

Abstract
Helicobacter pylori, the dominant member of the human gastric microbiota, elicits immunoregulatory responses implicated in protective versus pathological outcomes. To evaluate the role of macrophages during infection, we employed a system with a shifted pro-inflammatory macrophage phenotype by deleting PPARγ in myeloid cells, and found a 5 to 10-fold decrease in gastric bacterial loads. Higher levels of colonization in wild-type (WT) mice were associated with increased presence of mononuclear phagocytes (MNPs) and in particular with the accumulation of CD11b+F4/80hiCD64+CX3CR1+ macrophages in the gastric lamina propria. Depletion of phagocytic cells by clodronate liposomes in WT resulted in a reduction of gastric H. pylori colonization compared to non-treated mice. PPARγ-deficient and macrophage-depleted mice presented decreased IL-10-mediated myeloid and T cell regulatory responses early post-infection. IL-10 neutralization during H. pylori infection led to increased IL-17-mediated responses and increased neutrophil accumulation at the gastric mucosa. In conclusion, we report the induction of IL-10-driven regulatory responses mediated by CD11b+F4/80hiCD64+CX3CR1+ MNPs that contribute to maintaining high levels of H pylori loads in the stomach by modulating effector T cell responses at the gastric mucosa.
Introduction
Helicobacter pylori is a microaerophilic, Gram-negative, spiral-shaped bacterium that selectively establishes lifelong colonization of the gastric mucosa in over 50% of the human population (1, 2). Approximately 10–15% of H. pylori-infected individuals will eventually develop gastro-duodenal ulcers and H. pylori carriers have over a two-fold greater risk of developing gastric cancer in the form of B-cell lymphoma of mucosal-associated lymphoid tissue (MALT) lymphoma or adenocarcinoma (3, 4). In spite of the reported links between gut pathologies and H. pylori, its role as a beneficial and dominant member of the human gastric microbiota is emerging through epidemiological, clinical and experimental data illustrating that it might actually protect from esophageal cancer, asthma, obesity-induced insulin resistance and inflammatory bowel disease (IBD) (5–10). This dual role of H. pylori as commensal and pathogenic organism denotes a complex, context-dependent interaction with its host and provides a means to tracking the induction of mucosal effector or regulatory responses to a single organism.
H. pylori is mainly found free-floating on the thick mucus layer of the stomach or on the apical side of epithelial cells. However, a small fraction of the H. pylori population can invade the lamina propria (LP) following disruption of tight junctions. While effector immune mechanisms of elimination have not been dissected in depth, the type of immune response elicited may depend on the location and kind of cell that first comes in contact with H. pylori. Colonization of the gastric mucosa by H. pylori induces mixed effector and regulatory immune responses (11). However, its chronic persistence in the host suggests that the regulatory immune responses might predominate over effector mechanisms (12–20). Computational modeling of immune responses to H. pylori predicted that macrophages are central regulators of the mucosal immune response (21–23). Interestingly, in line with our computational prediction, the loss of protein-activated receptor 1, MMP7 or hemoxygenase results in lower rates of colonization, more severe pathology and changes in macrophages towards a pro-inflammatory or classically activated state (24–26). Thus, macrophages could be critical in tipping the balance between pro-inflammatory/effector and regulatory responses and significantly affect the outcome of this bacteria-host interaction. Macrophages and dendritic cells (DC) belong to the mononuclear phagocytic compartment, which comprises a heterogeneous class of cells that perform functions ranging from tissue development, remodeling and repair, to pathogen recognition and initiation of inflammation and antigen-specific immune responses (27, 28). Functional characterization based on phenotypic traits and the establishment of a clear division of labor among mononuclear phagocytes (MNPs) has been challenging because these cells arise from common progenitors and undergo radical reprogramming in the presence of danger signals (29–31). Here, we show that during H. pylori infection phagocytic cells promote high H. pylori loads rather than contributing to bacterial clearance. However, disruption of MNPs phenotype through either genetic ablation of PPARγ or depletion via clodronate liposomes results in more efficient bacterial elimination although not complete clearance. By performing a detailed immunological profiling of MNPs in the stomach of H. pylori-infected mice, we have identified and traced a subset of CD11b+F4/80hiCD64+CX3CR1+ macrophages that begin to accumulate in the gastric LP between days 21 and 24 post-infection in WT but not in mice lacking PPARγ in myeloid cells. These cells produce IL-10 and thus could be responsible for establishing a microenvironment that facilitates H. pylori colonization of the stomach. We also show that IL-10 deficiency leads to low colonization and significant infiltration by neutrophils. Our studies demonstrate the presence of a very complex system of MNPs associated with the gastric mucosa that is predominantly regulatory and highly susceptible to modulation by environmental changes. Furthermore, the state of the gastric MNP system can also impact the microbial composition by facilitating colonization by certain bacterial species, like H. pylori. Moreover, we have identified a novel MNP subset that could provide new insights on the mechanisms of mucosal immunoregulation underlying the protective versus pathogenic behavior of gastrointestinal bacteria.
Methods
Mouse strains and infection
C57BL/6J wild type (WT) (fl/fl, cre-) and mice lacking PPARγ in T cells (PPARγfl/fl; CD4-Cre+) (32) or in myeloid cells (PPARγfl/fl; Lysozyme M Cre-) (33) were used in this study. CX3CR1-GFP+/+ reporter and IL-10−/− mice were obtained from Jackson and bred in our mouse facilities for 6 months and 2 years, respectively. WT, CD4-cre and LysMcre mice used in these experiments originated from a colony kept for 10 years at Virginia Tech’s animal facilities. All mice used in these experiments were bred and maintained in the same colony. Mice were kept in the same room for breeding/maintenance under ABSL1 conditions and in a separate room under ABSL2 conditions for H. pylori challenge studies. For H. pylori infection, mice were challenged after a 6-hour fasting period with freshly prepared 5×107 colony forming units (CFU) of strain SS1 given in sterile PBS through orogastric gavage on days 0 and 2. A non-infected group that received sterile PBS without any bacteria was included for each genotype.
Helicobacter pylori culture and inoculum preparation
The European mouse-adapted CagA positive strain H. pylori SS1 (kindly provided by Dr. Richard Peek, Vanderbilt University) was used in this study. H. pylori was grown on plates prepared with Difco Columbia agar base (BD Biosciences) supplemented with 7% of lacked horse blood (Lampire) and Helicobacter pylori selective supplement (containing 10 mg/L vancomycin, 5 mg/L trimethoprim, 5 mg/L amphotericin, and 5 mg/L polymyxin from Oxoid) at 37°C under microaerophilic conditions. The challenge inoculum was prepared by harvesting bacteria into sterile 1X PBS and adjusting to Optical Density (OD)=1.0 at 600nm which was estimated as a concentration of 1×108 CFU/ml as previously determined by a growth curve correlating OD measurements with colony counts on blood agar plates.
Bacterial reisolation from murine gastric tissue
Stomachs were opened along the large curvature, rinsed in sterile 1X PBS and total CFU were determined by plate counting (21). Briefly, weighted gastric specimens were homogenized in Brucella broth using a grinder. Homogenates and serial dilutions (1:10, 1:100, 1:1,000 and 1:10,000) were plated onto Difco Columbia agar base plates supplemented with 7% of lacked horse blood and H. pylori selective antibiotic supplement (containing 10 mg/L vancomycin, 5 mg/L trimethoprim, 5 mg/L amphotericin, and 5 mg/L polymyxin). Plates were incubated for 4 days at 37°C under microaerophilic conditions (21, 34). Bacterial numbers are reported as the mean and SD of the number of CFU/g of stomach tissue.
Isolation of lymphocytes from gastric lamina propria and lymph nodes
Isolation of lymphocytes from the stomach was performed by digestion with collagenase (300 U/ml) and DNaseI (50 U/ml) in RPMI. Cells were further purified by centrifugation in discontinuous Percoll gradient (44%/67%), washed and resuspended in cRPMI. Cells from gastric lymph nodes (GLN) were isolated by digestion in collagenase (300 U/ml) and DNaseI (50 U/ml) in RPMI. Cells were then washed and resuspended in cRPMI.
Cytometric Bead Array
Cell suspensions from GLN were seeded at 1×106/ml in 96-well plates and stimulated with 5 μg/ml of formalin-inactivated H. pylori SS1 antigen. Cultures were incubated for 72h at 37°C, 95% humidity and 5% CO2. Cytokines in supernatants were measured with the Th1, Th2, and Th17 CBA kit (BD Biosciences) following manufacturer’s instructions.
Flow cytometry
Cells (3×105–5×105/well) were fist incubated in FcBlock (BD Pharmingen) and then with cocktails of antibodies using CD45-APCCy7 (BD Pharmingen, 30-F11), F4/80-PECy5 (eBioscience, BM8), CD11b-AlexaFluor700 (BD Pharmingen, M1/70), MHCII-biotin (eBiosciences, M5/114.15.2) followed by streptavidin-PETexasRed (BD Pharmingen), CD64-PE (BD Pharmingen, X54-5/7.1.1), CX3CR1-unconjugated (AbD Serotec, polyclonal) followed by anti-IgG(H+L)-FITC (Southern Biotech), and anti-IL10-APC (eBiosciences, JES5-16E3), CD3-PECy5 (eBiosciences, 145-2C11), CD4-PECy7 (eBiosciences, GK1.5), CD19-PE (eBiosciences, MB19-1), FoxP3-PE (eBiosciences, FJK-16s), and PD1-PE (eBiosciences, J43). For intracellular staining, cells were fixed and permeabilized with Cytofix-Cytoperm solution (eBiosciences). Flow results were computed with a BD LSR II flow cytometer and data analyses was performed by using the FACS Diva software (BD).
Macrophage depletion
Macrophages were depleted by intraperitoneal injection of clodronate-containing liposomes (Anionic Clophosome, FormulaMax) 11 days after H. pylori infection (correlating with the observed upregulation of macrophages in the gastric lamina propria after infection) following the manufacturer’s instructions. Control mice received clodronate-free liposomes on the same days.
BrdU administration in vivo
WT mice were injected IP with 1mg of BrdU (BD Biosciences) followed by 3 days of 0.8mg/mL BrdU (Sigma) in drinking water. Following stomach collection and isolation of gastric LP leukocytes, BrdU uptake was assessed using BD BrdU Flow kit (BD Bioscience).
IL-10 neutralization
WT mice received 100μg of rat anti-IL-10 antibody (R&D systems) intraperitoneally in PBS on days 17, 19 and 21 post H. pylori infection. Control mice received 100μg of Rat IgG1 isotype control (R&D systems).
Statistics
Data are expressed as mean and standard error of the mean. Parametric data were analyzed by using the Analysis of Variance (ANOVA) followed by Scheffe’s multiple comparison test as previously described (35). Analysis of variance (ANOVA) was performed by using the general linear model procedure of SAS, release 9.2 (SAS Institute Inc., Cary, NC). A 2×2 factorial arrangement comparing genotype and infection treatment was employed. Statistical significance was determined at P ≤ 0.05.
Study approval
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Tech and met or exceeded requirements of the Public Health Service/National Institutes of Health and the Animal Welfare Act. The IACUC approval ID for the study was 12-074-VBI and 15-147-VBI.
Results
Loss of PPARγ in myeloid cells results in low H. pylori colonization and changes in the macrophage compartment
To determine the impact of macrophages on the outcome of H. pylori infection, we used a cre-lox tissue-specific peroxisome proliferator activated receptor γ (PPARγ) mouse with deletion targeted to myeloid cells (PPARγfl/fl; LysMcre+). These mice have been used extensively and characterized in models of IBD (35–37), with the deletion affecting mainly macrophages and partially dendritic cells (DC) (38). While we did not aim to elucidate the role of PPARγ during H. pylori infection per se, we selected this regulatory transcription factor because it down-regulates pro-inflammatory cytokine expression (38, 39). Prior to our work, others had evaluated the role of specific macrophage genes (i.e., MMP7, PAR-1, HO-1) whose deficiency leads to enhanced inflammation and pathology following H. pylori infection in mice (40–42). Wild-type (WT), PPARγfl/fl;CD4-cre (CD4cre) and PPARγfl/fl;LysM-cre (LysMcre) mice in a C57BL/6J background were infected with H. pylori strain SS1 and gastric bacterial loads were measured weekly for 6 months (Figure 1A). All mice were colonized to similar levels based on re-isolation data from weeks 1 and 2 post-infection. However, between weeks 2 and 3, there was an abrupt drop in bacterial burden in the stomachs of LysMcre mice that led to a significant and sustained 5 to 10- fold lower colonization levels when compared to WT and CD4-cre mice. Similar results were obtained when mice were infected with H. pylori strain PMSS1 (Figure S1). We performed a detailed profiling of myeloid cells present in the gastric LP using a broad selection of MNPs markers (31, 43–45), including CD11b, CD11c, MHC-II, CX3CR1, F4/80, CD103 and CD64, which revealed substantial alterations due to the loss of PPARγ in the myeloid compartment. The stomach mucosa of WT mice was enriched in a population of F4/80+CD11b+ myeloid cells, as opposed to LysMcre mice (Figure 1C). Within these F4/80+CD11b+ cells, we characterized two gastric mucosal subsets based on the level of expression of F4/80: an F4/80hi subset corresponding to macrophages based on the expression of CD64, and an F4/80lo. The percentage of F4/80hi, corresponding to macrophages, was suppressed in mice lacking PPARγ in myeloid cells (Figure 1B). In addition, within the CD64-MHC-II+ fraction, we characterized two major DC subsets based on CD11c and CD103 expression: CD11c+ CD103+, and CD11c+CD103−, which constituted the majority of DC. Of note, while the CD11c+CD103+ were negative in CD11b and F4/80, the CD11c+CD103− had heterogeneous expression of F4/80 and CD11b, although F4/80 was always expressed in low levels (Figure S2), in contrast to the CD64+ macrophages, which expressed high levels of F4/80. On the other hand, neutrophils could be easily identified as CD11bhi cells that expressed also high levels of GR1. This phenotypic analysis showed significant differences between WT and LysMcre mice with regards to the percentage of F4/80+CD11b+ cells, which were significantly higher in WT. In particular, PPARγ deletion affected the proportion of macrophages in the gastric mucosa (Figure 1B). We further characterized the myeloid compartment in the stomach mucosa of naïve mice using CX3CR1-GFP+/+ reporter mice (Figure 1D). We show that the F4/80hiCD11b+CD64+ cells were all in the CX3CR1+ fraction, while the CD11b+F4/80lo, including CD11c+MHCII+CD64− DC could be either CX3CR1+ or CX3CR1−. These analyses demonstrate the presence of a complex system of myeloid cells in the stomach mucosa that has not been previously described, and its composition is significantly altered due to the loss of PPARγ in LysMcre mice.
Figure 1. Loss of peroxisome proliferator-activated receptor γ (PPARγ) in myeloid cells results in lower colonization with Helicobacter pylori and altered myeloid compartment.
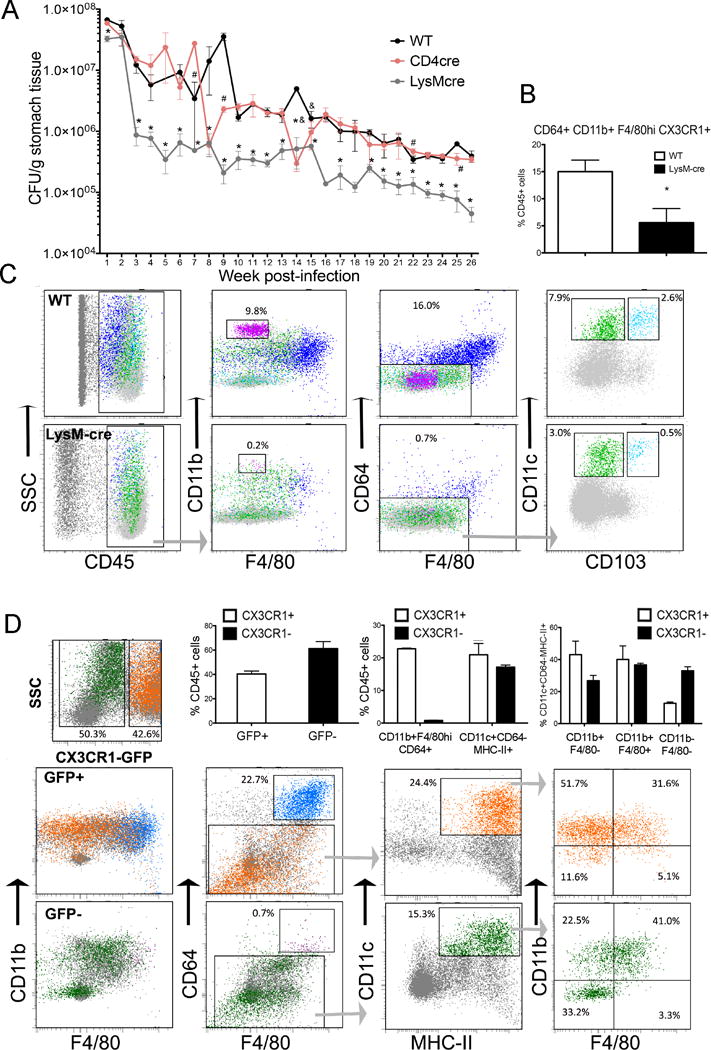
(A) WT, PPARγ-myeloid cell deficient (LysMcre) or PPARγ-T cell deficient (CD4cre) mice were infected with H. pylori SS1. Bacterial burden measured weekly up to 6 months post-infection show a significant reduction in colonization due to the loss of PPARγ in myeloid cells. Data represents mean ± SEM (n=10). (B) Loss of PPARγ in myeloid cells results in significantly lower percentage of CD64+CD11b+F4/80hiCX3CR1+ macrophages. Data represents mean ± SEM (n=5). (C) Phenotypic analysis of gastric myeloid cells in WT and LysMcre shows a significant depletion in myeloid cells in LysMcre mice including CD11b+F4/80hiCD64+, neutrophils (CD11bhi) and DC. (D) Phenotypic analysis of myeloid cells in naïve CX3CR1-GFP+/+ reporter mice. Macrophages, defined by CD64 expression, were found to be CX3CR1+, DC were analyzed in the CD64− fraction, after negative gating of CD3 and CD19 expression, and defined based on MHCII, CD11c and CD103 expression. Top panel represents CX3CR1+ cells and bottom panel the CX3CR1− cells. Results from B, C and D are representative of 3 independent replicate experiments with same results. Points with an asterisk are significantly different when compared to the control (WT) group (P<0.05).
Macrophages accumulate in the gastric mucosa of H. pylori-infected mice
We performed a time-course study spanning the first 7 weeks post-infection since the drop in H. pylori loads in LysMcre mice consistently occurs between weeks 2 and 3 post-infection. The results show a significant increase in numbers of F4/80hiCD11b+CD64+CX3CR1+ cells in WT mice in comparison with LysMcre mice (Figure 2A, E). These cells accumulated in the stomach mucosa starting on day 14 post-infection in the WT but not in the LysMcre mice. We also found increased percentages of neutrophils on days 14 and 28 and total numbers on day 28 post-infection in WT mice (Figure 2B, F), as well as in the percentages of CD11c+CD103+ and CD11c+CD103− DC (gated within CD64−MHC-II+), which were increased in WT mice when compared to LysMcre (Figure 2C, D, G, H), although no differences were found in absolute numbers. Overall, our data indicate that the major sustained difference between strains was in the accumulation of F4/80hiCD11b+CD64+CX3CR1+ in the stomach of WT mice following infection. We then determined whether macrophages proliferate in situ in the gastric mucosa via Brdu staining. WT and LysMcre mice infected with H. pylori SS1 received 1mg of BrdU intraperitoneally followed by 0.8 mg/mL in drinking water for 3 days. Brdu was withdrawn and incorporation was measured on the same day, corresponding to day 15 post-infection, and on days 18 and 21 in cells isolated from the stomach. The results show that F4/80hiCD64+ cells, which correspond to F4/80hiCD11b+CD64+CX3CR1+, incorporated BrdU with a slightly higher increase in WT compared to LysMcre mice on day 18 (Figure 2I). We also detected positive BrdU staining in F4/80loCD64− cells, although only an average 15% was positive by day 21 (Figure 2J), whereas the percentages of F4/80hiCD64+ stained with BrdU remained higher in both WT and LysMcre. These results indicate that F4/80hiCD11b+CD64+CX3CR1+ cells can proliferate in situ in the gastric mucosa during H. pylori infection.
Figure 2. Helicobacter pylori infection causes accumulation of CD11b+F4/80hiCD64+CX3CR1+ macrophages in the gastric mucosa.
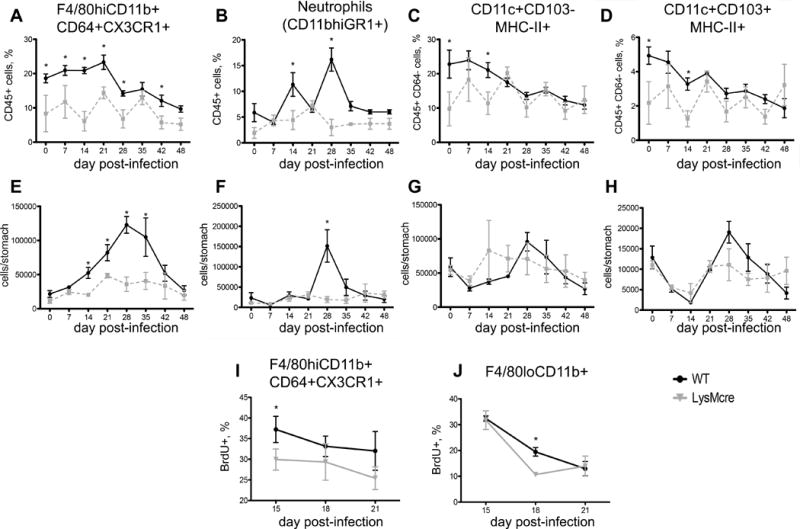
Time-course FACS analysis of gastric myeloid cells after H. pylori infection of WT and LysMcre mice. The analysis was performed at the indicated times in cells isolated from mouse stomachs. Plots represent percentages (top row) and absolute numbers (middle row) of CD11b+F4/80hiCD64+CX3CR1+ macrophages (A, E) neutrophils, (B, D) CD11c+CD103− (C, G) and CD11c+CD103+ DC (D, H) subsets. DC gating was done on MHCII+CD64− cells. Results are averages of 5 mice per time-point and are presented as mean ± SEM (n=5/time point). (I, J) BrdU incorporation was measured by flow cytometry in CD11b+F4/80hiCD64+CX3CR1+ and F4/80loCD11b+ cells isolated from the stomach of H. pylori infected mice on days 15, 18 and 21 post-infection. Points with an asterisk are significantly different when compared to the control group (P<0.05). Results are representative of 5 independent replicate experiments with same results. Points with an asterisk are significantly different when compared to the control (WT) group (P<0.05).
We also analyzed changes in the lymphocyte compartment due to infection in both WT and LysMcre strains. The results show higher percentages and numbers of CD4+ T cells, CD8+ T cells and B cells in PPARγ mice (Figure S3), suggesting that defects derived from the loss of PPARγ in myeloid cells of the stomach lead to suppressed numbers of F4/80hiCD11b+CD64+CX3CR1+ and secondary increase of lymphocytes in the gastric mucosa.
Phagocytic cells facilitate colonization of the gastric mucosa by H. pylori
In order to determine whether MNPs were required for the high colonization phenotype of WT mice, we depleted phagocytic cells by using clodronate liposomes. More specifically, WT mice were infected with H. pylori SS1 and treated with either PBS liposomes (negative control) or clodronate-containing liposomes. A group of non clodronate-treated, H. pylori SS1-infected LysMcre mice was included for comparison with a strain with low colonization phenotype. Liposomes were administered 4 days before macrophages started to accumulate in the stomach based on the initial time-course results. To maintain macrophage depletion, liposomes were administered every 3 days and changes in MNPs populations in the stomach were determined 24h after each injection (Figure 3A). Measurement of stomach bacterial burden during depletion clearly shows a progressive decline in H. pylori colonization to levels similar to LysMcre mice on day 21 post-infection (Figure 3B). Suppressed bacterial loads coincided with effective depletion of F4/80hiCD11b+CD64+CX3CR1+ macrophages, and were similar to those observed in LysMcre mice, on days 18 and 21 (Figure 3C).
Figure 3. Macrophage depletion reduces Helicobacter pylori loads and suppresses IL-10-mediated regulatory responses in the stomach of WT mice.
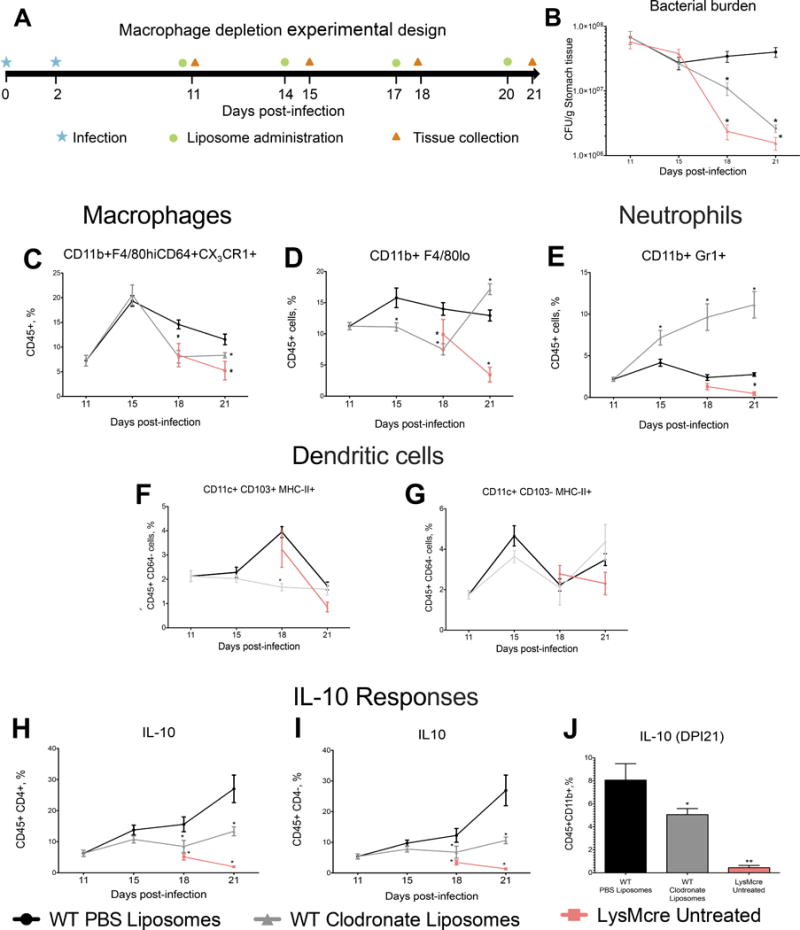
(A) WT mice were infected with SS1 and received three doses of either clodronate liposomes or PBS liposomes on days 11, 14, 17, and 20 post-infection. Analyses were performed prior to the first injection, day 11 (before treatment started), or one day after each injection. SS1-infected, non-treated LysMcre mice were used for reference. (B) Macrophage depletion suppressed bacterial loads to levels of untreated LysMcre mice. (C) Clodronate treatment depleted F4/80hiCD11b+CX3CR1+ and (D) F4/80hiCD11b+CD64+ cells from the stomach of WT mice. (E) Neutrophil levels ware significantly increased in clodronate-treated mice. (F, G) Differences were observed in CD11c+CD103+, but not on CD11c+CD103+ DC (gated on CD64−MHCII+ cells). IL-10 production was measured in CD4+ T cells (H), CD4− cells (I) during the time course study, and on (J) CD11b+ cell on day 18 post-infection. Data represents mean ± SEM (n=8). Points with an asterisk are significantly different when compared to the control group (P<0.05). Results are representative of 2 independent replicate experiments with same results. Points with an asterisk are significantly different when compared to the control (WT) group (P<0.05).
Phenotype based on results from our previous data. CD11b+F4/80lo cells were also affected by clodronate administration, although they quickly recovered on the last time-point measured despite the administration of clodronate one day prior to the analysis (Figure 3D). Interestingly, loss of MNP in the stomach mucosa during H. pylori infection resulted in a dramatic influx of neutrophils, which was not detected in either WT PBS-liposome controls or LysMcre mice (figure 3E). On the other hand, clodronate administration only had a small effect on the CD11c+CD103− but significantly affected CD11c+CD103+ DC on day 18 (Figures 3F, G).
In view of these results, we proposed that MNPs favor H. pylori colonization by promoting a mucosal regulatory microenvironment. We then assayed the presence of cells with regulatory phenotype and if they were affected by MNPs depletion. Flow cytometry results revealed a significant decrease in IL-10 production in both CD45+CD4+ T cells (Figure 3H) and CD45+CD4− cells (Figure 3I) in depleted mice. Because we found a significant reduction in IL-10 production in CD4− cells during depletion, we assessed IL-10 in CD11b+ cells from PBS-liposome, clodronate-treated and LysMcre mice on day 21 post-infection, and confirmed that the expression of this cytokine was significantly suppressed in MNPs-depleted and in LysMcre mice (Figure 3J).
H. pylori infection induces IL-10 production by gastric CD4+ T and B cells
WT and LysMcre mice were infected with H. pylori SS1 and stomachs were collected before infection (day 0) and on days 18 and 24 post-infection to better characterize and trace the source of H. pylori-induced IL-10. As expected, IL-10 production by F4/80loCD11b+ and F4/80hiCD11b+CD64+CX3CR1+ cell subsets were significantly increased in WT mice when compared to LysMcre mice (Figures 4A and 4B). In addition, IL-10 was constitutively expressed by these cells, since it was already detectable before infection, and H. pylori did not significantly augment the fraction of cells producing it in WT. In contrast, H. pylori infection suppressed IL-10 production in myeloid cells from LysMcre mice on a time-dependent manner (Figure 4B). A plausible scenario is that MNPs, as a key source of IL-10, promote the induction of T cells with a regulatory phenotype in the gastric mucosa. In fact, IL-10 expression by overall CD4+ T cells and CD19+ lymphocytes were higher in WT when compared to LysMcre mice (Figure 4C), particularly on day 24 post-infection, when we detected a sharp increase of IL-10 production by both cell types (Figure 4D and 4E). LysMcre mice showed higher levels of both CD4+ T cells and CD19+ B cells, as described before (Figure S3). Further phenotypic characterization of the CD4+ T cell compartment revealed increased CD4+FoxP3+ Treg and CD4+PD1+ Tr1-like cells due to infection. H. pylori increased FoxP3+ Treg cells in WT on day 24, while only a small difference was observed between genotypes with regards to PD1+ cells on day 18 post-infection. In any case, the fraction of IL-10-secreting cells was higher in WT when compared to LysMcre for both cell types, and the differences were more accentuated in the CD4+PD1+ subset (Figures 4F and 4G). These data suggest that the increase in IL-10-producing MNPs that follows colonization of the gastric mucosa by H. pylori, could condition the tissue environment to favor the induction of CD4+PD1+ Tr1-like and FoxP3+ Treg cell-mediated regulatory responses.
Figure 4. Phenotypic characterization of IL-10-producing cells during Helicobacter pylori infection.
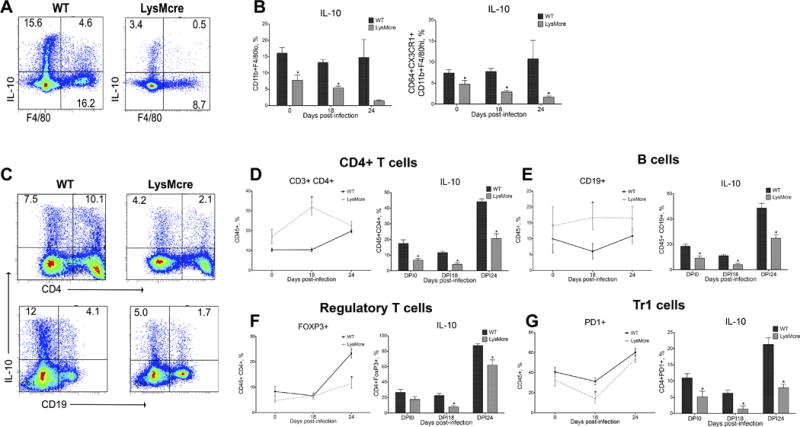
WT and LysMcre mice were infected with H. pylori SS1 strain. Stomachs were collected at days 0 (prior to infection), 18 and 24 post-infection. (A) Representative dot plots of IL-10 production by F8/40+ cells in WT and LysMcre mice. (B) Average IL-10 producing cells within CD11b+F4/80lo and CD11b+F4/80hiCD64+CX3CR1+ macrophages. (C) Representative plots of IL-10 production by CD4+ T cells and B cells from H. pylori SS1-infected WT and LysMcre mice. Average IL-10 production by (D) CD4 T cells, (E) B cells, (F) Foxp3+ regulatory CD4 T cells and (G) PD-1+ Treg cells. Average result data represent mean ± SEM (n=8). Points with an asterisk are significantly different when compared to the control group (P<0.05). Results are representative of 4 independent replicate experiments with same results. Points with an asterisk are significantly different when compared to the control (WT) group (P<0.05).
IL-10 is required for high levels of H. pylori colonization
To further evaluate the role of IL-10 during the initial phases of colonization, we performed IL-10 neutralization studies in mice infected with H. pylori and compared their response to control mice that received an isotype antibody. Mice received 3 doses of either control or neutralizing antibody (100 μg/mouse) on days 17, 19 and 21. Measurement of bacterial loads on day 22 post-infection showed that indeed IL-10 is required for optimal gastric colonization, since gastric H. pylori burden was significantly lower in the group of mice in which IL-10 was neutralized (Figure 5A). IL-10 blockade also significantly increased neutrophils (Figure 5C) in the gastric mucosa but did not affect the levels of F4/80hiCD11b+CD64+CX3CR1+ macrophages (Figure 5B), which indicates that IL-10 is dispensable for the accumulation of these cell type in the stomach. On the other hand, MHCII+CD11c+CD64− DCs were slightly but significantly suppressed by IL-10 depletion (Figure 5D). In addition to IL-10 neutralization, we infected IL-10−/− mice and obtained the same results: H. pylori loads were suppressed and neutrophil influx increased (Figure S4).
Figure 5. IL-10 neutralization during Helicobacter pylori infection.
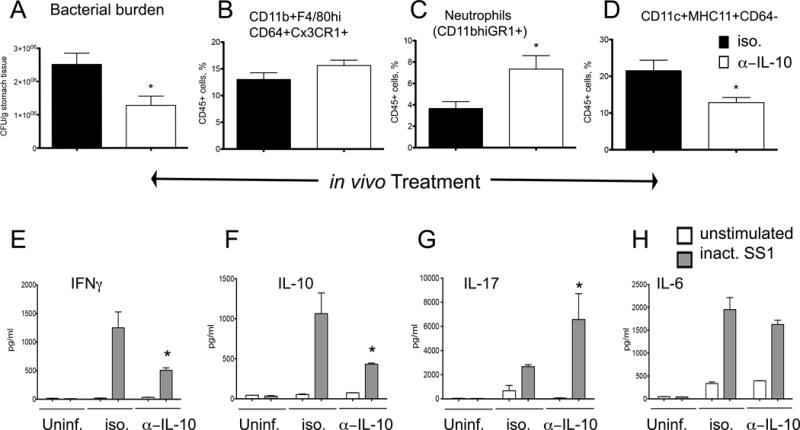
WT mice were infected with H. pylori and on days 17, 19 and 21 post-infection they were treated with 100 μg of either neutralizing anti-mouse IL-10 or Rat IgG1 isotype control antibodies. Mice (n=6), were euthanized on day 22 post-infection to measure (A) bacterial burden in the stomach, and percentages of (B) CD11b+F4/80hiCD64+CX3CR1+, (C) neutrophils, and (D) DC. Cells from the gastric lymph nodes of 3–4 mice of the same gender and treatment were stimulated ex-vivo with RPMI (unstimulated) or 5 μg/ml of formalin-inactivated H. pylori SS1. Production of (E) IFNγ, (F) IL-10, (G) IL-17 and (H) IL-6 were measured in 72-hour cell culture supernatant using a cytometric bead array. Results are expressed as average of 3–5 samples of pooled cells and data represent mean ± SEM. Points with an asterisk are significantly different when compared to the control group (P<0.05). Results are representative of 2 independent replicate experiments with same results. Points with an asterisk are significantly different when compared to the control (WT) group (P<0.05).
IL-10-producing MNP accumulate in the stomach of H. pylori-infected mice, and this initial response influences CD4+ T cell and B cell compartments. To assess how the absence of IL-10 could affect downstream effector responses, we cultured cells obtained from gastric lymph nodes of H. pylori-infected mice that were either treated with IL-10 neutralizing or control antibodies, and stimulated them ex vivo with inactivated whole H. pylori SS1. Supernatants were collected 72-hours post-stimulation and cytokine levels measured using a cytometric bead array. The results (Figure 5E–H) show that ex vivo stimulation with inactivated H. pylori induces significant production of IFNγ, IL-10, IL-17 and IL-6 in gastric lymph nodes from infected mice. However, IL-10 neutralization was associated with a suppression of IFNγ (Figure 5E, F) and increased production of IL-17 (Figure 5G).
Discussion
H. pylori has colonized the human stomach since early evolution, diverged with prehistoric human migrations (46–49), and co-evolved with its human host for over 60,000 years (1, 2). However, its identification as the main etiologic agent of gastro-duodenal ulcers, and gastric cancer (50) set the stage for the pre-conceived notion of H. pylori as pathogen. Thirty years after this discovery, it is broadly accepted that H. pylori can predispose carriers to develop serious gastric pathologies (51, 52). However, emerging clinical and epidemiological data support the theory that H. pylori might also be a beneficial commensal organism and its disappearance has been linked to increased incidence of diseases like asthma or IBD (53, 54). Indeed, H. pylori’s ability to establish life-long chronic colonization of the gastric mucosal niche has been linked to the induction of potent regulatory responses that dampen effector mechanisms of bacterial eradication, although the induction of these responses has been attributed mainly to the modulation of DC, as opposed to macrophages (55–57). In any case, the mechanisms underlying the induction of these protective responses are not fully understood. This study reports a complex network of MNPs, which include DCs and a subset of CD11b+F4/80hiCD64+CX3CR1+ macrophages not previously described in the stomach. We provide evidence that MNPs can facilitate H. pylori colonization by promoting IL-10 responses.
The prevailing theory is that the regulatory/suppressor responses associated with of H. pylori gastric infection are induced by DCs (18, 19, 55, 58) and not by macrophages. Moreover, it has been suggested that macrophages contribute to the initiation of gastritis. For instance, Shumacher and colleagues identified a subset of CD11b+F4/80+Ly6Chi cells that is recruited to the stomach of mice as early as 2 days post-infection, and the loss of this subset was associated with reduced gastritis (59). Others have shown that the loss of matrix metalloproteinase 7, heme oxygenase and protease-activated receptor 1 worsens H. pylori-induced gastritis through the regulation of pro-inflammatory gene expression in macrophages (40–42). To investigate how macrophage phenotype influences the outcome of infection, we used myeloid-specific PPARγ deficient mice with inflammatory-prone macrophages driven by the deletion of PPARγ, an important regulatory transcription factor. Interestingly, our data shows that loss of PPARγ results in a significant drop in bacterial loads, which consistently occurs between weeks 2 and 3 post-infection, and is paralleled mainly by impaired accumulation of CD11b+F4/80hiCD64+CX3CR1+ macrophages at the gastric mucosa when compared to WT mice. Our finding that mice with a targeted deletion of PPARγ in myeloid cells failed to expand and maintain this population was unexpected, since loss of PPARγ in myeloid cells does not affect their differentiation or survival (38, 39). Of note, iNos levels are higher in H. pylori-associated atrophic gastritis compared to uncomplicated gastritis, indicating the potential contribution of M1-like macrophages to lesion development (60). Furthermore, PPARγ polymorphisms in humans are associated with an increased risk of developing H. pylori-related gastric cancer (61). Although the importance of PPARγ in the host response to H. pylori has been established in previous studies, the mechanisms underlying the protective actions of gastric H. pylori colonization remain incompletely understood. Here we provide novel evidence that PPARγ is essential for mounting a regulatory immune response to H. pylori via accumulation of CD11b+F4/80hiCD64+CX3CR1+ macrophages and IL-10 production. A possible mechanism is that H. pylori infection favors endogenous PPARγ agonist production locally. Interestingly, differentiation of monocytes into macrophages in the presence of the endogenous PPARγ agonists 9-HODE and 13-HODE, two major oxidized linoleic acid metabolite components of oxLDL, induced a shift from CCR2 to CX3CR1 surface expression and upregulation of CD36. This phenotypic switch occurred in the presence of lipid-induced TNFα, IFNγ and IL-1β, and it was inhibited by RNAi-mediated knockdown of PPARγ and treatment with its antagonist GW9662. Although CX3CR1 was constitutively expressed in monocytes, only PPARγ activation upregulated CX3CR1 expression by directly binding to PPARγ response element (PPRE) consensus sites on the CX3CR1 promoter (62). Indeed, infection with H. pylori CagA+ strains has been associated with increased levels of oxidized low-density lipoprotein (oxLDL) in the plasma of human subjects with more severe coronary atherosclerosis (63). In line with this, our unpublished observations demonstrate that co-culture of H. pylori with BMDM upregulates CX3CR1 although mRNA expression is suppressed in PPARγ-deficient BMDM.
To further investigate the mechanisms by which MNPs are implicated in facilitating bacterial colonization, we first performed a depletion study to confirm that temporary loss phagocytic cells results in suppressed H. pylori burden in the stomach. We then hypothesized that MNPs could facilitate gastric colonization by promoting an IL-10-mediated regulatory microenvironment. Consistent with this hypothesis, clodronate-induced depletion indeed decreased IL-10 production by CD3+CD4+ T cells and CD11b+ cells, which resulted in a sharp increase in the recruitment of neutrophils. Similar results have been previously reported by Oertli et al., who observed a significant reduction in H. pylori loads after depletion of CD11c+ cells using DTR-CD11c mice due to enhanced IFNγ responses (64). Our phenotypic analysis shows that in addition to DC (MHCII+CD11c+CD103+ and MHCII+CD11c+CD103−), some CD11b+F4/80hiCD64+CX3CR1+ macrophages also express CD11c (not shown), and thus could have also been affected by the depletion. Taken together, these data indicate that MNPs contribute to high levels of bacterial colonization in the gastrointestinal tract by inducing IL-10-mediated regulatory responses at the mucosa and creating a host tolerant environment that favors colonization. Follow-up challenge studies in mice revealed that F4/80loCD11b+ and F4/80hiCD11b+CD64+CX3CR1+ cells from WT mice produce significant amounts of IL-10. Notably, IL-10 levels on a per cell basis were not augmented in WT mice following infection with H. pylori, which suggests that these cells have innate regulatory function and produce IL-10 in the steady state to maintain mucosal homeostasis. As opposed to MNPs, H. pylori infection induced IL-10 production by gastric CD19+ B cells and CD4+ T cells on day 24 post-infection, particularly from PD-1+ Tr1-like cells, and FoxP3+ iTreg cells. This increase in IL-10 production by lymphocytes in response to H. pylori, suggests that IL-10-producing MNPs are the main source and promoters of IL-10-mediated responses at the gastric mucosa. Interestingly, in an attempt to characterize resident MNPs in the kidneys, Duffield and colleagues have identified a CD11bint CD11cint F4/80hi subpopulation that expresses high levels of CX3CR1 and IL-10 (45). Also, depletion of CX3CR1+ MNPs results in more severe colitis in the Citrobacter rodentium infection colitis model. The mechanism is mediated by CX3CR1+ MNP’s ability to promote IL-22 secretion from ILC3. An equivalent CX3CR1+ cell subset was identified in the human intestine, which additionally expressed CD64 and CD68 (65).
To evaluate the potential mechanism by which IL-10-producing MNPs could modulate the gastric environment early post-infection, we neutralized IL-10 in WT mice infected with H. pylori. As expected, IL-10 ablation lead to suppressed colonization and, similarly to our results during MNPs depletion, to a significant influx of neutrophils. Surprisingly, IL-10 neutralization did not affect numbers of F4/80hiCD11b+CD64+CX3CR1+ cells, which suggests that IL-10 is dispensable for their accumulation in the stomach. We also report that H. pylori infection induced IFNγ, IL-10, IL-6 and IL-17 responses in gastric lymph nodes, which is in line with the mixed response to infection that has been reported previously. However, IL-10 blockade led to increased IL-17 and diminished IFNγ and IL-10 production. This response fits well with the enhancement of TH17 responses and can explain the effect of neutrophil accumulation after neutralization of IL-10 or depletion of IL-10-producing MNPs.
In summary, we provide data showing that MNPs facilitate H. pylori colonization early post-infection. We show that the gastric mucosa hosts a large and heterogeneous population of myeloid cells, including DC and a newly identified CD11b+F4/80hiCD64+CX3CR1+ macrophage subset. Although our profiling data does not provide definitive evidence of this subset being responsible for the induction of the regulatory responses that facilitate optimal colonization, the fact that they: 1) accumulate in the stomach following H. pylori infection, 2) are the subset most affected cell by clodronate treatment, and 3) produce IL-10 constitutively, makes them the cell type most likely responsible for inducing a regulatory microenvironment in the gastric mucosa during H. pylori infection. It is tempting to speculate that CD11b+F4/80hiCD64+CX3CR1+ MNPs are resident cells of the gastric immune system that promote tolerogenic responses. These macrophages expand following H. pylori infection, most likely by proliferating locally based on BrdU data, and impose an IL-10-dominated tissue microenvironment that dampens effector responses, particularly TH17, against H. pylori and thereby facilitate a more effective colonization. An interesting finding of this study is the very critical role of PPARγ in macrophages for the maintenance of regulatory homeostasis in the stomach. The mechanism by which the loss of PPARγ results in more efficient H. pylori elimination does not seem to be related to increased neutrophil influx, although it was associated with lower production of IL-10. While neutralization of IL-10 did not affect accumulation of CD11b+F4/80hiCD64+CX3CR1+ MNPs in the stomach, the myeloid compartment was severely affected by genetic ablation of PPARγ. Three immediate implications derived from this work that deserve further investigation are: i) To what extent do these host tolerance mechanisms contribute to the role of H. pylori as a beneficial commensal that protects from immune-mediated diseases?; ii) Do CD11b+F4/80hiCD64+CX3CR1+ MNPs facilitate colonization by other members of the gastrointestinal microbiota; and iii) Are there other master regulators of homeostasis in the GI mucosa that target this cell type and can the used for therapeutic development?
Supplementary Material
Acknowledgments
We would like to thank Dr. Richard Peek from Vanderbilt University for kindly providing Helicobacter pylori strain SS1.
This work was supported in part by National Institute of Allergy and Infectious Diseases Contract No. HHSN272201000056C to JB-R and funds from the Nutritional Immunology and Molecular Medicine Laboratory (www.nimml.org). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors have declared that no conflict of interest exists.
References
- 1.Stolte M. Helicobacter pylori gastritis and gastric MALT-lymphoma. Lancet. 1992;339:745–746. doi: 10.1016/0140-6736(92)90645-j. [DOI] [PubMed] [Google Scholar]
- 2.Pernitzsch SR, Sharma CM. Transcriptome complexity and riboregulation in the human pathogen Helicobacter pylori. Front Cell Infect Microbiol. 2012;2:14. doi: 10.3389/fcimb.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 4.Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306–323. doi: 10.1053/j.gastro.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Pacifico L, Anania C, Osborn JF, Ferraro F, Chiesa C. Consequences of Helicobacter pylori infection in children. World J Gastroenterol. 2010;16:5181–5194. doi: 10.3748/wjg.v16.i41.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, Taube C, Muller A. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest. 2011;121:3088–3093. doi: 10.1172/JCI45041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selgrad M, Bornschein J, Kandulski A, Hille C, Weigt J, Roessner A, Wex T, Malfertheiner P. Helicobacter pylori but not gastrin is associated with the development of colonic neoplasms. Int J Cancer. 2014;135:1127–1131. doi: 10.1002/ijc.28758. [DOI] [PubMed] [Google Scholar]
- 8.Bassaganya-Riera J, Dominguez-Bello MG, Kronsteiner B, Carbo A, Lu P, Viladomiu M, Pedragosa M, Zhang X, Sobral BW, Mane SP, Mohapatra SK, Horne WT, Guri AJ, Groeschl M, Lopez-Velasco G, Hontecillas R. Helicobacter pylori Colonization Ameliorates Glucose Homeostasis in Mice through a PPAR gamma-Dependent Mechanism. PLoS One. 2012;7:e50069. doi: 10.1371/journal.pone.0050069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook KW, Crooks J, Hussain K, O’Brien K, Braitch M, Kareem H, Constantinescu CS, Robinson K, Gran B. Helicobacter pylori infection reduces disease severity in an experimental model of multiple sclerosis. Front Microbiol. 2015;6:52. doi: 10.3389/fmicb.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engler DB, Reuter S, van Wijck Y, Urban S, Kyburz A, Maxeiner J, Martin H, Yogev N, Waisman A, Gerhard M, Cover TL, Taube C, Muller A. Effective treatment of allergic airway inflammation with Helicobacter pylori immunomodulators requires BATF3-dependent dendritic cells and IL-10. Proc Natl Acad Sci U S A. 2014;111:11810–11815. doi: 10.1073/pnas.1410579111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherdantseva LA, Potapova OV, Sharkova TV, Belyaeva YY, Shkurupiy VA. Association of Helicobacter pylori and iNOS production by macrophages and lymphocytes in the gastric mucosa in chronic gastritis. J Immunol Res. 2014;2014:762514. doi: 10.1155/2014/762514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun. 2003;71:1755–1762. doi: 10.1128/IAI.71.4.1755-1762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raghavan S, Suri-Payer E, Holmgren J. Antigen-specific in vitro suppression of murine Helicobacter pylori-reactive immunopathological T cells by CD4CD25 regulatory T cells. Scand J Immunol. 2004;60:82–88. doi: 10.1111/j.0300-9475.2004.01447.x. [DOI] [PubMed] [Google Scholar]
- 14.Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, Reindl W, Dossumbekova A, Friedrich M, Saur D, Wagner H, Schmid RM, Prinz C. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131:525–537. doi: 10.1053/j.gastro.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, Johnsson E, Suri-Payer E, Larsson P, Rudin A, Svennerholm AM, Lundin BS. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. 2005;73:523–531. doi: 10.1128/IAI.73.1.523-531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kandulski A, Wex T, Kuester D, Peitz U, Gebert I, Roessner A, Malfertheiner P. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter. 2008;13:295–303. doi: 10.1111/j.1523-5378.2008.00612.x. [DOI] [PubMed] [Google Scholar]
- 17.Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, Pena A, Rollan A, Viviani P, Guiraldes E, Schmitz JM, Lorenz RG, Novak L, Smythies LE, Smith PD. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134:491–499. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao JY, Rathinavelu S, Eaton KA, Bai L, Zavros Y, Takami M, Pierzchala A, Merchant JL. Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of ineffective host defense. Am J Physiol Gastrointest Liver Physiol. 2006;291:G73–81. doi: 10.1152/ajpgi.00139.2005. [DOI] [PubMed] [Google Scholar]
- 20.Bimczok D, Clements RH, Waites KB, Novak L, Eckhoff DE, Mannon PJ, Smith PD, Smythies LE. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 2010;3:260–269. doi: 10.1038/mi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carbo A, Bassaganya-Riera J, Pedragosa M, Viladomiu M, Marathe M, Eubank S, Wendelsdorf K, Bisset K, Hoops S, Deng X, Alam M, Kronsteiner B, Mei Y, Hontecillas R. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PloS One. 2013;8:e73365. doi: 10.1371/journal.pone.0073365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carbo A, Olivares-Villagomez D, Hontecillas R, Bassaganya-Riera J, Chaturvedi R, Piazuelo MB, Delgado A, Washington MK, Wilson KT, Algood HM. Systems modeling of the role of interleukin-21 in the maintenance of effector CD4+ T cell responses during chronic Helicobacter pylori infection. MBio. 2014;5:e01243–01214. doi: 10.1128/mBio.01243-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wendelsdorf K, Alam M, Bassaganya-Riera J, Bisset K, Eubank S, Hontecillas R, Marathe M. ENteric Immunity SImulator: A tool for in silico study of gastroenteric infections. IEEE Transactions on NanoBioScience. 2012;11:273–288. doi: 10.1109/TNB.2012.2211891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hitkova I, Yuan G, Anderl F, Gerhard M, Kirchner T, Reu S, Rocken C, Schafer C, Schmid RM, Vogelmann R, Ebert MP, Burgermeister E. Caveolin-1 protects B6129 mice against Helicobacter pylori gastritis. PLoS Pathog. 2013;9:e1003251. doi: 10.1371/journal.ppat.1003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li SL, Zhao JR, Ren XY, Xie JP, Ma QZ, Rong QH. Increased expression of matrix metalloproteinase-9 associated with gastric ulcer recurrence. World J Gastroenterol. 2013;19:4590–4595. doi: 10.3748/wjg.v19.i28.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barton SG, Rampton DS, Winrow VR, Domizio P, Feakins RM. Expression of heat shock protein 32 (hemoxygenase-1) in the normal and inflamed human stomach and colon: an immunohistochemical study. Cell Stress Chaperones. 2003;8:329–334. doi: 10.1379/1466-1268(2003)008<0329:eohsph>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. 2015;33:643–675. doi: 10.1146/annurev-immunol-032414-112220. [DOI] [PubMed] [Google Scholar]
- 28.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DW, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wohlfert EA, Nichols FC, Nevius E, Clark RB. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol. 2007;178:4129–4135. doi: 10.4049/jimmunol.178.7.4129. [DOI] [PubMed] [Google Scholar]
- 33.Hontecillas R, Horne WT, Climent M, Guri AJ, Evans C, Zhang Y, Sobral BW, Bassaganya-Riera J. Immunoregulatory mechanisms of macrophage PPAR-gamma in mice with experimental inflammatory bowel disease. Mucosal Immunol. 2011;4:304–313. doi: 10.1038/mi.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffman PS, Vats N, Hutchison D, Butler J, Chisholm K, Sisson G, Raudonikiene A, Marshall JS, Veldhuyzen van Zanten SJ. Development of an interleukin-12-deficient mouse model that is permissive for colonization by a motile KE26695 strain of Helicobacter pylori. Infection and immunity. 2003;71:2534–2541. doi: 10.1128/IAI.71.5.2534-2541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui Y, Hennighausen L, Gonzalez F, Rohrer J, Benninghoff AU, Hontecillas R. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 2004;127:777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB, Jr, Gonzalez FJ. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol. 2002;22:2607–2619. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res. 2001;10:545–553. doi: 10.1023/a:1013063514007. [DOI] [PubMed] [Google Scholar]
- 38.Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc Natl Acad Sci U S A. 2003;100:6712–6717. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, Barak Y, Schwabe J, Nagy L. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712. doi: 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gobert AP, Verriere T, Asim M, Barry DP, Piazuelo MB, de Sablet T, Delgado AG, Bravo LE, Correa P, Peek RM, Jr, Chaturvedi R, Wilson KT. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J Immunol. 2014;193:3013–3022. doi: 10.4049/jimmunol.1401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krakowiak MS, Noto JM, Piazuelo MB, Hardbower DM, Romero-Gallo J, Delgado A, Chaturvedi R, Correa P, Wilson KT, Peek RM., Jr Matrix metalloproteinase 7 restrains Helicobacter pylori-induced gastric inflammation and premalignant lesions in the stomach by altering macrophage polarization. Oncogene. 2015;34:1865–1871. doi: 10.1038/onc.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wee JL, Chionh YT, Ng GZ, Harbour SN, Allison C, Pagel CN, Mackie EJ, Mitchell HM, Ferrero RL, Sutton P. Protease-activated receptor-1 down-regulates the murine inflammatory and humoral response to Helicobacter pylori. Gastroenterology. 2010;138:573–582. doi: 10.1053/j.gastro.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 43.De Calisto J, Villablanca EJ, Mora JR. FcgammaRI (CD64): an identity card for intestinal macrophages. Eur J Immunol. 2012;42:3136–3140. doi: 10.1002/eji.201243061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ, Woltman AM, Reyal Y, Bonnet D, Sichien D, Bain CC, Mowat AM, Reis e Sousa C, Poulin LF, Malissen B, Guilliams M. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. 2012;42:3150–3166. doi: 10.1002/eji.201242847. [DOI] [PubMed] [Google Scholar]
- 45.Kawakami T, Lichtnekert J, Thompson LJ, Karna P, Bouabe H, Hohl TM, Heinecke JW, Ziegler SF, Nelson PJ, Duffield JS. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J Immunol. 2013;191:3358–3372. doi: 10.4049/jimmunol.1300342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wirth T, Wang X, Linz B, Novick RP, Lum JK, Blaser M, Morelli G, Falush D, Achtman M. Distinguishing human ethnic groups by means of sequences from Helicobacter pylori: lessons from Ladakh. Proc Natl Acad Sci U S A. 2004;101:4746–4751. doi: 10.1073/pnas.0306629101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, Yamaoka Y, Megraud F, Otto K, Reichard U, Katzowitsch E, Wang X, Achtman M, Suerbaum S. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 48.Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, Yamaoka Y, Graham DY, Perez-Trallero E, Wadstrom T, Suerbaum S, Achtman M. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mane SP, Dominguez-Bello MG, Blaser MJ, Sobral BW, Hontecillas R, Skoneczka J, Mohapatra SK, Crasta OR, Evans C, Modise T, Shallom S, Shukla M, Varon C, Megraud F, Maldonado-Contreras AL, Williams KP, Bassaganya-Riera J. Host-interactive genes in Amerindian Helicobacter pylori diverge from their Old World homologs and mediate inflammatory responses. J Bacteriol. 2010;192:3078–3092. doi: 10.1128/JB.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marshall BJ. The pathogenesis of non-ulcer dyspepsia. Med J Aust. 1985;143:319. [PubMed] [Google Scholar]
- 51.Fonseca-Nunes A, Agudo A, Aranda N, Arija V, Cross AJ, Molina E, Sanchez MJ, Bueno-de-Mesquita B, Siersema P, Weiderpass E, Krogh V, Mattiello A, Tumino R, Saieva C, Naccarati A, Ohlsson B, Sjoberg K, Boutron-Ruault MC, Cadeau C, Fagherazzi G, Boeing H, Steffen A, Kuhn T, Katzke V, Tjonneland A, Olsen A, Khaw KT, Wareham N, Key T, Lu Y, Riboli E, Peeters PH, Gavrila D, Dorronsoro M, Quiros JR, Barricarte A, Jenab M, Zamora-Ros R, Freisling H, Trichopoulou A, Lagiou P, Bamia C, Jakszyn P. Body iron status and gastric cancer risk in the EURGAST study. Int J Cancer. 2015 doi: 10.1002/ijc.29669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu N, Wang Z, Song X, Wei L, Kim BS, Freedman ND, Baek J, Burdette L, Chang J, Chung C, Dawsey SM, Ding T, Gao YT, Giffen C, Han Y, Hong M, Huang J, Kim HS, Koh WP, Liao LM, Mao YM, Qiao YL, Shu XO, Tan W, Wang C, Wu C, Wu MJ, Xiang YB, Yeager M, Yook JH, Yuan JM, Zhang P, Zhao XK, Zheng W, Song K, Wang LD, Lin D, Chanock SJ, Goldstein AM, Taylor PR, Abnet CC. Genome-wide association study of gastric adenocarcinoma in Asia: a comparison of associations between cardia and non-cardia tumours. Gut. 2015 doi: 10.1136/gutjnl-2015-309340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schulfer A, Blaser MJ. Risks of Antibiotic Exposures Early in Life on the Developing Microbiome. PLoS Pathog. 2015;11:e1004903. doi: 10.1371/journal.ppat.1004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.von Hertzen L, Beutler B, Bienenstock J, Blaser M, Cani PD, Eriksson J, Farkkila M, Haahtela T, Hanski I, Jenmalm MC, Kere J, Knip M, Kontula K, Koskenvuo M, Ling C, Mandrup-Poulsen T, von Mutius E, Makela MJ, Paunio T, Pershagen G, Renz H, Rook G, Saarela M, Vaarala O, Veldhoen M, de Vos WM. Helsinki alert of biodiversity and health. Ann Med. 2015;47:218–225. doi: 10.3109/07853890.2015.1010226. [DOI] [PubMed] [Google Scholar]
- 55.Shiu J, Blanchard TG. Dendritic cell function in the host response to Helicobacter pylori infection of the gastric mucosa. Pathog Dis. 2013;67:46–53. doi: 10.1111/2049-632X.12014. [DOI] [PubMed] [Google Scholar]
- 56.Shiu J, Czinn SJ, Kobayashi KS, Sun Y, Blanchard TG. IRAK-M expression limits dendritic cell activation and proinflammatory cytokine production in response to Helicobacter pylori. PLoS One. 2013;8:e66914. doi: 10.1371/journal.pone.0066914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kronsteiner B, Bassaganya-Riera J, Philipson CW, Viladomiu M, Carbo A, Abedi V, Hontecillas R. Systems-wide analyses of mucosal immune responses to Helicobacter pylori. World Journal of Gastroenterology. 2015 doi: 10.1080/19490976.2015.1116673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang M, Liu M, Luther J, Kao JY. Helicobacter pylori directs tolerogenic programming of dendritic cells. Gut Microbes. 2010;1:325–329. doi: 10.4161/gmic.1.5.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Engler DB, Leonardi I, Hartung ML, Kyburz A, Spath S, Becher B, Rogler G, Muller A. Helicobacter pylori-specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and IL-18. Inflamm Bowel Dis. 2015;21:854–861. doi: 10.1097/MIB.0000000000000318. [DOI] [PubMed] [Google Scholar]
- 60.Quiding-Jarbrink M, Raghavan S, Sundquist M. Enhanced M1 macrophage polarization in human helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS One. 2010;5:e15018. doi: 10.1371/journal.pone.0015018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JM, Kim SS, Cho YS. The Role of PPARgamma in Helicobacter pylori Infection and Gastric Carcinogenesis. PPAR Res. 2012;2012:687570. doi: 10.1155/2012/687570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barlic J, Zhang Y, Foley JF, Murphy PM. Oxidized lipid-driven chemokine receptor switch, CCR2 to CX3CR1, mediates adhesion of human macrophages to coronary artery smooth muscle cells through a peroxisome proliferator-activated receptor gamma-dependent pathway. Circulation. 2006;114:807–819. doi: 10.1161/CIRCULATIONAHA.105.602359. [DOI] [PubMed] [Google Scholar]
- 63.Huang B, Chen Y, Xie Q, Lin G, Wu Y, Feng Y, Li J, Zhuo Y, Zhang P. CagA-positive Helicobacter pylori strains enhanced coronary atherosclerosis by increasing serum OxLDL and HsCRP in patients with coronary heart disease. Dig Dis Sci. 2011;56:109–114. doi: 10.1007/s10620-010-1274-6. [DOI] [PubMed] [Google Scholar]
- 64.Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Jarbrink M, Muller A. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. 2012;122:1082–1096. doi: 10.1172/JCI61029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211:1571–1583. doi: 10.1084/jem.20140678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.