MCF-7 cells, IGFBP-1 transcription was not induced in TamR cells (Fig. 1B) suggesting that this mechanism of action is altered during the development of tamoxifen resistance. Our previous work established GPER1 and the cAMP-response element-binding protein (CREB) as mediators of IGFBP-1 transcription in Tam-treated MCF-7 cells (6), therefore we determined if GPER1 and p-CREB (S133) levels were decreased TamR cells before or after Tam treatment. Consistent with previous reports (33), GPER1 expression was not altered in TamR cells (Supplementary Fig. S3A). However, p-CREB levels were increased in untreated TamR cells and p-CREB accumulation did not increase after Tam treatment (Supplementary Fig. S3B) compared to parental MCF-7 cells. To identify a potential mechanism for dysregulated IGFBP-1 transcription in TamR cells, the protein levels of FoxO1, a known regulator of IGFBP-1 expression, were determined (18, 20). Both FoxO1 expression and the expression of p21, a FoxO1-regulated cell cycle inhibitor (21), were decreased in TamR cells compared to MCF-7 cells suggesting that decreased FoxO1 protein levels result in dysregulation of IGFBP-1 transcription (Fig. 1C). Exogeneous FoxO1 rescued Tam-induced IGFBP-1 transcription in TamR cells (Fig. 1D). These indicate that IGFBP-1 transcription is dysregulated during the development of Tam resistance and suggest that the observed alteration in IGFBP-1 regulation is not due to the loss of GPER1 or CREB expression.
Fig. 1. Tam-induced IGFBP-1 transcription and FoxO1 expression are dysregulated in TamR cells.
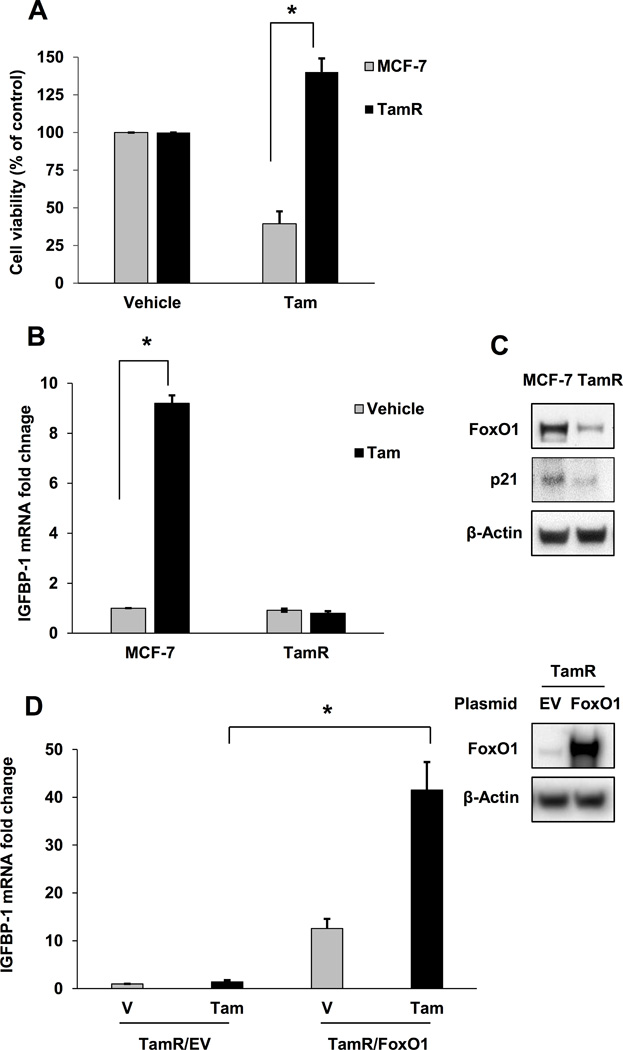
A, Relative cell viability of MCF-7 and TamR cells after 5 days of 1 μM Tam treatment. B, Relative IGFBP-1 transcription in MCF-7 and TamR cells after 24 h after treatment with 1 μM Tam. C, FoxO1 and p21 expression in MCF-7 and TamR cells. D, Relative IGFBP-1 transcription in TamR cells after exogenous FoxO1 expression and 1 μM Tam treatment. * = p < 0.05 and results are average or representative of three independent experiments. Error bars are SEM.
FoxO1 mediated Tam-stimulated IGFBP-1 transcription and inhibition of IGF-1-dependent cell signaling in MCF-7 cells
To demonstrate the functional relevance of FoxO1 expression for the IGFBP-1-depdendent mechanism of Tam action, FoxO1 knockdown was performed in MCF-7 cells as previously described (6). FoxO1 knockdown resulted in reduced IGFBP-1 transcription in vehicle and Tam-treated cells (Fig. 2A and B). Additionally, FoxO1 knockdown reduced the ability of Tam to inhibit the accumulation of p-AKT after IGF-1 stimulation (Fig. 2C, compare 2nd and 4th lanes). We previously showed that CREB mediates IGFBP-1 transcription downstream of GPER1 in Tam-treated MCF-7 cells (6). To determine if FoxO1 protein levels modulated Tam-induced accumulation of p-CREB, Tam-stimulated p-CREB accumulation was determined after FoxO1 knockdown. Tam-stimulated pCREB (S133) accumulation was reduced in MCF-7 cells after FoxO1 knockdown (Fig. 2D). These data suggest that FoxO1 and CREB cooperate to stimulate IGFBP-1 transcription in Tam-treated MCF-7 cells. Further evidence for the involvement of FoxO1 is the observed increase in transcription of FoxO1 in Tam-treated MCF-7 cells (Fig. 2E). These data indicate FoxO1 is a critical component of Tam-stimulated IGFBP-1 expression, and provide preliminary evidence that FoxO1 modulates CREB activation after Tam treatment in breast cancer cells.
Fig. 2. FoxO1 mediates Tam-stimulated IGFBP-1 transcription and inhibition of IGF-1-dependent cell signaling in MCF-7 cells.
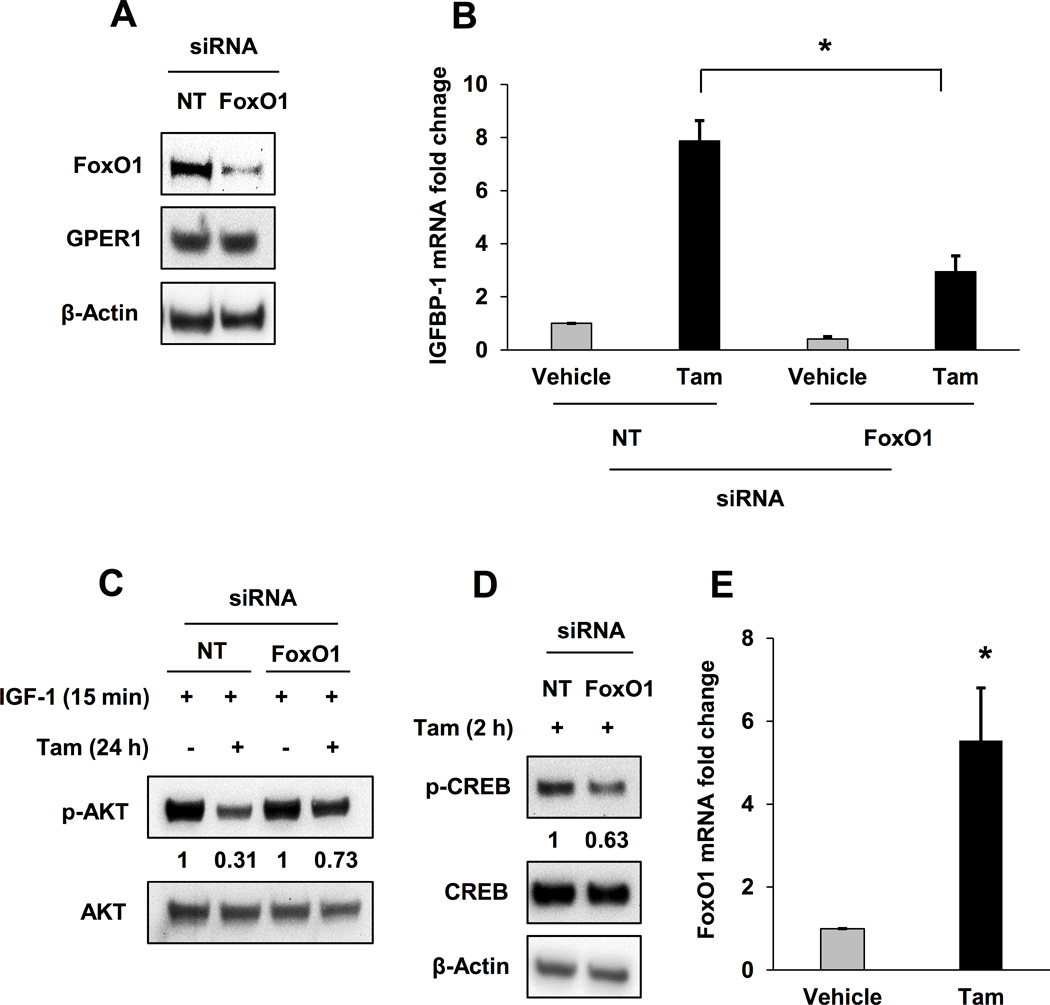
A, FoxO1 expression after siRNA knockdown MCF-7 cells. B, qPCR analysis of 1 µM Tam-induced IGFBP-1 transcription after FoxO1 knockdown in MCF-7 cells. C, Tam-dependent inhibition of IGF-1-stimulated phospho-AKT accumulation is decreased after FoxO1 knockdown in MCF-7 cells. D, CREB expression and accumulation of p-CREB after 1 µM Tam treatment after FoxO1 knockdown in MCF-7 cells. E, Relative qPCR analysis of FoxO1 transcription in 1 μM Tam-treated MCF-7 cells. * = p < 0.05 and results are average or representative of three independent experiments. Error bars are SEM.
Decreased Tam sensitivity in MCF-7 cells after IGF-1R knockdown
Loss or decreased IGF-1R expression is observed in tamoxifen-resistant breast cancer cells (15–17) and reduced IGF-1R expression results in poor prognosis for postmenopausal breast cancer patients treated with tamoxifen (14). However, the role that IGF-1R plays during tamoxifen treatment is not clearly understood. Consistent with previous studies, the TamR cells developed for this study had reduced IGF-1R expression compared with parental MCF-7 cells (Fig. 3A). To determine if decreased IGF-1R expression modulates Tam-stimulated IGFBP-1 transcription in breast cancer cells, MCF-7 cells were treated with Tam after IGF-1R knockdown and IGFBP-1 transcription was measured. After IGF-1R knockdown, Tam-induced IGFBP-1 transcription was not observed in MCF-7 cells (Fig. 3B and C). Similar to the observations in TamR cells, decreased IGF-1R expression in MCF-7 cells did not alter GPER1 expression suggesting that mechanisms other than loss of GPER1 expression result in decreased IGFBP-1 transcription after IGF-1R knockdown. These results indicate that IGF-1R expression is necessary for Tam-induced IGFBP-1 transcription in breast cancer cells.
Fig. 3. IGF-1R knockdown decreased Tam sensitivity MCF-7 cells.
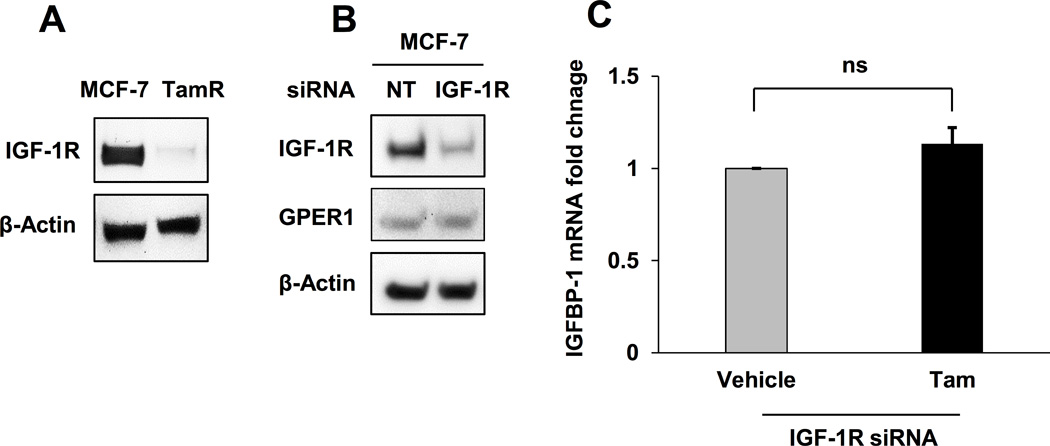
A, IGF-1R expression in MCF-7 and TamR cells. B, IGF-1R expression after IGF-1R knockdown in MCF-7 cells. C, qPCR analysis of 1 µM Tam-induced IGFBP-1 transcription after IGF-1R knockdown in MCF-7 cells. * = p < 0.05, ns = not significant, and results are average or representative of three independent experiments. Error bars are SEM.
Exogenous IGF-1R expression restored FoxO1 and IGFBP-1 expression in TamR and SK-BR-3 cells
To determine if FoxO1 protein levels are a result of dysregulated IGF-1R expression in breast cancer cells, FoxO1 protein expression was measured in TamR and SK-BR--3, breast cancer cells characterized by low IGF-1R expression, after exogenous expression of IGF-1R. (34, 35). In TamR cells, exogenous IGF-1R expression resulted in increased FoxO1 protein levels and increased IGFBP-1 transcript levels compared to cells transfected with an empty vector (Fig. 4A and B). To demonstrate that this mechanism is not unique to TamR cells, SK-BR-3 cells were used in similar experiments. Low IGF-1R expression and the ERα-null status were confirmed and compared to MCF-7 cells (Fig. 4C). Additionally, the IGFBP-1 transcript levels were determined for SK-BR-3 cells and shown to be significantly lower when compared with MCF-7 cells (Fig. 4D). Similar to results obtained in TamR cells, exogenous expression of IGF-1R increased FoxO1 and IGFBP-1 expression in SK-BR-3 cells (Fig. 4E). Taken together, these data provide evidence that FoxO1 protein levels correlate with IGF-1R expression in breast cancer cells.
Fig. 4. Exogenous IGF-1R expression restores FoxO1 and IGFBP-1 expression in TamR and SK-BR-3 cells.

A, IGF-1R and FoxO1 expression after exogenous IGF-1R expression in TamR cells. B, qPCR analysis of IGFBP-1 transcription in TamR cells after exogenous expression of IGF-1R. C, IGF-1R and ERα expression in MCF-7 and SK-BR-3 cells. D, qPCR analysis of IGFBP-1 transcription in MCF-7 and SK-BR-3 cells. E, IGF-1R, FoxO1, and IGFBP-1 expression after exogenous IGF-1R expression in SK-BR-3 cells. * = p < 0.05, EV = empty vector, and results are average or representative of three independent experiments. Error bars are SEM.
IGF-1R knockdown decreased FoxO1 protein levels in MCF-7 cells
To provide evidence that IGF-1R expression modulates FoxO1 protein levels and identify potential mechanisms of the observed decrease in FoxO1, IGF-1R knockdown was performed in MCF-7 cells and FoxO1 protein levels were measured. Knockdown of IGF-1R resulted in reduced FoxO1 protein in MCF-7 cells that occurred ~24h after reduced IGF-1R levels were observed (Fig. 5A) suggesting that FoxO1 levels are associated with IGF-1R expression. To determine if the observed decrease in FoxO1 levels are associated with loss or decrease in IGF-1R expression rather than a loss of IGF-1R-mediated cell signaling, IGF-1R signaling was inhibited in MCF-7 cells using IGF-1R-specific tyrosine kinase inhibitor picropodophyllin (PPP) (36, 37). Data from these experiments show that the inhibition of IGF-1R activity indicated by decreased p-IGF-1R levels did not result in decreased FoxO1 protein levels. The slight increase in FoxO1 protein levels after PPP treatment observed in these experiments was not statistically significant compared to vehicle treatment (Supplementary Fig. S4). Since ERK1/2 destabilizes FoxO1 resulting in decreased FoxO1 protein levels (24), the accumulation of p-ERK1/2 was measured after IGF-1R knockdown in MCF-7 cells to determine if hyperactivation may be associated with decreased FoxO1 levels. In MCF-7 cells, IGF-1R knockdown resulted in the accumulation of p-ERK1/2 (Fig. 5B). Additionally, p-ERK1/2 levels were greater in TamR cells when compared to MCF-7 cells (Fig. 5C), consistent with results from other studies (17, 38, 39). Taken together, these results suggested that inhibition of IGF-1R activation is not sufficient to decrease FoxO1 protein levels and that loss or decrease of IGF-1R expression is necessary for the observed decrease in FoxO1 protein levels. Furthermore, these experiments suggest that decreased FoxO1 protein levels associate with increased ERK1/2 activity in breast cancer cells after IGF-1R knockdown.
Fig. 5. IGF-1R knockdown induces the accumulation decreased FoxO1 protein levels.
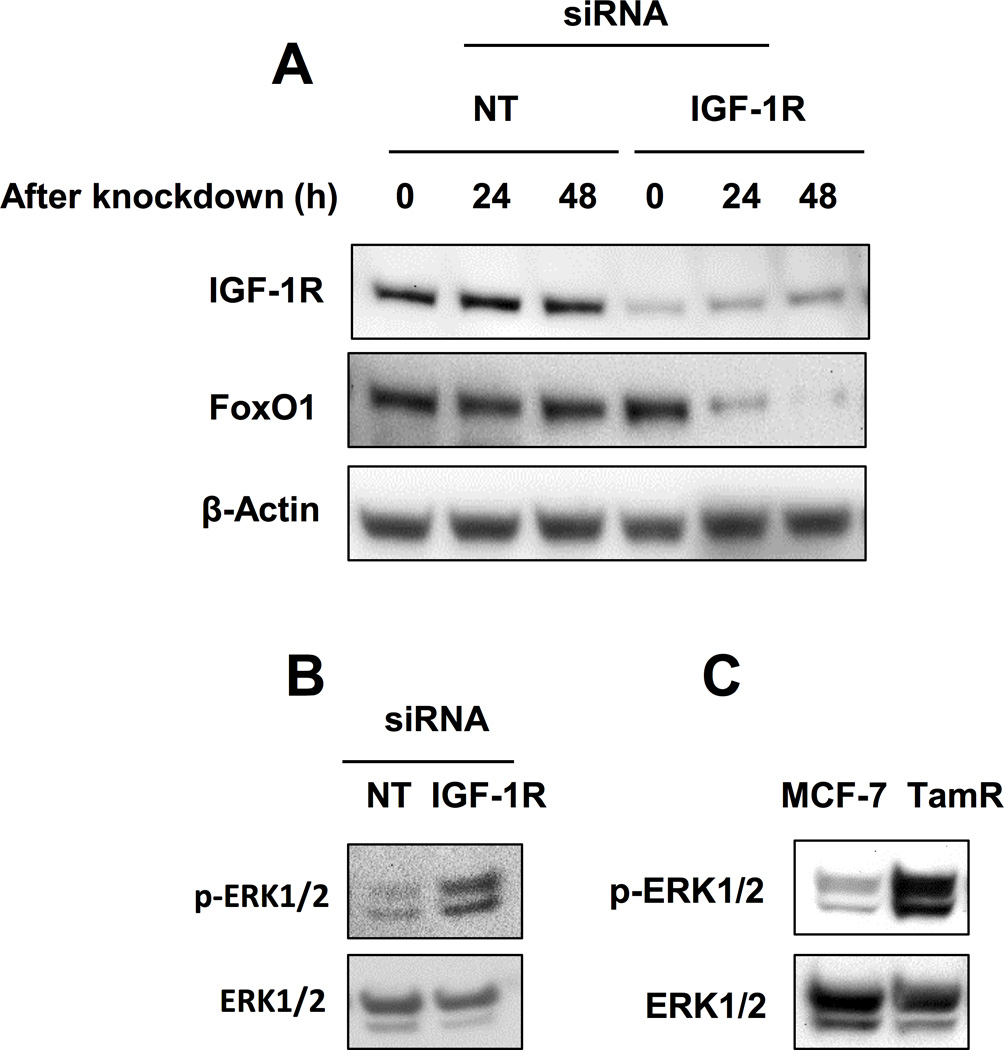
A, FoxO1 expression over 48 h after IGF-1R knockdown in MCF-7. B, Accumulation of p-ERK1/2 after IGF-1R knockdown in MCF-7 cells. C, Accumulation of p-ERK1/2 in MCF-7 and TamR cells. * = p < 0.05 and results are average or representative of three independent experiments. Error bars are SEM.
Altered ERα phosphorylation and loss of E2-induced progesterone receptor (PR) and FoxO1 expression in TamR cells
Another potential mechanism for reduced FoxO1 protein levels in breast cancer cells with decreased IGF-1R expression (i.e. TamR) is dysregulation of phosphorylation-dependent ERα activation downstream of IGF-1R. Cross-talk between ERα and IGF-1R in ERα-positive breast cancer cells has been well documented (40–42). In breast cancer cells characterized by insufficient IGF-1R expression, phosphorylation-dependent ERα activity that requires IGF-1R may be reduced. If this phosphorylation-dependent ERα activity is required to induce FoxO1 expression, then loss of IGF-1R would impact FoxO1 induction. Progesterone receptor (PR) regulates FoxO1 expression in endometrial and ovarian cells (43–45), however this regulation has not been studied in breast cancer cells. It is also well documented that PR expression is induced upon ERα activation (46). Since our results indicate that FoxO1 protein levels are modulated by IGF-1R expression in TamR cells, we determined if E2-dependent ERα phosphorylation/activity is also compromised in these cells. We observed E2 and IGF-1 stimulated ERα phosphorylation (pERα) at S118 and S167, respectively, in MCF-7 cells consistent with previous observations (42), however, increased phosphorylation was not observed in TamR cells (Fig. 6A). Whereas E2-stimulated PR expression in MCF-7 cells, PR expression was not induced in TamR cells (Fig. 6B). Lack of PR transcription in E2-treated TamR cells was previously reported (17). Concurrent with PR expression, the expression of FoxO1 was induced in E2-treated in MCF-7 cells. Furthermore, expression of FoxO1-modulated p21 and IGFBP-1 were induced upon E2 treatment in MCF-7 cells but not induced in TamR cells (Fig. 6C). E2 has previously been shown to stimulate p21 expression in MCF-7 cells (47). Interestingly, E2-induced accumulation of p-ERα (S118) correlates with IGFBP-1 transcription while IGF-1-induced accumulation of p-ERα (S167) is associated with decreased IGFBP-1 transcription in MCF-7 cells (Supplementary Fig. S5). These results suggest that dysregulation of FoxO1 expression may result from decreased IGF-1R-dependent p-ERα activity in breast cancer cells (i.e. TamR).
Fig. 6. Altered ERα phosphorylation and loss of E2-induced progesterone receptor (PR) and FoxO1 expression in TamR cells.
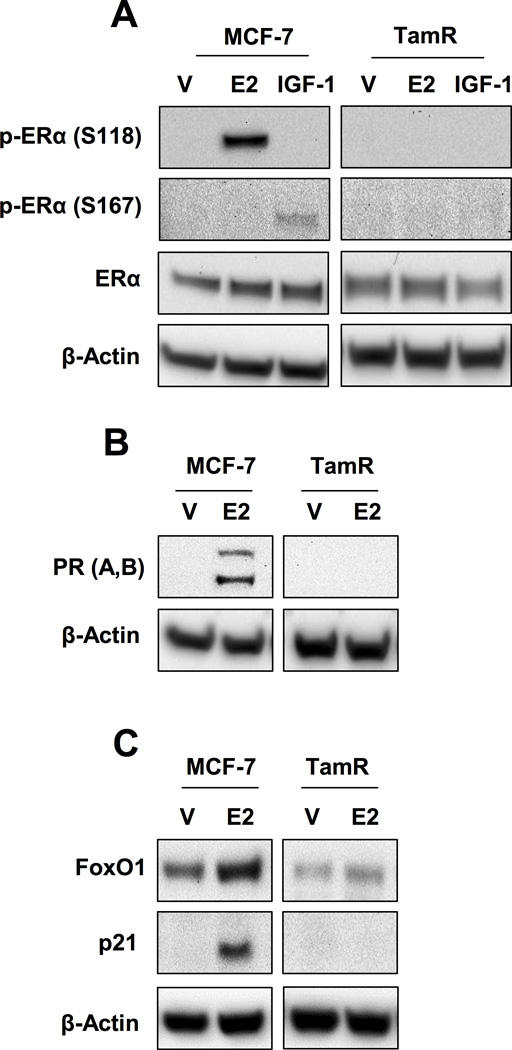
A, p-ERα (S118 and S167) in MCF-7 and TamR cells treated after 10 nM E2 or 50 ng/mL IGF-1 treatment. B, PRA and PRB expression in MCF-7 and TamR cells treated with 10 nM E2. C, FoxO1 and p21 expression in MCF-7 and TamR cells treated with 10 nM E2. * = p < 0.05 and results are average or representative of three independent experiments. Error bars are SEM.
Decreased Tam sensitivity in MCF-7 cells after IGF-1R or FoxO1 knockdown
Cell viability assays were performed using MCF-7 cells after IGF-1R or FoxO1 knockdown. Data from these experiments indicate that decreased IGF-1R or FoxO1 protein levels de-sensitize MCF-7 cells to Tam suggestingc that both IGF-1R and FoxO1 mediate Tam-induced breast cancer cell death (Fig. 7A and B). The results reported herein suggest that IGF-1R expression is required for Tam to induce cell death in breast cancer cells and provide a molecular mechanistic understanding of IGF-1R-dependent tamoxifen sensitivity in breast cancer cells.
Fig. 7. Decreased Tam sensitivity in MCF-7 cells after IGF-1R or FoxO1 knockdown.

A, Cell viability on day 5 of 1 μM Tam-treated MCF-7 cells after FoxO1 knockdown. B, Cell viability on day 5 of 1 μM Tam-treated MCF-7 cells after IGF-1R knockdown. * = p < 0.05 and results are average or representative of three independent experiments. Error bars are SEM.
Discussion
Data presented in this contribution provide mechanistic evidence describing how IGF-1R expression affects the FoxO1-mediated tamoxifen response in breast cancer cells. Consistent with previous observations, decreased IGF-1R expression was observed in Tam-resistant breast cancer cells (TamR) developed for this study. Decreased IGF-1R expression was correlated with attenuated IGFBP-1 induction subsequent to Tam treatment; IGFBP-1 has been previously identified as a mediator of Tam action in MCF-7 cells (6). Expression of the FoxO1 transcription factor, a known regulator of IGFBP-1, was significantly reduced in TamR cells compared to MCF-7 cells suggesting that reduced FoxO1 activity contributed to decreased IGFBP-1 induction in TamR cells. Exogenous expression of FoxO1 in TamR cells rescued the ability of Tam to induce IGFBP-1 transcription providing additional evidence that FoxO1 mediates this specific tamoxifen response. Knockdown of FoxO1 in MCF-7 cells resulted in a significant decrease in Tam-stimulated IGFBP-1 induction further suggesting that FoxO1 mediates IGFBP-1 transcription in Tam-treated breast cancer cells. Furthermore, knockdown of IGF-1R in MCF-7 cells reduced FoxO1 expression and Tam-stimulated IGFBP-1 induction resulting in decreased sensitivity of these cells to Tam. The correlation between the expression of IGF-1R and FoxO1 was also demonstrated in SK-BR-3 cells, a breast cancer cell line characterized by relatively low IGF-1R expression. Taken together these results provide mechanistic evidence to support the conclusion that IGF-1R expression is a critical determinant of FoxO1 and the IGFBP-1-dependent Tam response in breast cancer cells.
FoxO1, a member of the FoxO transcription factor family, regulates many cellular events such as differentiation, growth, and metabolism. Post-translational modifications of FoxO1 via phosphorylation (22) and acetylation (48) regulate biological activities. Attenuated expression of FoxO1 via upregulation of microRNAs or hyperactivation of kinases downstream of growth factor signaling pathways is observed in many cancers and during development of resistance to chemotherapeutic agents, including tamoxifen (25, 49–51). In the TamR cells IGF-1R depleted MCF-7 cells, increased phosphorylation of ERK1/2 was observed. Future work will need to be completed to determine if the hyperactivaiton of ERK1/2 in breast cancer cells results in decreased FoxO1 stability and the observed FoxO1 protein levels. FoxO1 regulates IGFBP-1 expression in hepatic cells (18, 20), and our data showed that IGFBP-1 expression was regulated by FoxO1 in breast cancer cells. In addition to the modulation of IGFBP-1 gene expression and inhibition of IGF-1 dependent, Tam treatment also induced FoxO1 transcription in MCF-7 cells. The current results suggest that FoxO1 is a key mediator of tamoxifen action and diminished FoxO1 expression attenuates the effectiveness of this SERM.
The data presented here also suggest that CREB and FoxO1 cooperate to induce IGFBP-1 transcription in Tam-treated breast cancer cells. Cooperation between CREB and FoxO1 has been observed in other cell types (52), but not in breast cancer cells. cAMP-dependent activation of CREB induces FoxO1 transcription in hepatocytes (53) and this could be one possible mechanism of cooperation. Tam-stimulated GPER1 signaling may activate CREB leading to increased FoxO1 transcription. Future studies will be aimed at defining the role of FoxO1 induction during Tam treatment in breast cancer cells. The IGFBP-1 promoter contains both CREB and FoxO1-binding sites (20, 54, 55) and the current results indicated that CREB phosphorylation in TamR cells is constitutive and not induced by Tam. In MCF-7 cells, CREB is phosphorylated after Tam treatment and CREB mediates IGFBP-1 transcription in these cells (6). In the current studies, p-CREB was not sufficient to induce IGFBP-1 in TamR cells and we conclude that this is due to the lack of FoxO1 expression. Additionally, the ability of Tam to induce p-CREB accumulation was reduced in MCF-7 cells after FoxO1 knockdown. These data suggest that FoxO1 expression is required for CREB to induce IGFBP-1 transcription and more work will need to be completed to identify the molecular mechanisms of IGFBP-1 co-modulation by FoxO1 and CREB in Tam treated breast cancer cells.
IGF-dependent signaling plays a critical role in breast carcinogenesis (8, 56, 57), however, low IGF-1R expression results in poor prognosis for postmenopausal breast cancer patients treated with tamoxifen (14). Additionally, loss of IGF-1R expression is associated with the development of acquired tamoxifen-resistance in breast cancer cells and patients (8, 56, 57). With regard to the clinical significance of ERα-phosphorylation and tamoxifen sensitivity, conflicting evidence has been published. Whereas higher levels of p-ERα (S118) are associated with tamoxifen resistance in one study (58), another report shows an enhanced tamoxifen benefit in patients expressing higher levels of pERα (S118) (59). Both hyperactivation of ERK1/2 in MCF-7 cells after IGF-1R knockdown and the dysregulation of ERα phosphorylation in TamR cells are reported in the current study. Taken together, these findings provide preliminary evidence for previously unidentified mechanisms of decreased FoxO1 expression in breast cancer cells. More work will need to be completed to address the relative contribution of these distinct mechanisms of FoxO1 regulation in breast cancer cells.
The involvement of GPER1 in E2-induced rapid cell signaling has previously been reported (32, 60) and GPER1 dysfunction is observed in Tam-resistant breast cancer cells (33). Selective activation of GPER1 induces p21 expression (61) supporting a potential GPER1-mediated mechanism for observed E2-induced p21 expression in MCF-7 cells. The current results indicate that either Tam or E2 treatment induce IGFBP-1 transcription in MCF-7 cells. In summary, these results provide a mechanistic understanding for the observation that loss of IGF-1R expression decreases tamoxifen sensitivity resulting from reduced FoxO1 expression in breast cancer cells.
Supplementary Material
Implications.
Loss of IGF-1R expression is associated with decreased tamoxifen efficacy in breast cancer patients and the development of tamoxifen resistance. This contribution identifies potential molecular mechanisms of altered tamoxifen sensitivity in breast cancer cells resulting from decreased IGF-1R expression.
Acknowledgments
The authors extend gratitude to Amanda K. Ashley, Ph.D. for critical evaluation of this manuscript.
Grant Support
This work was supported by the NM-INBRE grant from the National Institutes of Health (8 P20 GM103451).
Footnotes
Disclosure statement: The authors have nothing to disclose.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: A. Vaziri-Gohar, K.D. Houston
Development of methodology: A. Vaziri-Gohar, K.D. Houston
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A. Vaziri-Gohar, Y. Zheng
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A. Vaziri-Gohar, K.D. Houston
Writing, review, and/or revision of the manuscript: A. Vaziri-Gohar, K.D. Houston
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): A. Vaziri-Gohar, K.D. Houston
Study supervision: K.D. Houston
References
- 1.Jemal A, Siegel R, Ward E, Hao YP, Xu JQ, Thun MJ. Cancer Statistics, 2009. Ca-a Cancer Journal for Clinicians. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacological Reviews. 2001;53(1):25–71. [PubMed] [Google Scholar]
- 3.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocrine-Related Cancer. 2004;11(4):643–658. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 4.Vivacqua A, Romeo E, De Marco P, De Francesco EM, Abonante S, Maggiolini M. GPER mediates the Egr-1 expression induced by 17 beta-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Research and Treatment. 2012;133(3):1025–1035. doi: 10.1007/s10549-011-1901-8. [DOI] [PubMed] [Google Scholar]
- 5.Catalano S, Giordano C, Panza S, Chemi F, Bonofiglio D, Lanzino M, et al. Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Research and Treatment. 2014;146(2):273–285. doi: 10.1007/s10549-014-3017-4. [DOI] [PubMed] [Google Scholar]
- 6.Vaziri-Gohar A, Houston KD. GPER1-mediated IGFBP-1 induction modulates IGF-1-dependent signaling in tamoxifen-treated breast cancer cells. Mol Cell Endocrinol. 2016;422:160–171. doi: 10.1016/j.mce.2015.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgaud JL, Resnicoff M, Baserga R. MUTANT IGF-I RECEPTORS AS DOMINANT NEGATIVES FOR GROWTH AND TRANSFORMATION. Biochemical and Biophysical Research Communications. 1995;214(2):475–481. doi: 10.1006/bbrc.1995.2311. [DOI] [PubMed] [Google Scholar]
- 8.Cullen KJ, Yee D, Sly WS, Perdue J, Hampton B, Lippman ME, et al. INSULIN-LIKE GROWTH-FACTOR RECEPTOR EXPRESSION AND FUNCTION IN HUMAN-BREAST CANCER. Cancer Research. 1990;50(1):48–53. [PubMed] [Google Scholar]
- 9.Lee AV, Yee D. Insulin-like growth factors and breast cancer. Biomedicine & Pharmacotherapy. 1995;49(9):415–421. doi: 10.1016/0753-3322(96)82678-3. [DOI] [PubMed] [Google Scholar]
- 10.Sachdev D, Yee D. The IGF system and breast cancer. Endocr Relat Cancer. 2001;8(3):197–209. doi: 10.1677/erc.0.0080197. [DOI] [PubMed] [Google Scholar]
- 11.Zeng X, Yee D. Insulin-like growth factors and breast cancer therapy. Breast Cancer Chemosensitivity. 2007;608:101–112. doi: 10.1007/978-0-387-74039-3_7. [DOI] [PubMed] [Google Scholar]
- 12.Yee D, Jackson JG, Kozelsky TW, Figueroa JA. INSULIN-LIKE GROWTH-FACTOR BINDING PROTEIN-1 EXPRESSION INHIBITS INSULIN-LIKE GROWTH FACTOR-I ACTION IN MCF-7 BREAST-CANCER CELLS. Cell Growth & Differentiation. 1994;5(1):73–77. [PubMed] [Google Scholar]
- 13.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocrine Reviews. 2002;23(6):824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 14.Aaltonen KE, Rosendahl AH, Olsson H, Malmstrom P, Hartman L, Ferno M. Association between insulin-like growth factor-1 receptor (IGF1R) negativity and poor prognosis in a cohort of women with primary breast cancer. Bmc Cancer. 2014;14 doi: 10.1186/1471-2407-14-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frogne T, Jepsen JS, Larsen SS, Fog CK, Brockdorff BL, Lykkesfeldt AE. Antiestrogen-resistant human breast cancer cells require activated Protein Kinase B/Akt for growth. Endocrine-Related Cancer. 2005;12(3):599–614. doi: 10.1677/erc.1.00946. [DOI] [PubMed] [Google Scholar]
- 16.Drury SC, Detre S, Leary A, Salter J, Reis-Filho J, Barbashina V, et al. Changes in breast cancer biomarkers in the IGF1R/PI3K pathway in recurrent breast cancer after tamoxifen treatment. Endocrine-Related Cancer. 2011;18(5):565–577. doi: 10.1530/ERC-10-0046. [DOI] [PubMed] [Google Scholar]
- 17.Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res. 2012;72(13):3372–3380. doi: 10.1158/0008-5472.CAN-12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall RK, Yamasaki T, Kucera T, Waltner-Law M, O'Brien R, Granner DK. Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein-1 gene expression by insulin - The role of winged helix/forkhead proteins. Journal of Biological Chemistry. 2000;275(39):30169–30175. doi: 10.1074/jbc.M004898200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang WW, Patil S, Chauhan B, Guo SD, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver - Effects on gluconeogenic, glycolytic, and lipogenic gene expression. Journal of Biological Chemistry. 2006;281(15):10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 20.Durham SK, Suwanichkul A, Scheimann AO, Yee D, Jackson JG, Barr FB, et al. FKHR binds the insulin response element in the insulin-like growth factor binding protein-1 promoter. Endocrinology. 1999;140(7):3140–3146. doi: 10.1210/endo.140.7.6856. [DOI] [PubMed] [Google Scholar]
- 21.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 22.Biggs WH, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96(13):7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H, Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochimica Et Biophysica Acta-Molecular Cell Research. 2011;1813(11):1961–1944. doi: 10.1016/j.bbamcr.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cellular Signalling. 2007;19(3):519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 25.Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284(35):23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coser KR, Wittner BS, Rosenthal NF, Collins SC, Melas A, Smith SL, et al. Antiestrogen-resistant subclones of MCF-7 human breast cancer cells are derived from a common monoclonal drug-resistant progenitor. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(34):14536–14541. doi: 10.1073/pnas.0907560106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Entingh-Pearsall A, Kahn CR. Differential roles of the insulin and insulin-like growth factor-I (IGF-I) receptors in response to insulin and IGF-I. Journal of Biological Chemistry. 2004;279(36):38016–38024. doi: 10.1074/jbc.M313201200. [DOI] [PubMed] [Google Scholar]
- 28.Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. Journal of Biological Chemistry. 1999;274(24):16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 29.Xie Y. Regulation of insulin-like growth factor signaling by metformin in endometrial cancer cells. ONCOLOGY LETTERS. 2014;8:1993–1999. doi: 10.3892/ol.2014.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, Gui LS, Zhao XL, Zhu LL, Li QW. FOXO1 is a tumor suppressor in cervical cancer. Genetics and Molecular Research. 2015;14(2):6605–6616. doi: 10.4238/2015.June.18.3. [DOI] [PubMed] [Google Scholar]
- 31.de Jonge HJM, Fehrmann RSN, de Bont E, Hofstra RMW, Gerbens F, Kamps WA, et al. Evidence Based Selection of Housekeeping Genes. Plos One. 2007;2(9) doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 33.Mo ZQ, Liu MR, Yang FF, Luo HJ, Li ZH, Tu G, et al. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Research. 2013;15(6) doi: 10.1186/bcr3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu YH, Zi XL, Zhao YH, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin) Journal of the National Cancer Institute. 2001;93(24):1852–1857. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 35.Hartog H, Van Der Graaf WT, Boezen HM, Wesseling J. Treatment of breast cancer cells by IGF1R tyrosine kinase inhibitor combined with conventional systemic drugs. Anticancer Res. 2012;32(4):1309–1318. [PubMed] [Google Scholar]
- 36.Girnita A, Girnita L, del Prete F, Bartolazzi A, Larsson O, Axelson M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004;64(1):236–242. doi: 10.1158/0008-5472.can-03-2522. [DOI] [PubMed] [Google Scholar]
- 37.Vasilcanu D, Girnita A, Girnita L, Vasilcanu R, Axelson M, Larsson O. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene. 2004;23(47):7854–7862. doi: 10.1038/sj.onc.1208065. [DOI] [PubMed] [Google Scholar]
- 38.Fagan D, Yee D. Crosstalk Between IGF1R and Estrogen Receptor Signaling in Breast Cancer. Journal of Mammary Gland Biology and Neoplasia. 2008;13(4):423–429. doi: 10.1007/s10911-008-9098-0. [DOI] [PubMed] [Google Scholar]
- 39.Thomas NB, Hutcheson IR, Campbell L, Gee J, Taylor KM, Nicholson RI, et al. Growth of hormone-dependent MCF-7 breast cancer cells is promoted by constitutive caveolin-1 whose expression is lost in an EGF-R-mediated manner during development of tamoxifen resistance. Breast Cancer Res Treat. 2010;119(3):575–591. doi: 10.1007/s10549-009-0355-8. [DOI] [PubMed] [Google Scholar]
- 40.Huynh H, Nickerson T, Pollak M, Yang XF. Regulation of insulin-like growth factor I receptor expression by the pure antiestrogen ICI 182780. Clinical Cancer Research. 1996;2(12):2037–2042. [PubMed] [Google Scholar]
- 41.Lee AV, Weng CN, Jackson JG, Yee D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. Journal of Endocrinology. 1997;152(1):39–47. doi: 10.1677/joe.0.1520039. [DOI] [PubMed] [Google Scholar]
- 42.Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF Pathway Regulates ER alpha through a S6K1-Dependent Mechanism in Breast Cancer Cells. Molecular Endocrinology. 2011;25(3):516–528. doi: 10.1210/me.2010-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyo S, Sakaguchi J, Kiyono T, Shimizu Y, Maida Y, Mizumoto Y, et al. Forkhead Transcription Factor FOXO1 is a Direct Target of Progestin to Inhibit Endometrial Epithelial Cell Growth. Clinical Cancer Research. 2011;17(3):525–537. doi: 10.1158/1078-0432.CCR-10-1287. [DOI] [PubMed] [Google Scholar]
- 44.Diep CH, Charles NJ, Gilks CB, Kalloger SE, Argenta PA, Lange CA. Progesterone receptors induce FOXO1-dependent senescence in ovarian cancer cells. Cell Cycle. 2013;12(9):1433–1449. doi: 10.4161/cc.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura M, Takakura M, Fujii R, Maida Y, Bono Y, Mizumoto Y, et al. The PRB-dependent FOXO1/IGFBP-1 axis is essential for progestin to inhibit endometrial epithelial growth. Cancer Letters. 2013;336(1):68–75. doi: 10.1016/j.canlet.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 46.N PL, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143(12):4583–4591. doi: 10.1210/en.2002-220369. [DOI] [PubMed] [Google Scholar]
- 47.Mandal S, Davie JR. Estrogen regulated expression of the p21 Waf1/Cip1 gene in estrogen receptor positive human breast cancer cells. Journal of Cellular Physiology. 2010;224(1):28–32. doi: 10.1002/jcp.22078. [DOI] [PubMed] [Google Scholar]
- 48.Gan LX, Han YS, Bastianetto S, Dumont Y, Unterman TG, Quirion R. FoxO-dependent and -independent mechanisms mediate SirT1 effects on IGFBP-1 gene expression. Biochemical and Biophysical Research Communications. 2005;337(4):1092–1096. doi: 10.1016/j.bbrc.2005.09.169. [DOI] [PubMed] [Google Scholar]
- 49.Shang X, Wu Y, Sarkissyan M, Koeffler HP, Vadgama JV. Overexpression of FOXO1 increased sensitivity to Tamoxifen in MCF7 cells, and is associated with estrogen receptor α (ERα) expression. 2010 p 8 Suppl. [Google Scholar]
- 50.Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, et al. MicroRNA-221/222 Confers Tamoxifen Resistance in Breast Cancer by Targeting p27Kip1. Journal of Biological Chemistry. 2008;283(44):29897–29903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Washington E, Sarkissyan M, Sarkissyan S, We Y, Vadgama J. Involvment of FOXO1 in E2/ER pathway and tamoxifen resistance in MCF-7 cells. Miami, FL: American Association for Cancer Research; 2010. [Google Scholar]
- 52.Oh KJ, Han HS, Kim MJ, Koo SH. CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. Bmb Reports. 2013;46(12):567–574. doi: 10.5483/BMBRep.2013.46.12.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wondisford AR, Xiong L, Chang E, Meng S, Meyers DJ, Li M, et al. Control of Foxo1 Gene Expression by Co-activator P300. Journal of Biological Chemistry. 2014;289(7):4326–4233. doi: 10.1074/jbc.M113.540500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugawara J, Tazuke SI, F-Suen L, Powell DR, Kaper F, Giaccia AJ, et al. Regulation of insulin-like growth factor-binding protein 1 by hypoxia and 3 ',5 '-cyclic adenosine monophosphate is additive in HepG2 cells. Journal of Clinical Endocrinology & Metabolism. 2000;85(10):3821–3827. doi: 10.1210/jcem.85.10.6866. [DOI] [PubMed] [Google Scholar]
- 55.Shin D-J, Joshi P, Hong S-H, Mosure K, Shin D-G, Osborne TF. Genome-wide analysis of FoxO1 binding in hepatic chromatin: Potential involvement of FoxO1 in linking retinoid signaling to hepatic gluconeogenesis. Nucleic Acids Research. 2012;40(22):11499–11509. doi: 10.1093/nar/gks932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arteaga CL. INTERFERENCE OF THE IGF SYSTEM AS A STRATEGY TO INHIBIT BREAST-CANCER GROWTH. Breast Cancer Research and Treatment. 1992;22(1):101–106. doi: 10.1007/BF01833338. [DOI] [PubMed] [Google Scholar]
- 57.Yu H, Rohan T. Role of the Insulin-Like Growth Factor Family in Cancer Development and Progression. Journal of National Cancer Institute. 2000;92(18):1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 58.Chen M, Cui Y-K, Huang W-H, Man K, Zhang G-J. Phosphorylation of estrogen receptor α at serine 118 is correlated with breast cancer resistance to tamoxifen. Oncol Letters. 2013;6(1):118–124. doi: 10.3892/ol.2013.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kok M, Holm-Wigerup C, Hauptmann M, Michalides R, Stål O, Linn S, et al. Estrogen receptor-alpha phosphorylation at serine-118 and tamoxifen response in breast cancer. J Natl Cancer Inst. 2009;101(24):1725–1729. doi: 10.1093/jnci/djp412. [DOI] [PubMed] [Google Scholar]
- 60.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GrPR30. Annual Review of Physiology. Volume 70, Annual Review of Physiology. 2008:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 61.Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010;70(3):1184–1194. doi: 10.1158/0008-5472.CAN-09-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.