

Abstract
Chinese Mongolian sheep are an important ruminant raised for wool and meat production. However, little is known about the microbiota of the gastrointestinal tract (GIT) of Chinese Mongolian sheep. To increase our understanding of the microbial community composition in the GIT of Chinese Mongolian sheep, microbiota of five sheep is investigate for the first time using the Illumina MiSeq platform. High microbial diversity was obtained from the GIT, and the microbiota exhibited a higher biodiversity in the stomach and large intestine than in the small intestine. Firmicutes (44.62%), Bacteroidetes (38.49%), and Proteobacteria (4.11%) were the three most abundant phyla present in the GIT of the sheep. The present study also revealed the core genera of Prevotella, Bacteroides, Ruminococcus, Oscillospira, Treponema, and Desulfovibrio in the GIT. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States indicated that the metabolic pathway related to carbohydrate metabolism was the richest in the sheep GIT. In addition, a series of metabolic pathways related to plant secondary metabolism was most abundant in the stomach and large intestine than in the small intestine. Overall, the present study provides insight into the microbial community composition in GIT of the Chinese Mongolian sheep which is highly diverse and needs to be studied further to exploit the complex interactions with the host.
Electronic supplementary material
The online version of this article (doi:10.1186/s13568-017-0378-1) contains supplementary material, which is available to authorized users.
Keywords: Chinese Mongolian sheep, Gastrointestinal tract, Illumina MiSeq, Microbiota, Metabolic pathways
Introduction
Microbiota of mammalian gastrointestinal tract (GIT) is a complex ecosystem constitute of diverse bacterial populations (Falony et al. 2016). The intra- and interpersonal variation in the composition of the human microbiome significantly complicates the analysis of microbiome data (Consortium HMP 2012; Taglialatela et al. 2009). The recently proposed concept of enterotypes or stool community types has overcome this difficulty (Arumugam et al. 2011; Koren et al. 2013). Gut microbiota assists in intestinal homeostasis and other aspects of the host, including intestinal immune response, digestion, physiology, and disease treatment (Donia et al. 2014; Koboziev et al. 2014). The microflora in the GIT of ruminants plays a critical role in fiber degradation (Nyonyo et al. 2014; Thoetkiattikul et al. 2013). Ruminants can efficiently digest dietary fiber and absorb nutrients because of their unique stomachs, including the rumen, reticulum, omasum, and abomasum, particularly the rumen (Morgavi et al. 2013). Due to the easiest sampling procedure feces and rumen of ruminants were most studied (Kittelmann et al. 2013; Lee et al. 2011). The microbiota in the stomach, small intestine, and large intestine of koalas, hoatzins, Brazilian Nelore steer, and mice has been recently explored through high-throughput next-generation sequencing (Barker et al. 2013; de Oliveira et al. 2013; Godoy-Vitorino et al. 2012; Gu et al. 2013).
Chinese Mongolian sheep are an important ruminant raised for wool and meat production; this animal is the source of one of the three most common varieties of coarse wool sheep in China (Zhang et al. 2008). In our previous study, we successfully characterized the cellulolytic bacterial communities along the GIT of Chinese Mongolian sheep through polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) and real-time PCR analyses (Zeng et al. 2015). Various bacteria thrive along the GIT, and these bacteria are more abundant in the stomach and large intestine than in the small intestine. We thus hypothesized that abundant gut microbiota information can be obtained from the GIT of Chinese Mongolian sheep through high-throughput next-generation sequencing. The samples in the Zeng et al. (2015) is identical to the ones in this study. Therefore, Illumina MiSeq platform was used to explore the bacteria diversity and composition in the GIT of Chinese Mongolian sheep.
Materials and methods
Animals and sampling
Samples were collected from five two-year-old male healthy Chinese Mongolian sheep (48.16 ± 1.48 kg body weight). These animals were reared and maintained in Gansu, China, in accordance with the standard livestock management practices. A diet of corn silage was provided in accordance with the agricultural industry standard of the People’s Republic of China (NYT816-2004). Animals were butchered in accordance with the approved by the Sichuan Agricultural University Committee on Ethics in the Care and Use of Laboratory Animals (Permit No. DKY-S20123517). Fresh samples (20 g) were collected from different segments of the GIT, namely, they were the stomach (rumen, reticulum, omasum, and abomasum), small intestine (duodenum, jejunum, and ileum), and large intestine (cecum, colon, and rectum). Finally, 50 samples were placed in sterile centrifuge tubes and immediately frozen in liquid nitrogen containers. The samples were then stored at −80 °C until further analysis. All samples were analyzed within a month.
DNA extraction
Bacteria DNA was extracted from GIT samples (100 mg each) in accordance with the instructions of the EZNA Stool DNA Kit (Omega Bio-tek). Sterile zirconia beads were used to increase the extraction yield and the quality of the bacteria DNA (Yu and Morrison 2004). All procedures were performed on ice. The final elution volume was 200 μL, and DNA concentration was conducted on a Nano Drop spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA). DNA samples were then stored frozen (−20 °C) until further analysis.
16S rRNA amplification and MiSeq sequencing
The V4 region of the 16S rRNA gene was amplified from genomic DNA using the following primers: 515F, 5′-GTGCCAGCMGCCGCGGTAA-3′; 806R, 5′-GGACTA CHVGGGTWTCTAAT-3′ (Caporaso et al. 2011). The reverse primer was barcoded with a unique 6 bp error-correcting to each sample. In brief, PCR was performed in triplicate in a 20 μL reaction mixture containing 10 ng of template DNA, 0.2 μM of each primer, 4 μL of 5× FastPfu Buffer, 0.4 μL of FastPfu Polymerase, and 2 μL of 2.5 mM dNTPs (MBI Fermentas, Waltham, MA, USA). The following thermal cycling conditions were used: 3 min of initial denaturation at 94 °C; 35 cycles of denaturation at 94 °C for 45 s, annealing at 50 °C for 60 s, and elongation at 72 °C for 90 s; and a last step at 72 °C for 10 min. The amplified products were evaluated by electrophoresis in 2% agarose gel and purified with the QIAquick Gel Extraction Kit (Qiagen, Dusseldorf, Germany). After purification, the samples were quantified using a Nano Drop spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA). Sequencing was performed on an Illumina MiSeq 2 × 250 platform (Illumina, Inc. San Diego) at the Beijing Genomics Institute (Shanghai, China) in accordance with a previously described protocol (Caporaso et al. 2012).
Data analysis
Bioinformatics analysis was performed on the website (http://www.mothur.org/wiki/MiSeq_SOP). The bacterial sequence reads were assembled using mothur v 1.32 (Schloss et al. 2009). To increase the analysis quality, the USEARCH software was used to remove chimeric sequences (Edgar et al. 2011). Operational Taxonomic Units (OTUs) were selected by using sequences with a 3% dissimilarity level. The microbial diversity structures (alpha and beta diversity) in different samples were analyzed using the QIIME software (Caporaso et al. 2010) with Python scripts. Alpha diversity was performed using the Shannon index, Chao 1, Observed species index and Simpson index. The taxonomy assignment of OTUs was investigated by comparing sequences to the Green-gene (http://rdp.cme.msu.edu/). Gene prediction was performed using PICRUSt 1.0.0 and Greengenes database v13.5 (Langille et al. 2013). The Venn diagrams were constructed on the basis of the relative abundance of bacteria on the level of genus. The R packages “Biom”, “Phyloseq”, and “Pheatmap” were used for data analysis and plotting (Mcdonald et al. 2012; Mcmurdie and Holmes 2013). The original sequencing data of raw reads were deposited in the sequence read archive of the National Center for Biotechnology (Aaccession Nos. PRJNA294127, SRR2242800, and SRS1048835).
Results
Metadata and sequencing
Using the Illumian MiSeq platform of 16S rRNA gene amplicons, a total of 557,657 sequences with a median length of 252 base pairs (bp) (V4~533–786 bp) assigned to 16,252 OTUs were obtained from all samples (Additional file 1: Table S1). Each sample has 15,933 sequences and 464 OTUs on average (Additional file 1: Table S2). The bacterial communities in the GIT samples from sheep were divided into three clear groups, namely, stomach, small intestine, and large intestine, through principal coordinate analysis (PCoA) by using the UniFrac tool (Fig. 1). We evaluated the alpha (Chao, Ace, Shannon, and Simpson) and beta diversities of the bacterial community in the GIT (Additional file 1: Table S2; Fig. 2). The indices of alpha diversity were analyzed on the basis of OTUs. The bacterial diversity was higher in the stomach and large intestine than in the small intestine. The Chao, Ace, Shannon, and Simpson indices of bacterial communities ranged from 190 to 882, 200 to 863, 2.55 to 5.33, and 0.0111 to 0.1929, respectively. No significant difference was observed among the samples obtained from the same compartment in five individuals (P < 0.05). Meanwhile, the beta diversity showed a degree of diversity discrepancy in all samples.
Fig. 1.
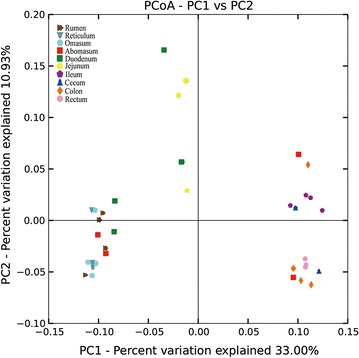
The PCoA analysis of the GIT samples (unweighted UniFrac metric). The colored circles represent the gut microbiota from the rumen, reticulum, omasum, abomasum, duodenum, jejunum, ileum, cecum, colon, and rectum, respectively
Fig. 2.
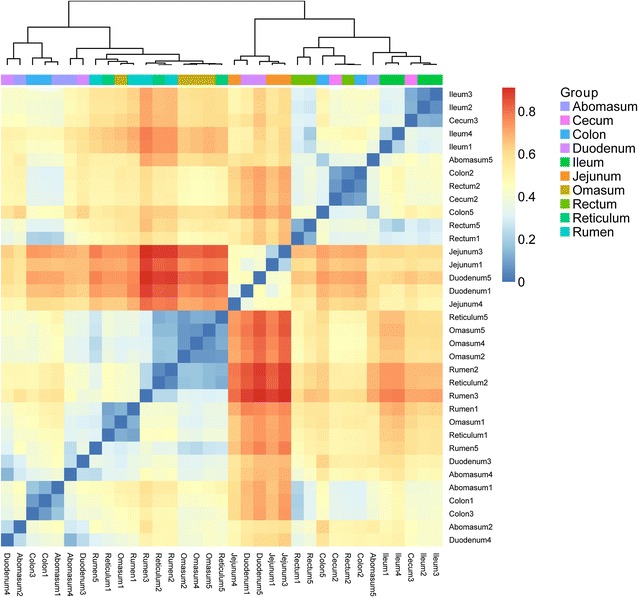
The heatmap of beta diversity of samples. The different color intensities represent the relative bacteria abundance in each sample. The number following the sample names stand for the sheep number. For example, reticulum 1, reticulum 2, reticulum 3, reticulum 4, and reticulum 5 stands for the reticulum samples from the 1st, 2nd, 3rd, 4th, and 5th sheep
According to the SILVA taxonomic database, all sequences of the samples were classified from phylum to species by using the QIIME program. In this study, the classifications of relative abundance of bacterial OTUs from phylum, class, order, family, genus, and species were shown with heatmaps (Additional file 1: Figure S1a–f). On the basis of the classifications (>1%), the segments of GIT from Chinese Mongolian sheep harbored bacteria from 11 phyla (e.g., Firmicutes, 44.62%; Bacteroidetes, 38.49%; Proteobacteria, 4.11%; Spirochaetes, 3.44%; and Euryarchaeota, 1.78%), 19 classes (e.g., Clostridia, 42.02%; Bacteroidia, 37.30%; Spirochaetes, 3.36%; Unclassified, 2.80%; and Fibrobacteria, 0.58%), 20 orders (e.g., Clostridiales, 42.01%; Bacteroidales, 37.30%; Spirochaetales, 3.26%; Unclassified, 2.90%; and Methanobacteriales, 1.83%), 38 families (e.g., Ruminococcaceae, 20.76%; Prevotellaceae, 16.60%; Lachnospiraceae, 8.37%; Unclassified, 6.88%; and Bacteroidaceae, 5.23%), 40 genera (e.g., Unknown, 20.76%; Unclassified, 19.92%; Prevotella, 15.56%; Ruminococcus, 6.35%; and Treponema, 3.26%), and 18 species (e.g., Unknown, 63.42%; Unclassified, 21.70%; Prevotella ruminicola, 5.45%; Ruminococcus flavefaciens, 3.63%; and Ruminobacter albus, 1.72%).
The results shown in Fig. 3a describe the composition of the bacterial communities in the GIT at the phylum level. Among these phyla, Firmicutes and Bacteroidetes were the most predominant. The number of Bacteroidetes was higher in the stomach and large intestine than in the small intestine. Meanwhile, reverse results were obtained in Firmicutes. To analyze further the composition of the bacterial communities, we demonstrated the genera from Firmicutes and Bacteroidetes (Fig. 3b, c), respectively. The 10 most abundant genera from Firmicutes were Ruminococcus, SMB53, Oscillospira, Clostridium, Mogibacterium, Butyrivibrio, Faecalibacterium, Lactococcus, Bulleidia, and Coprococcus. The seven most abundant genera from Bacteroidetes were Prevotella, Bacteroides, 5-7N15, [Prevotella], Parabacteroides, CF231, and YRC22.
Fig. 3.
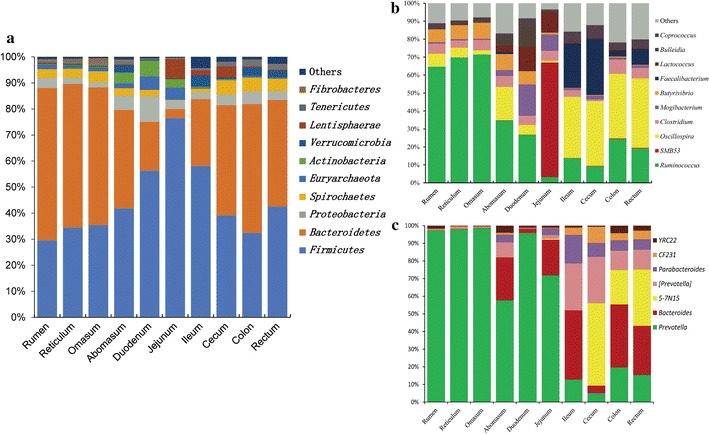
GIT microbiota at the phylum and genus level. Relative abundance of OTUs at the phylum level in individuals (a). Relative abundance of OTUs from Firmicutes (b) and Bacteroidetes (c) at the genus level in individuals. Only phyla or genera with greater than 1% representation are shown
Unique and shared bacterial genera in the sheep GIT
We detected unique and shared bacterial genera along the GIT from sheep at the genus level by using our sequencing data. The genera with an average abundance >0.1% were analyzed using Venn diagrams (Fig. 4a). Surprisingly, a large amount of unknown genera (15.65–40.44%) was discovered in the GIT from Chinese Mongolian sheep (Fig. 4b). A total of 27 genera were observed, and only 25.93% of them belonged to the shared bacterial genera, including three genera (Prevotella, Bacteroides, and Parabacteroides) from Bacteroidetes, two genera (Ruminococcus and Oscillospira) from Firmicutes, one genus (Treponema) from Spirochaetes, and one genus (Desulfovibrio) from Proteobacteria. We found four unique bacterial genera (Methanobrevibacter, Bulleidia, Butyrivibrio, and Succinivibrio) between the stomach and small intestine. In addition, two unique bacterial genera (Akkermansia and Faecalibacterium) were observed between the small intestine and large intestine. However, we did not find unique bacterial genera between the stomach and large intestine.
Fig. 4.
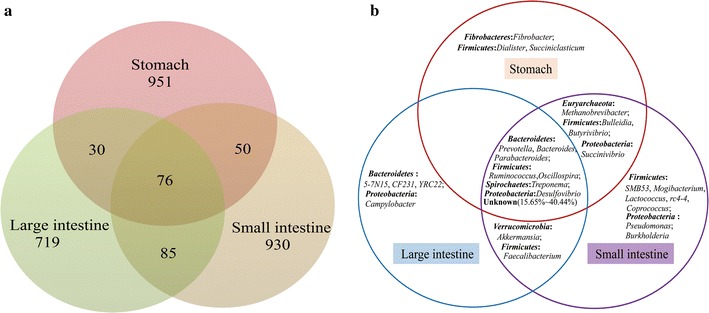
Venn diagrams of shared OTUs and bacterial genera. The shared OTUs between the stomach, small intestine, and large intestine microbiomes (a). The unique and shared bacterial genera (with the percentage of >1% colonized in segment) at the genus level in the sheep GIT (b)
Every segment from the GIT of sheep harbors a complicated ecological bacterial community. Additional file 1: Figure S2a and Table S3 shows that Oscillospira was observed only in the rumen, reticulum, and abomasum. Succinivbrio was observed only in the rumen and abomasum, and Fibrobacter was discovered only in the reticulum and abomasum. In the small intestine (Additional file 1: Figure S2b, Table S4), Prevotella and Ruminococcus were shared only by the duodenum, jejunum, and ileum. Methanobrevibacter, Lactococcus, Mogibacterium, and Pseudomonas were discovered only in the duodenum and jejunum. Treponema and Oscillospira were discovered only in the duodenum and ileum. As to the large intestine (Additional file 1: Figure S2c, Table S5), eight bacterial genera, namely, Prevotella, 5-7N15, Bacteroides, CF231, Parabacteroides, Treponema, Oscillospira, and Ruminococcus, were shared with the cecum, colon, and rectum. Faecalibacterium was observed only in the cecum and rectum, and Campylobacter was discovered only in the rectum.
Bacterial function prediction in the GIT of sheep
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to predict the functional composition of the gut microbiota genomes in Chinese Mongolian sheep. The functional profiles are shown in Fig. 5. A total of 24 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were found abundant in the stomach, small intestine, and large intestine (Fig. 5a, P < 0.01). Among these 24 KEGG pathways, eight (“Carbohydrate metabolism”, “Peptidoglycan biosynthesis”, “Ethylbenzene degradation”, “Geraniol degradation”, “Primary immunodeficiency”, “Arachidonic acid metabolism”, “Biosynthesis of siderophore group nonribosomal peptides”, and “Flagellar assembly”) primarily related to carbohydrate metabolism and bacterial flagellar assembly were more abundant in the stomach and small intestine than in the large intestine; three (“Membrane and intracellular structural molecules”, “Ubiquinone and other terpenoid-quinone biosynthesis”, and “Adipocytokine signaling pathway”) primarily related to the metabolism of cofactors and vitamins were significantly abundant only in the stomach; and two (“Ether lipid metabolism” and “RIG-I-like receptor signaling pathway”) were significantly abundant only in the small intestine. Careful analysis of each segment showed that 23 KEGG pathways were significantly more abundant in the rumen, reticulum, omasum, abomasum, duodenum, jejunum, ileum, cecum, colon, and rectum (Fig. 5b, P < 0.05). Three KEGG pathways (“One carbon pool by folate”, “Nicotinate and nicotinamide metabolism”, and “Ubiquinone and other terpenoid-quinone biosynthesis”) were significantly abundant only in the reticulum. However, five KEGG pathways (“Transporters”, “Bacterial motility proteins”, “Bacterial chemotaxis”, “Flagellar assembly”, and “Phosphotransferase system”) were significantly abundant only in the jejunum.
Fig. 5.
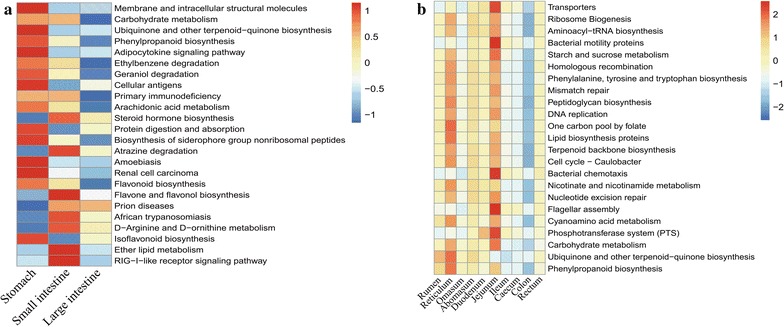
Predicted function of the gut micorbiota in the sheep of GIT. KEGG pathways were shown in two heatmaps. The bootstrap Mann–Whitney u-test was used to detect the gene distribution with cutoffs of P < 0.05, FDR <0.2, Mean counts >10,000 (a) and P < 0.01, FDR <0.1, Mean counts >10 (b)
Discussion
This study aimed to provide new insights into the diverse symbiotic bacterial communities along the GIT of Chinese Mongolian sheep. The gut microbiota co-developed with the host from birth is involved in the regulation of mammal’s immune function, digestion, physiology, and disease treatment (Koboziev et al. 2014). However, insufficient information is available about the microbial flora along the GIT of ruminants. In this study, we successfully characterized the microbiota in all segments of the GIT for the first time by using Illumina MiSeq. A remarkable microbiota composition was obtained from the GIT, and the microbiota showed a higher biodiversity in the stomach and large intestine than in the small intestine (Fig. 3; Additional file 1: Table S2). In this study, the microbiota varied along the GIT and was similar in the same segment of individual animals, which is in agreement with a previous finding (Koren et al. 2013). A recent study has reported that the foregut and hindgut of hoatzin and cow possess a relatively similar microbiota composition regardless of host species (Godoy-Vitorino et al. 2012). A similar result was obtained from our study; samples from adjacent parts of the GIT were clustered together (Fig. 1). The recently proposed concept of enterotypes and stool community types has overcome the difficulty in analyzing microbiome data because of intra- and interpersonal variation (Armstrong and Smithard 1979; Holmes et al. 2012; Koren et al. 2013; Turnbaugh et al. 2007).
In the present study, Firmicutes, Bacteroidetes, and Proteobacteria were predominantly abundant in all samples on average. These findings paralleled those of other studied on the microbiota in the GIT of ruminants (Cunha et al. 2011; Li et al. 2012b). Interestingly, the number of Bacteroidetes was higher in the stomach and large intestine than in the small intestine. Consistently, Bacteroidetes was found predominantly in the rumen, reticulum, and omasum of bovine (Peng et al. 2015), whereas reverse results were obtained in Firmicutes. Bacteroidetes aids in the digestion of complex carbohydrates (Spence et al. 2006), and Firmicutes is the dominant species in the GIT of ruminants and mainly consists of diverse fibrolytic and cellulolytic bacterial genera (Evans et al. 2011). In the present study, Firmicutes was more abundant in the small intestine than in the stomach and large intestine. This result is consistent with the findings in Brazilian Nelore steer (de Oliveira et al. 2013). However, reverse results were obtained in our previous study using real-time PCR (Zeng et al. 2015). This discrepancy is mainly attributed to the different primers used in real-time PCR and Illumina MiSeq. Proteobacteria comprises a large amount of bacteria that can catabolize feedstuff components (Evans et al. 2011), including corn and grass (Callaway et al. 2010). In the present study, Proteobacteria were predominantly abundant in the duodenum. However, another study on the South American folivorous hoatzin found that the number of Proteobacteria is lower in the foregut than in the hindgut (Godoy-Vitorino et al. 2012). In addition, Fibrobacteres is predominantly abundant in the omasum and the reticulum. These findings are consistent with our previous study (Zeng et al. 2015). In a previous study, the number of dominant fibrolytic bacteria, including Ruminococcus albus, Fibrobacter succinogenes, and Ruminococcus flavefaciens, is consistently higher in the stomach than in the large and small intestine (Zeng et al. 2015).
The results of 454 pyrosequencing showed that Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes are predominantly abundant in all fecal samples of mammals, including 6 pigs, 14 healthy adult humans, 6 cows, 6 chickens, and 6 geese (Lee et al. 2011). In the present study, the genera Prevotella, Bacteroides, Ruminococcus, Oscillospira, Treponema, and Desulfovibrio were found in all samples. These genera belong to Bacteroidetes, Firmicutes, and Proteobacteria. Prevotella aids in the utilization of feed proteins in the rumen of ruminants (Xu and Gordon 2003) and can increase in abundance if the animal is fed a grain-based diet (Li et al. 2012a). Prevotella are sometimes believed to work in conjunction with the cellulolytic species Fibrobacter succinogenes in utilizing hemicellulose (Osborne and Dehority 1989). Ruminococcus plays a critical role in the digestion and metabolism of dietary fiber in ruminants (Han et al. 2015). Previous studies reported that Treponema is a genus of the primary bacterial community in the rumen; this genus reportedly disintegrates plant polysaccharides from ingested food (Avguštin et al. 1997; Bekele et al. 2011). Meanwhile, Desulfovibrio plays a significant role in the sulfate reduction of rumen and is more abundant in developing rumen than in mature rumen (Wu et al. 2012). Importantly, Mogibacterium, Lactococcus, Pseudomonas, and Burkholderia are abundant in the small intestine. Mogibacterium is a group of Gram-positive anaerobic bacteria that predominates the rumen of goats (Patel et al. 2011). The relative abundance of Mogibacterium could increase with high-grain feeding (Liu et al. 2015). Coprococus, which is abundant in the ileum, is an Enterococcus that can digest xylanolytic (Valdez-Vazquez et al. 2015). Our study provides evidence that the ileum may be another important segment of the GIT for dietary fiber. This observation agrees with the previous finding that Coprococus is a ubiquitous genus in Nelore GIT (de Oliveira et al. 2013). Surprisingly, Campylobacter species are abundant in the large intestine, especially in the cecum. Although this genus is usually known to comprise pathogenic bacteria, some species isolated from cattle and starlings show a high resistance to multiple antimicrobial drugs, including ciprofloxacin, gentamicin, and erythromycin (Sanad et al. 2013). Finally, Fibrobacter, Dialister, and Succiniclasticum were found more abundant in the stomach than in the small and large intestine. As reported, Succiniclasticum represents the majority of the sequence tags of the family Veillonellaceae, which belongs to the class Clostridia from Firmicutes in the three stomachs of bovine (Peng et al. 2015).
Microbiota function prediction revealed that most of the metabolic pathways in the GIT are related to carbohydrate metabolism. This finding is consistent with the observation that Firmicutes and Bacteroidetes are predominantly abundant in the GIT. Importantly, Bacteroidetes and Firmicutes are the dominant species aiding the digestion of complex carbohydrates in the GIT of ruminants (Evans et al. 2011; Spence et al. 2006). In the present study, higher diversity was detected in the stomach and large intestine than in the small intestine. Similarly, a previous research detected that the microbial fermentation and absorption of indigestible dietary substrates primarily occur in the rumen and colon and not in the small intestine (Abbeele et al. 2011). Another research reported that the rumen and colon of the North American moose are distinct environments (Ishaq and Wright 2012). In general, feed and fodder are first ingested and absorbed in the stomach of ruminants, the rest are ingested in the small and large intestine. In addition, the rumen is the most important segment of nutrient utilization (Kebreab et al. 2009), which primarily involves protein metabolism and plant secondary metabolism. Nevertheless, main nutrients, particularly proteins, are absorbed in the small intestine (Klieve 2005). Importantly, the rapid uptake and conversion of simple carbohydrates help maintain the micro-ecological balance of the small intestine (Zoetendal et al. 2012). In addition, indigested feed including some cellulose and starch can be completely but slowly assimilated in the large intestine (Armstrong and Smithard 1979). In the present study, we detected that some metabolic pathways related to microbial decomposition were significantly more abundant in the stomach than in the small intestine and large intestine. In addition, the metabolic pathways related to microbial synthesis (“Transporters”, “Ribosome Biogenesis”, and “Aminoacyl-tRNA biosynthesis”) were abundant in the reticulum. The reticulum is the second stomach along the GIT of ruminants and is conducive to the uniformity in the rumen fluid microbiome through the churning action with rumen (Braun 2009). A recent study has analyzed the bacterial composition of the rumen, reticulum, omasum, and abomasum of bovine for the first time by using a metagenomic approach (Peng et al. 2015). The primary composition of the microbiome was determined in the rumen, reticulum, and omasum. In addition, the metabolic pathways related to lipid metabolism and pattern-recognition receptors were significantly more abundant in the small intestine than in the stomach and large intestine. In general, lipid metabolism is primarily determined in the small intestine. Genes involved in the lipid metabolism are expressed in response to changes in the barrier lipids of the skin of sheep (Ovis aries) for their more significant role of volatile fatty acids (Jiang et al. 2014). We also detected that the metabolic pathways related to the motility of bacterial proteins and the chemotaxis of bacteria are significantly more abundant in the jejunum in other segments of the GIT. A previous in vitro study showed that the intestinal contents from the jejunum can digest cellulose and neutral detergent fiber (Jiao et al. 2013).
In the present study, according to the classifications (>1%) of relative abundance of bacterial OTUs from phylum, class, order, family, genus, and species, bacteria from 2.80% phyla, 2.90% class, 2.90% order, 19.92% family, 6.88% genera, and 63.42% species were unknown. Although we obtained a huge number of microbiota members (>93.12%) along the GIT of sheep on the level of genus, a large number of microbiota members (>63.42%) cannot be classified or remain unknown on the level of species. Recently, a metagenomic data analysis of the human gut has shown extensive strain-level variation across species, and differences in gene copy number affect specific adaptive functions (Greenblum et al. 2015). Upon the completion of the 1000 Genomes Project, scientists have proposed an interdisciplinary Unified Microbiome Initiative to discover and advance tools for understanding and harnessing the capabilities of various ecosystems of microbial communities, such as the human gut and marine ecosystems, to improve human health, agriculture, bio-energy, and the environment (Alivisatos et al. 2015; Consortium et al. 2015). Meanwhile, a similar study for animals must also be conducted in the future. Therefore, further analyses through metagenomics, metabolomics, and transcriptomics are needed to identify completely the microbiota along the GIT of Chinese Mongolian sheep.
In summary, we successfully for the first time characterized the bacterial taxa and metabolic pathways in all intestinal segments of Chinese Mongolian sheep by using Illumina MiSeq. Nevertheless, the obtained functional profiles are merely a prediction; detailed analyses are still needed to elucidate this aspect. Further studies are warranted to determine the contributions of the bacterial taxa and metabolic pathways to the health, development, and physiology of Chinese Mongolian sheep.
Authors’ contributions
All authors contributed to the design of the experiment. YZ performed and wrote the experiments. PJ and SX analyzed the experimental data. All authors read and approved the final manuscript.
Acknowledgements
We are very grateful the Beijing Genomics Institute (Shanghai, China) offered an Illumina MiSeq platform.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
This study does not contain any individual person’s data.
Ethical approval
All animal experiment procedures were conducted in accordance with the guidelines of the Animal Welfare Act and all procedures and protocols were approved by the Institutional Animal Care and Use Committee of the Sichuan Agricultural University.
Funding
Financial support was from the Natural Science Foundation of China (31672318).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- GIT
gastrointestinal tract
- PCR-DGGE
polymerase chain reaction-denaturing gradient gel electrophoresis
- Real-time PCR
real-time polymerase chain reaction
- OTUs
operational taxonomic units
- QIIME
quantitative insights into microbial ecology
- PCoA
principal coordinate analysis
Additional file
Additional file 1. Additional tables and figures.
Contributor Information
Yan Zeng, Email: zengyan88@stu.sicau.edu.cn.
Dong Zeng, Email: zend@sicau.edu.cn.
Xueqin Ni, Email: xueqinni@foxmail.com.
Hui Zhu, Email: 462591336@qq.com.
Ping Jian, Email: 1247872121@qq.com.
Yi Zhou, Email: 15283523916@163.com.
Shuai Xu, Email: 422262026@qq.com.
Yicen Lin, Email: 120213268@qq.com.
Yang Li, Email: 1292753853@qq.com.
Zhongqiong Yin, Email: yinzhongq@163.com.
Kangcheng Pan, Email: pankangcheng71@126.com.
Bo Jing, Email: jingbooo@163.com.
References
- 1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, Mccarthy S, Mcvean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alivisatos AP, Blaser MJ, Brodie EL, Chun M, Dangl JL, Donohue TJ, Dorrestein PC, Gilbert JA, Green JL, Jansson JK. MiICROBIOME. A unified initiative to harness Earth’s microbiomes. Science. 2015;350:507–508. doi: 10.1126/science.aac8480. [DOI] [PubMed] [Google Scholar]
- Armstrong DG, Smithard RR. The fate of carbohydrates in the small and large intestines of the ruminant. Proc Nutr Soc. 1979;3:283–294. doi: 10.1079/PNS19790050. [DOI] [PubMed] [Google Scholar]
- Arumugam M, Jeroen R, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin JJ, Sicheritzponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, Consortium M. Weissenbach J, Ehrlich SD, Bork P. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avguštin G, Wallace RJ, Flint HJ. Phenotypic diversity among ruminal isolates of Prevotella ruminicola: proposal of Prevotella brevis sp. nov., Prevotella bryantii sp. nov., and Prevotella albensis sp. nov. and redefinition of Prevotella ruminicola. Int J Syst Bacteriol. 1997;2:284–288. doi: 10.1099/00207713-47-2-284. [DOI] [PubMed] [Google Scholar]
- Barker CJ, Gillett A, Polkinghorne A, Timms P. Investigation of the koala (Phascolarctos cinereus) hindgut microbiome via 16S pyrosequencing. Vet Microbiol. 2013;167:554–564. doi: 10.1016/j.vetmic.2013.08.025. [DOI] [PubMed] [Google Scholar]
- Bekele AZ, Koike S, Kobayashi Y. Phylogenetic diversity and dietary association of rumen Treponema revealed using group-specific 16S rRNA gene- based analysis. FEMS Microbiol Lett. 2011;1:51–60. doi: 10.1111/j.1574-6968.2010.02191.x. [DOI] [PubMed] [Google Scholar]
- Braun U. Ultrasonography of the gastrointestinal tract in cattle. Vet Clin N Am Food Anim. 2009;3:567–590. doi: 10.1016/j.cvfa.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Callaway TR, Dowd SE, Edrington TS, Anderson RC, Krueger N, Bauer N, Kononoff PJ, Nisbet DJ. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci. 2010;12:3977–3983. doi: 10.2527/jas.2010-2900. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;5:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108(Supplement 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;8:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium HMP Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha IS, Barreto CC, Costa OY, Bomfim MA, Castro AP, Kruger RH, Quirino BF. Bacteria and Archaea community structure in the rumen microbiome of goats (Capra hircus) from the semiarid region of Brazil. Anaerobe. 2011;3:118–124. doi: 10.1016/j.anaerobe.2011.04.018. [DOI] [PubMed] [Google Scholar]
- de Oliveira MNV, Jewell KA, Freitas FS, Benjamin LA, Totola MR, Borges AC, Moraes LA, Suen G. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet Microbiol. 2013;3:307–314. doi: 10.1016/j.vetmic.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Donia MS, Cimermancic P, Schulze CJ, Wielandbrown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158:1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;16:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans NJ, Brown JM, Murray RD, Getty B, Birtles RJ, Hart CA, Carter SD. Characterization of novel bovine gastrointestinal tract Treponema isolates and comparison with bovine digital dermatitis treponemes. Appl Environ Microbiol. 2011;1:138–147. doi: 10.1128/AEM.00993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, Tito RY, Chaffron S, Rymenans L, Verspecht C, De Sutter L, Lima-Mendez G, D’hoe K, Jonckheere K, Homola D, Garcia R, Tigchelaar EF, Eeckhaudt L, Fu J, Henckaerts L, Zhernakova A, Wijmenga C, Raes J. Population-level analysis of gut microbiome variation. Science. 2016;352:560. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- Godoy-Vitorino F, Goldfarb KC, Karaoz U, Leal S, Garcia-Amado MA, Hugenholtz P, Tringe SG, Brodie EL, Dominguez-Bello MG. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012;3:531–541. doi: 10.1038/ismej.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblum S, Carr R, Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell. 2015;160:583–594. doi: 10.1016/j.cell.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Chen D, Zhang JN, Lv X, Wang K, Duan LP, Nie Y, Wu XL. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE. 2013;10:e74957. doi: 10.1371/journal.pone.0074957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Yang Y, Yan H, Wang X, Qu L, Chen Y. Rumen bacterial diversity of 80 to 110-day-old goats using 16S rRNA sequencing. PLoS ONE. 2015;2:e0117811. doi: 10.1371/journal.pone.0117811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS ONE. 2012;7:e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishaq SL, Wright ADG. Insight into the bacterial gut microbiome of the North American moose (Alces alces) BMC Microbiol. 2012;1:212. doi: 10.1186/1471-2180-12-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T, Wu C, Muzny DM, Li Y, Zhang W, Stanton JA, Brauning R, Barris WC, Hourlier T, Aken BL, Searle SM, Adelson DL, Bian C, Cam GR, Chen Y, Cheng S, DeSilva U, Dixen K, Dong Y, Fan G, Franklin IR, Fu S, Fuentes-Utrilla P, Guan R, Highland MA, Holder ME, Huang G, Ingham AB, Jhangiani SN, Kalra D, Kovar CL, Lee SL, Liu W, Liu X, Lu C, Lv T, Mathew T, McWilliam S, Menzies M, Pan S, Robelin D, Servin B, Townley D, Wang W, Wei B, White SN, Yang X, Ye C, Yue Y, Zeng P, Zhou Q, Hansen JB, Kristiansen K, Gibbs RA, Flicek P, Warkup CC, Jones HE, Oddy VH, Nicholas FW, McEwan JC, Kijas JW, Wang J, Worley KC, Archibald AL, Cockett N, Xu X, Wang W, Dalrymple BP. The sheep genome illuminates biology of the rumen and lipid metabolism. Science. 2014;6188:1168–1173. doi: 10.1126/science.1252806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao JJ, Wang PP, He ZX, Tang SS, Zhou CS, Han XF, Wang M, Wu DQ, Kang JH, Tan ZL. In vitro evaluation on neutral detergent fiber and cellulose digestion by post-ruminal microorganisms in goats. J Sci Food Agric. 2013;9:1745–1752. doi: 10.1002/jsfa.6485. [DOI] [PubMed] [Google Scholar]
- Kebreab E, Dijkstra J, Bannink A, France J. Recent advances in modeling nutrient utilization in ruminants. J Anim Sci. 2009;87(E.Suppl.):E111–E122. doi: 10.2527/jas.2008-1313. [DOI] [PubMed] [Google Scholar]
- Kittelmann S, Seedorf H, Walters WA, Clemente JC, Knight R, Gordon JI, Janssen PH. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE. 2013;2:1112–1126. doi: 10.1371/journal.pone.0047879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klieve AV. Rumen microbiology. Queensland: University of Queensland; 2005. [Google Scholar]
- Koboziev I, Webb CR, Furr KL, Grisham MB. Role of the enteric microbiota in intestinal homeostasis and inflammation. Free Radic Biol Med. 2014;68:122–133. doi: 10.1016/j.freeradbiomed.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput Biol. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Zaneveld J, Caporaso JG, Mcdonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Lee S, Sung J, Ko G. Analysis of human and animal fecal microbiota for microbial source tracking. ISME J. 2011;2:362–365. doi: 10.1038/ismej.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhou M, Adamowicz E, Basarab JA, Guan LL. Characterization of bovine ruminal epithelial bacterial communities using 16SrRNA sequencing, PCR-DGGE and qRT-PCR analysis. Vet Microbiol. 2012;1:72–80. doi: 10.1016/j.vetmic.2011.08.007. [DOI] [PubMed] [Google Scholar]
- Li RW, Connor EE, Li C, Baldwin VI, Ransom L, Sparks ME. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbial. 2012;1:129–139. doi: 10.1111/j.1462-2920.2011.02543.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Bian G, Zhu W, Mao S. High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front Microbiol. 2015;6:167. doi: 10.3389/fmicb.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdonald D, Clemente JC, Kuczynski J, Rideout JR, Stombaugh J, Wendel D, Wilke A, Huse S, Hufnagle J, Meyer F. The biological observation matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience. 2012;1:7. doi: 10.1186/2047-217X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;4:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgavi DP, Kelly WJ, Janssen PH, Attwood GT. Rumen microbial (meta) genomics and its application to ruminant production. Animal. 2013;7(s1):184–201. doi: 10.1017/S1751731112000419. [DOI] [PubMed] [Google Scholar]
- Nyonyo T, Shinkai T, Mitsumori M. Improved culturability of cellulolytic rumen bacteria and phylogenetic diversity of culturable cellulolytic and xylanolytic bacteria newly isolated from the bovine rumen. FEMS Microbiol Ecol. 2014;88:528–537. doi: 10.1111/1574-6941.12318. [DOI] [PubMed] [Google Scholar]
- Osborne JM, Dehority BA. Synergism in degradation and utilization of intact forage cellulose, hemicellulose, and pectin by three pure cultures of ruminal bacteria. Appl Environ Microbiol. 1989;9:2247–2250. doi: 10.1128/aem.55.9.2247-2250.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JKM, Jhala MK, Soni P, Shabir N, Pandya PR, Singh KM, Rank DN, Joshi CG. Molecular characterization and diversity of rumen bacterial flora in Indian goat by 16S rDNA sequencing. Vetscan. 2011;6:77–82. [Google Scholar]
- Peng S, Yin J, Liu X, Jia B, Chang Z, Lu H, Jiang N, Chen Q. First insights into the microbial diversity in the omasum and reticulum of bovine using Illumina sequencing. J Appl Genet. 2015 doi: 10.1007/s13353-014-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanad YM, JrG Closs, Kumar A, LeJeune JT, Rajashekara G. Molecular epidemiology and public health relevance of Campylobacter isolated from dairy cattle and European starlings in Ohio, USA. Foodborne Pathoq Dis. 2013;3:229–236. doi: 10.1089/fpd.2012.1293. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Horn DJV, Weber CF. Introducing mothur: open-source, platform- independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb. 2009;23:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence C, Wells WG, Smith CJ. Characterization of the primary starch utilization operon in the obligate anaerobe Bacteroides fragilis: regulation by carbon source and oxygen. J Bacterial. 2006;13:4663–4672. doi: 10.1128/JB.00125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela JP, Russell JL, Pope SM, Morton T, Bogart S, Reamer LA, Schapiro SJ, Hopkins WD. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoetkiattikul H, Mhuantong W, Laothanachareon T, Tangphatsornruang S, Pattarajinda V, Eurwilaichitr L, Champreda V. Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing. Curr Microbiol. 2013;2:130–137. doi: 10.1007/s00284-013-0336-3. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett C, Knight R, Gordon JI. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature. 2007;7164:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez-Vazquez I, Pérez-Rangel M, Tapia A, Buitrón G, Molina C, Hernández G, Amaya-Delgado L. Hydrogen and butanol production from native wheat straw by synthetic microbial consortia integrated by species of Enterococcus and Clostridium. Fuel. 2015;159:214–222. doi: 10.1016/j.fuel.2015.06.052. [DOI] [Google Scholar]
- Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol Rev. 2011;35(4):681–704. doi: 10.1111/j.1574-6976.2011.00270.x. [DOI] [PubMed] [Google Scholar]
- Wu S, Baldwin RLV, Li W, Li C, Connor EE, Li RW. The bacterial community composition of the bovine rumen detected using pyrosequencing of 16S rRNA genes. Metagenomics. 2012;1:1–11. doi: 10.4303/mg/235571. [DOI] [Google Scholar]
- Xu J, Gordon JI. Honor Thy Symbionts. Proc Natl Acad Sci USA. 2003;18:10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;5:808–813. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Zeng D, Zhang Y, Ni XQ, Tang YR, Zhu H, Wang HS, Yin ZQ, Pan KC, Jing B. Characterization of the cellulolytic bacteria communities along the gastrointestinal tract of Chinese Mongolian sheep by using PCR-DGGE and real-time PCR analysis. World J Microb Biot. 2015;7:1103–1113. doi: 10.1007/s11274-015-1860-z. [DOI] [PubMed] [Google Scholar]
- Zhang XL, He XL, Wu RTY, Sa RL. The origin, domestication, differentiation, and preservation of Mongolian sheep. Anim Husb Feed Sci. 2008;3:36–38. [Google Scholar]
- Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, Kleerebezem M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;7:1415–1426. doi: 10.1038/ismej.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]